Abstract
Celiac disease (CD) is an immune-mediated enteropathy triggered by the ingestion of gluten in genetically susceptible individuals. It is one of the most common lifelong disorders on a worldwide basis. Celiac enteropathy is the final consequence of an abnormal immune reaction, showing features of both an innate and an adaptive response to gluten prolamins. The clinical spectrum is wide, including cases with either typical intestinal or atypical extraintestinal features, and silent forms. The only available treatment consists in dietary exclusion of grains containing gluten. New pharmacological treatment are currently under scrutiny.
INTRODUCTION
Celiac disease (CD) is an autoimmune enteropathy triggered by the ingestion of gluten-containing grains in susceptible individuals [Citation1]. It is the result of the interplay between environmental and genetic factors. The gliadin and glutenin fractions of wheat gluten and similar alcohol-soluble proteins in other grains are the environmental factors responsible for the development of the intestinal damage. The genetic predisposition is related to HLA (human leucocyte antigen) class II genes: most of CD patients are HLA-DQ2 positive, and the remaining patients are usually HLA-DQ8 positive [Citation2]. These genes are estimated to explain some 40% of the disease heritability; the remaining 60% of the genetic susceptibility to CD is shared between an unknown number of non-HLA genes, each of which is estimated to contribute only a small risk [Citation3].
The typical intestinal damage is characterized by loss of absorptive villi and hyperplasia of the crypts, and it completely resolves upon elimination of gluten-containing grains from the patient's diet [Citation1].
EPIDEMIOLOGY
In the past, CD was considered a rare disorder, mostly affecting individuals of European origin, and usually characterized by onset during the first years of life. At that time, diagnosis was entirely based on the detection of typical gastro-intestinal symptoms and confirmation by the small intestinal biopsy. The availability of highly sensitive and specific serological tools, first the anti-gliadin (AGA) and later the anti-endomysium (EMA) and the anti-transglutaminase (tTG) antibodies, made it possible to evaluate the true prevalence of CD (number of affected persons in a population at a given time), showing an unsuspected frequency of clinically atypical or even silent forms of CD. Approximately 20 years ago, Italy was the birth land of the new “era” of CD epidemiology, the one based on serological screening of general population samples. In a sample of 17,201 healthy Italian students, it was shown that CD is much more common than previously thought, and that most atypical cases remained undiagnosed unless actively searched by serological screening. The overall prevalence of CD (including known CD cases) was 5.44 per 1000 or 1 in 184 subjects. The ratio of known (previously diagnosed) to undiagnosed CD cases was as high as 1 to 7 [Citation4].
A recently published large international, multicenter study investigated a wide population sample in four different European countries: on average, the overall prevalence of CD was 1%, with large variations between countries (2.0% in Finland, 1.2% in Italy, 0.9% in Northern Ireland, and 0.3% in Germany). This study confirmed that many CD cases would remain undetected without active serological screening [Citation5].
Similar rates have been reported from the US population (1:133) [Citation6] and from developed countries mostly populated by individuals of European origin, e.g., Australia and New Zealand [Citation7, 8]. The presence of CD is long established in many South American countries that are mostly populated by individuals of European origin. Among Brazilian blood donors, the prevalence of CD ranged between 1:681 [Citation9] and 1:214 [Citation10]. It is worth noting that studies on blood donors tend to underestimate the prevalence of CD, as these individuals represent the “healthiest” segment of the population and are mostly males (while CD is more common among women). In Argentina, Gomez et al. found an overall prevalence of 1 in 167 in 2000 adults involved in a prenuptial examination [Citation11].
CD is not only frequent in developed countries. Recent epidemiological studies performed in areas of the developing world show prevalence rates overlapping European figures, especially in North Africa (i.e., 0.53% in Egypt, 0.79% in Libya, and 0.6% in Tunisia) [Citation12–14], Middle East (i.e., 0.88% in Iran and 0.6% in Turkey) [Citation15, 16], and India (i.e., 0.7%) [Citation17].
This widespread diffusion is not surprising at all, given that causal factors, HLA predisposing genotypes (DQ2 and DQ8) and consumption of gluten-containing cereals, show a worldwide distribution [Citation18].
The Saharawi population of Arab-Berber origin living in Algeria has the highest prevalence of CD (5.6%) among all world populations [Citation19]. High levels of consanguinity, high frequencies of HLA-DQ2 [Citation20], and heavy gluten ingestion may potentially explain this finding in this population.
There are no data about the prevalence of CD in Sub-Saharan Africa; however, there are reasons to believe that this condition is not common in those countries, as: (a) staple cereals are mostly naturally gluten free (e.g., millet and rice), and (b) the HLA-related predisposing genes DQ2 and DQ8 are much less frequent in those areas than in Western countries.
Finally, there are only anecdotic reports of CD in the Far East countries. Given the low prevalence of HLA-related predisposing genes DQ2 and DQ8 and the low/absent gluten consumption, reduced disease prevalence should be expected in those populations.
The epidemiology of CD is efficiently conceptualized by the iceberg model [Citation1] (). The overall number of CD cases is the size of the iceberg, which is influenced not only by the frequency of the predisposing genotypes in the population but also by the pattern of gluten consumption. Typical CD cases are usually diagnosed because of suggestive complaints. They make up the visible part of the celiac iceberg, expressed by the incidence of the disease in quantitative terms. In developed countries, however, for each diagnosed case of CD, an average of five cases remain undiagnosed (the submerged part of the iceberg), usually because of atypical, minimal, or even absent complaints. These undiagnosed cases remain untreated, leaving individuals exposed to the risk of long-term complications, such as infertility, osteoporosis, or cancer. Currently, the best approach to improving the diagnostic rate is a process of case finding focused on at-risk groups, a procedure that minimizes costs and is ethically appropriate. An increased awareness of the many clinical faces of CD, coupled with a higher inclination for using blood tests, could efficiently uncover a large portion of the submerged CD iceberg. The primary care physician's office would provide the most natural setting for selective screening to first identify individuals at risk for CD who need referral for definitive diagnosis.
FIGURE 1 The celiac iceberg model.
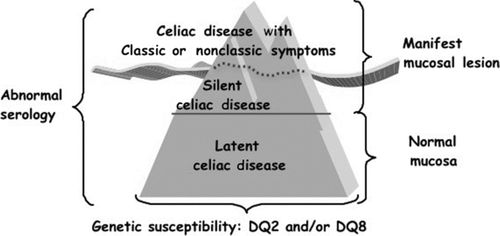
FIGURE 2 Prevalence of celiac disease in the US during the last decades [Citation22].
![FIGURE 2 Prevalence of celiac disease in the US during the last decades [Citation22].](/cms/asset/23d2b783-0f8a-4339-bd6a-a6f737d26aeb/iiri_a_602443_f0002_b.gif)
It is interesting to note that the prevalence of CD seems to be on the rise in developed countries. The total prevalence of CD has doubled in Finland during the last two decades (1.05% in 1978–80 and 1.99% in 2000–01) [Citation21], and the increase cannot simply be attributed to a better rate of detection. Recently, it has been shown that CD autoimmunity, within an American population followed since 1974, doubled between 1974 (1 in every 501 subjects) and 1989 (1 in every 219 subjects). This trend apparently continued in the following years. In a different sample of the adult American population in 2001, a CD prevalence of 1 in 105 subjects was reported. Therefore, during the last 30 years, the prevalence of CD among adults in the US appeared to increase by five-fold, doubling approximately every 15 years (), as shown by Catassi et al. (2010). Remarkably, this study showed that loss of gluten tolerance may occur at any time in life, for reasons that are currently unclear [Citation22]. The implications of this discovery could have a wide-ranging effect on the diagnosis and treatment of CD in elderly patients.
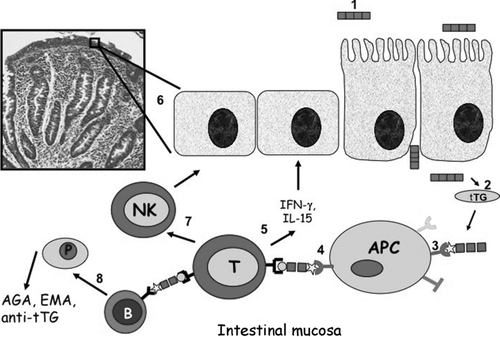
FIGURE 3 A simplified scheme of the adaptive immune response to gluten in celiac disease from gluten ingestion to intestinal damage. (1) Gluten peptides reach the lamina propria via either an increased epithelial tight junctional permeability or epithelial transcytosis. (2) Deamidation of gluten peptides by tissue transglutaminase creates potent immunostimulatory epitopes that (3) bind to HLA-DQ2 or HLA-DQ8 on antigen-presenting cells and (4) activate CD4+ T cells. (5) Activated CD4+ T cells secrete mainly Th1 cytokines, such as IFN-γ, which induces the release and activation of metalloproteases by myofibroblasts, (6) finally resulting in mucosal remodeling and villus atrophy. (7) CD4+ T cells also increase cytotoxicity of intraepithelial lymphocytes or natural killer (NK) T cells, thus leading to enterocyte apoptotic death. (8) Through the production of Th2 cytokines, activated CD4+ T cells also drive the activation and clonal expansion of B cells, which differentiate into plasma cells and produce antibodies to gluten and transglutaminase, contributing to the intestinal damage.
A steady rise in the incidence of autoimmune disorders as well as allergic disorders has been registered in industrialized countries during the last few decades. According to the “hygiene hypothesis,” the “cleaner” environment found nowadays in Western countries led to lower frequency of early childhood infections and differences in the spectrum of microorganisms populating the gut. These changes could modify the immune response and be responsible for higher risk of different autoimmune disorders [Citation23]. However, the rising prevalence of adult onset of CD that we observed in the US study can hardly be explained by hygienic changes occurring in childhood.
The amount and the quality of ingested gluten, the type and duration of wheat dough fermentation, the spectrum of intestinal microorganisms and how they change over time, intestinal infections, and stressors in general are all possible switches of the tolerance–immune response balance [Citation24–26]. However, more research is needed to determine whether and how these factors can cause loss of gluten tolerance. The results of these studies could be instrumental in determining how to prevent not only the onset of CD but also other autoimmune disorders.
PATHOGENESIS
The development of CD is determined by both environmental and genetic factors. In recent years, much has been discovered about the genetic and immunologic aspects of CD (). Under physiological circumstances, intestinal epithelia are almost impermeable to macromolecules such as gliadin. In CD, paracellular permeability is enhanced and the integrity of the tight junction (TJ) system is compromised. The upregulation of zonulin, an intestinal peptide involved in TJ regulation, appears to be responsible, at least in part, for the increased gut permeability characteristic of CD [Citation27]. After stimulation of normal rat intestinal cells with gliadin, zonulin is released, which induces a protein kinase C-mediated polymerization of intracellular actin filaments, which are directly connected to TJ complex proteins, thereby regulating epithelial permeability [Citation28]. Further, persistent presence of inflammatory mediators, such as tumor necrosis factor-α and interferon (IFN)-γ, has been shown to increase the permeability across the endothelial and epithelial layers, suggesting that the initial breach of the intestinal barrier function caused by zonulin can be perpetuated by the inflammatory process after the access of gliadin to the submucosa. Recent studies, which focused on the early effects of gliadin on the intestinal epithelial mucosa and the structures that dictate mucosal TJ competency, showed that gliadin activates the zonulin signaling, resulting in immediate reduction of intestinal barrier function and passage of gliadin into the subepithelial compartment [Citation29]. On the other hand, other studies suggest that gluten peptides can be transported across the intestinal epithelium via transcytosis [Citation30] or immunoglobulin A (IgA)-mediated retrotranscytosis [Citation31].
The main disease mechanism leading to CD is based on an adaptive immune response to gluten-derived peptides, taking place in the lamina propria of the intestinal mucosa. It is well established that gluten peptides presented by either HLA-DQ2 or HLA-DQ8 induce a CD4+ T-cell response in CD patients. Both HLA-DQ2 and HLA-DQ8 code for heterodimers located on antigen-presenting cells that preferentially bind peptides with negatively charged amino acids at anchor residues. Gluten peptides, however, are virtually devoid of negative charges, and native gluten peptides thus bind poorly to HLA-DQ2 or HLA-DQ8 [Citation32]. It has become clear that the enzyme tissue transglutaminase 2 (tTG2) can modify gluten peptides to fit the requirements for high-affinity binding to HLA-DQ2 and HLA-DQ8. tTG2 can convert noncharged glutamine into negatively charged glutamic acid, a process called deamidation [Citation33]. Recent studies have identified in the sequence motifs QXP (Q = glutamine, P = proline, X = any aminoacid), the glutamine residues that are the preferential substrate for tTG-mediated deamidation [Citation32, Citation34]. This represents an important tool for the prediction of toxic gliadin peptides. Similarly, this analysis might prove useful to screen wheat varieties to identify potential nontoxic grains.
Activated gluten-reactive CD4+ T cells produce high levels of pro-inflammatory cytokines, thus inducing a T-helper-cell–type-(Th)1 pattern dominated by IFN-γ [Citation35]. Th1 cytokines promote inflammatory effects, including fibroblast or lamina propria mononuclear cell secretion of matrix metalloproteinases, which are responsible for tissue remodeling that results in villus atrophy and crypt hyperplasia, which are characteristic of CD. Other cytokines such as interleukin (IL)-18, IFN-γ, and IL-21 seem to play a role in polarizing and maintaining the Th1 response [Citation36].
Additionally, through the production of Th2 cytokines, activated CD4+ T cells drive the activation and clonal expansion of B cells, which differentiate into plasmacells and produce anti-gliadin and anti-tTG antibodies. By interacting with the extracellular membrane-bound tTG, tTG–autoantibody deposits in the basement membrane region might induce enterocyte cytoskeleton changes with actin redistribution and consequent epithelial damage [Citation37].
In active CD, the number of CD8+ T-cell receptor (TCR)αβ+ and TCRγδ+ intraepithelial lymphocytes (IELs) is markedly increased. It is unclear whether this is a response to changes in the homeostasis of the epithelium or a consequence of the pro-inflammatory milieu created by the CD4+ T-cell response in the lamina propria [32]. IELs in the epithelium acquire activating natural killer (NK) receptors [Citation38] and the ability to lyse epithelial cells; at the same time, intestinal epithelial cells upregulate the expression of ligands for these activating receptors: the MICs [Citation39] and HLA-E [38], respectively. Therefore, by the Fas/Fas ligand system or the IL-15-induced perforin–granzyme and NFG2D–MIC signaling pathways, IELs induce the apoptotic death of epithelial cells, thus likely contributing to the typical tissue damage in CD [Citation40]. IL-15 secreted by epithelial cells and by dendritic cells could be the main factor orchestrating the selective expansion of IELs.
Indeed, gluten can also elicit an innate immune response. Upon stimulation with gliadin peptide p31-49 (and other peptides), epithelial cells, macrophages, and dendritic cells secrete IL-15, which in turn may stimulate cytotoxic lymphocytes, thus inducing increased epithelial apoptosis and permeability [Citation40–42]. It is currently unclear how gluten could have such a range of biological effects on innate immune cells and how it can bind to unrelated receptors; further study is therefore required to identify the molecular mechanisms involved. However, if confirmed, the possible role of gluten as an activator of the innate immune system might explain how inflammatory gluten-specific CD4+ T-cell responses are induced and how IELs become licensed to kill intestinal epithelial cells. Based on the current data, it has been suggested that in genetically susceptible individuals, gluten leads to the activation of cellular stress pathways or to the conversion of self molecules into ligands for immune receptors (such as Toll-like receptors), which in turn could trigger the release of proinflammatory mediators that promote the development of inflammatory T-cell responses [40].
CLINICAL PRESENTATION
The clinical manifestations of CD vary greatly, and although this was once perceived as a purely pediatric disorder, the diagnosis is increasingly made in adult life, although many adults are still misdiagnosed as having irritable bowel syndrome or other gastrointestinal syndromes ().
Table 1 Clinical manifestations of celiac disease (CD).
Typical CD
This form is characterized by villous atrophy and typical symptoms of intestinal malabsorption. It presents between 6 and 24 months of age with impaired growth, abnormal stools, abdominal distension, muscle wasting and hypotonia, poor appetite, and unhappy behavior. In developing countries, chronic diarrhea, abdominal distension, stunting (height for age lower than 2 standard deviations), and anemia are frequent findings [Citation1].
Atypical CD
Atypical CD is usually seen in older children and adults and features of overt malabsorption are absent. Intestinal features may be absent or include unusual complaints, such as recurrent abdominal pain, nausea, vomiting, bloating, dental enamel defects [Citation43], and recurrent aphthous stomatitis [Citation44]. Isolated increase in serum aminotransferase level caused by mild, nonprogressive liver inflammation is a common presentation in children [Citation45]. Several extraintestinal manifestations of the disease, alone or in association, have been described so far, potentially affecting any organ or body system.
Anemia is a frequent finding in patients with CD and may be the presenting feature; its prevalence varies greatly according to different reports and has been found in 12–69% of newly diagnosed patients with CD [Citation46]. Iron deficiency anemia is the most common in the setting of CD and has been reported in up to 46% of cases of subclinical CD, with a higher prevalence in adults than in children. Iron is absorbed in the proximal small intestine and the absorption is dependent upon several factors, including an intact mucosal surface. Iron deficiency is typically refractory to oral iron therapy and can be the sole manifestation of CD, especially in pediatric patients [Citation46]. Between 2% and 6% of patients with iron deficiency anemia attending a hematology clinic are found to have CD [Citation47]. The treatment of iron deficiency anemia associated with CD is primarily a gluten-free diet (GFD) and iron supplementation until the iron stores have been restored. The anemia seen in CD can also result from malabsorption of vitamin B12 (a small proportion of which is absorbed passively along the entire small bowel) and folic acid (absorbed in the jejunum) [Citation46].
Dermatitis herpetiformis is currently regarded as a variant of CD (“skin CD”). It is a blistering skin disease characterized by typical granular IgA deposition at the dermoepidermal junction, with stippling in the dermal papillae. The primary lesion consists of erythematous papules or vesicles; grouping of these vesicles in a herpetiform configuration may sometimes be observed. These cutaneous eruptions are usually symmetrically distributed over extensor surfaces, especially the elbows, knees, shoulders, sacrum, buttocks, and posterior nuchal area. In some cases, lesions may also involve the scalp, face, and groin. Generally, lesions heal without scarring unless secondary bacterial infection has set in. Postinflammatory pigmentary changes do occur. Suppressive therapy and a GFD should be considered to be complementary. The institution of a dietary therapy then provides long-term control of the underlying gluten sensitivity. Intermittent dapsone therapy could be used to suppress occasional outbreaks caused by lapses of the diet [Citation48].
Short stature can be the primary manifestation in an otherwise healthy child. It has recently been established that in 2–8% of children with short stature and no gastrointestinal symptoms, CD may be the underlying cause, being the most common organic cause of slow height velocity, far more common than growth hormone (GH) deficiency [Citation49]. The endocrinological pattern usually includes delayed bone age, either normal or blunted GH response to stimulatory tests, and low levels of insulin-like growth factor-1 (IGF-1). The pathogenesis of CD-associated short stature is still unclear: zinc deficiency is the basic nutritional deficit that impairs the production of IGF-1; together with other deficiencies characteristically found in patients with CD, it is deleterious for bone metabolism and growth [Citation50]. The impaired pituitary release of GH may be related to malnutrition, to the presence of circulating anti-pituitary antibodies in the central nervous system [Citation51], or to an abnormal brain monoamine metabolism [Citation50]. Treatment with a GFD often leads to complete catch-up growth within 2–3 years. If no catch-up growth occurs after 12 months of GFD, an associated and transient GH deficiency should be suspected [Citation52].
CD predisposes to low bone mineral density and osteoporosis. Of 86 consecutive patients with newly diagnosed, biopsy-confirmed CD, 40% had osteopenia and 26% osteoporosis [Citation53]. The recorded prevalence of CD among osteoporotic individuals is 3.4% [Citation54]. Bone alterations were once thought to derive from calcium and vitamin D deficiency secondary to intestinal malabsorption. Recently, other causes of bone metabolism impairment have been claimed, including the interaction between cytokines and local/systemic factors influencing bone formation and reabsorption. A lifelong GFD is the only effective measure to restore bone metabolism to an apparent normality. In the pediatric population, a prompt enforcement of a GFD can even lead to a satisfactory recovery of the bone mass. Contrary to pediatric cases, adults affected by osteoporosis secondary to CD do not experience spontaneous recovery, and there are no conclusive data on the efficacy of standard therapies for osteoporosis in reducing the fracture risk. This evidence stresses the need of a timely diagnosis as a preventive intervention to avoid CD complications [Citation55].
A growing number of studies have reported a wide spectrum of neurological conditions associated with CD, with an estimated prevalence in adults as high as 26% [Citation56]. This literature has become quite controversial because of variable selection criteria and patient characteristics. A recent meta-analysis indicates that CD patients have a risk of developing some neurological complications during childhood (i.e., headache, peripheral neuropathy, and white matter disease) [Citation57]. The overall prevalence of neurological involvement in childhood, as compared with adulthood, is lower. Actually, there is no compelling evidence that there is a casual relation between CD and epilepsy, autism, or cerebellar ataxia in the pediatric age group [Citation57].
Women with CD more frequently experience recurrent spontaneous miscarriage, early menopause, and amenorrhoea. Fertility problems can also be present in male patients. The real mechanism by which CD produces these changes is unclear, but such factors as malnutrition and iron, folate, and zinc deficiencies have all been implicated [Citation37].
Silent CD
Since the introduction of serological tests, silent (apparently symptomless) CD has been increasingly recognized because of occasional screening. This is often the case in subjects with a family history of CD, patients with associated autoimmune (e.g., type 1 diabetes) or genetic (Down, Turner, or Williams syndrome) disorders. A thorough history and investigation will, however, reveal a low-grade illness in many of these individuals. Common features are: (a) behavioral disturbances, such as irritability and impaired school performance; (b) impaired physical fitness and chronic fatigue; (c) iron deficiency with or without anemia; and (d) reduced bone mineral density. An improved psycho-physical well-being is often reported in children affected with apparently silent CD after starting treatment with a GFD [Citation1].
Potential CD
Potential CD is characterized by a normal intestinal mucosa or by subtle histological abnormalities, such as an increased number of IEL (type 1 lesion). Such patients are positive for anti-tTG antibodies and/or EMA and/or subepithelial deposits of anti-tTG IgA at the biopsy. They can either be well or have intestinal symptoms, which may respond to a GFD. In time, they may develop a flat mucosa, although there is no evidence to support managing these patients with a GFD until unequivocal mucosal flattening is recorded [Citation37].
Associated Diseases
CD prevalence is increased in at-risk conditions, such as autoimmune diseases, IgA deficiency, and some genetic syndromes (Down, Turner, and Williams syndrome). The average prevalence of CD among children with type 1 diabetes mellitus is 4.5% [Citation58]. A six- to seven-fold increase in CD prevalence has been reported in subjects with autoimmune thyroid disease [Citation59]. In children affected with Down syndrome, the reported prevalence of CD ranges between 3.2% and 10.3% [Citation60].
Given the wide variability in the clinical spectrum, a high index of clinical suspicion is essential to identify children with CD. Once the possibility has been recognized, tests should be carried out to confirm the diagnosis. This case-finding procedure is currently advocated as the most cost-effective approach to detect undiagnosed CD.
TREATMENT
The only currently available treatment for CD consists in dietary exclusion of grains containing gluten (i.e., wheat, rye, barley, triticale, couscous, spelt, and kamut) [Citation1]. Rice, maize, and buckwheat do not contain gluten and can be eaten. Potato, chestnut, tapioca, sorghum, millet teff, quinoa, and amaranth are other safe starchy food. In the past, oats were considered to be toxic to individuals with CD. Many recent studies, however, have shown that the ingestion of uncontamineated oats is not only safe but can also improve the quality of the diet in the majority of patients with CD or dermatitis herpetiformis [Citation61]. Other natural foods such as vegetables, salads, pulses, fruits, nuts, meat, fish, poultry, cheese, eggs, and milk can be consumed in a GFD without limitations. A wide range of attractive and palatable gluten-free products that guarantee the absence of gluten are specifically manufactured for celiac patients and may be labeled by an internationally recognized mark, the crossed ear of wheat. In many areas of the world, including Europe, America, Australasia, and North Africa, gluten-rich products such as bread and pasta are part of the staple diet. Gluten-containing foods make a substantial contribution to daily energy intake and are enjoyable to eat. The changes needed to begin and maintain a GFD are important and have a major impact on daily life. Supportive nutritional care in the case of iron, calcium, and vitamin deficiencies is also recommended [Citation62]. In the long term (1–2 years), a GFD is associated with clinical, serologic, and histological remission and seems to reduce the risk of complications [1]. However, it is almost impossible to maintain a diet with zero gluten content because gluten contamination is very common in food. “Hidden” gluten (used as protein filler) may be found in commercially available products, such as sausages, soups, soy sauces, and ice cream. Even products specifically targeted to dietary treatment of CD may contain tiny amounts of gluten proteins, either because of the cross-contamination of originally gluten-free cereals during their milling, storage, and manipulation or because of the presence of wheat starch as a major ingredient [Citation63]. It has been recently demonstrated in multicenter, double-blind, placebo-controlled trial that the safety threshold of prolonged exposure to trace amounts of gluten (i.e., contaminating gluten) is 50 mg/day, and that the threshold of 20 ppm (parts per million) keeps the intake of gluten from “special celiac food” well below the amount of 50 mg/day, which allows a safety margin for the variable gluten sensitivities and dietary habits of patients [Citation63].
As previously noted, both in-vivo challenges and in-vitro immunologic studies support the possibility that oats (once considered toxic for CD patients) can be ingested safely [63]. However, because of uncontrolled harvesting and milling procedures, cross-contamination of oats with gluten is a concern [Citation1].
A GFD is, often, difficult to sustain, owing to small levels of gluten contamination in food products, high costs and restricted availability of gluten-free food alternatives, low palatability, and cultural practices, leading to a substantial social burden. Moreover, in a minority of adult patients with so-called “refractory CD,” the disease does not respond to treatment with a GFD. Therefore, in the past decade, researchers have become increasingly interested in therapeutic alternatives for continuous or intermittent use of a GFD in patients with CD [Citation64].
Newly developed treatment modalities for CD are based on currently available insights into the pathogenesis of the disease () [Citation65–88]. These therapies focus on engineering gluten-free grains, degradation of immunodominant gliadin peptides that resist intestinal proteases by exogenous endopeptidases, decrease in intestinal permeability by blockage of the epithelial zonuline receptor, inhibition of intestinal tTG2 activity by transglutaminase inhibitors, inhibition of gluten peptide presentation by HLA-DQ2 antagonists, modulation or inhibition of proinflammatory cytokines, and induction of oral tolerance to gluten [Citation65–88].
Table 2 Potential novel therapies for celiac disease.
Although some of these goals are in an advanced state of development (i.e., engineering gluten-free grains), some are already in phase 1 or 2 clinical trials, and others are extremely challenging and will require an international task force to generate meaningful data.
In order to achieve the ambitious goal of CD prevention, future directions in CD research should be aimed to understand the role of environmental factors, such as the type, the amount, and the age of gluten introduction, the pattern of breast-milking, and finally, the potential role of intestinal microbiota [Citation89].
For the time being, strict adherence to a GFD should be advised for all patients with CD, as it remains the only effective and safe therapy.
Declaration of Interest
Elena Lionetti reports no conflict of interest. Carlo Catassi has served as a consultant for Menarini Diagnostics and Schär. The authors alone are responsible for the content and writing of the paper.
REFERENCES
- Fasano A, Catassi C. Current approaches to diagnosis and treatment of celiac disease: an evolving spectrum. Gastroenterology. 2001;120:636–651.
- Wolters VM, Wijmenga C. Genetic background of celiac disease and its clinical implications. Am J Gastroenterol. 2008;103:190–195.
- Romanos J, van Diemen CC, Nolte IM, Analysis of HLA and non-HLA alleles can identify individuals at high risk for celiac disease. Gastroenterology. 2009;137:834–840.
- Catassi C, Fabiani E, Rätsch IM, The coeliac iceberg in Italy. A multicentre antigliadin antibodies screening for coeliac disease in school-age subjects. Acta Paediatr Suppl. 1996;412:29–35.
- Mustalahti K, Catassi C, Reunanen A, The prevalence of CD in Europe: results of a centralized, international mass screening project. Ann Med. 2010;42:587–595.
- Fasano A, Berti I, Gerarduzzi T, Prevalence of CD in at-risk and non at-risk groups: a large, multicentre study. Arch Intern Med. 2003;163:286–292.
- Hovell CJ, Collett JA, Vautier G, High prevalence of coeliac disease in a population-based study from Western Australia: a case for screening? Med J Aust. 2001;175:247–250.
- Cook HB, Burt MJ, Collett JA, Whitehead MR, Frampton CM, Chapman BA. Adult coeliac disease: prevalence and clinical significance. J Gastroenterol Hepatol. 2000;15:1032–1036.
- Gandolfi L, Pratesi R, Cordoba JC, Tauil PL, Gasparin M, Catassi C. Prevalence of CD among blood donors in Brazil. Am J Gastroenterol. 2000;95:689–692.
- Oliveira RP, Sdepanian VL, Barreto JA, High prevalence of CD in Brazilian blood donor volunteers based on screening by IgA antitissue transglutaminase antibody. Eur J Gastroenterol Hepatol. 2007;19:43–49.
- Gomez JC, Selvaggio GS, Viola M, Pizarro B, la Motta G, de Barrio S. Prevalence of CD in Argentina: screening of an adult population in the La Plata area. Am J Gastroenterol. 2001;96:2700–2704.
- Abu-Zekry M, Kryszak D, Diab M, Catassi C, Fasano A. Prevalence of CD in Egyptian children disputes the east-west agriculture-dependent spread of the disease. J Pediatr Gastroenterol Nutr. 2008;47:136–140.
- Alarida K, Harown J, Ahmaida A, Coeliac disease in Libyan children: a screening study based on the rapid determination of anti-transglutaminase antibodies. Dig Liver Dis. 2011 (February 8) [Epub ahead of print].
- Ben Hariz M, Kallel-Sellami M, Kallel L, Prevalence of CD in Tunisia: mass screening study in schoolchildren. Eur J Gastroenterol Hepatol. 2007;19:687–694.
- Imanzadeh F, Sayyari AA, Yaghoobi M, Akbari MR, Shafagh H, Farsar AR. CD in children with diarrhea is more frequent than previously suspected. J Pediatr Gastroenterol Nutr. 2005;40:309–311.
- Ertekin V, Selimoğlu MA, Kardaş F, Aktaş E. Prevalence of CD in Turkish children. J Clin Gastroenterol. 2005;39:689–691.
- Sood A, Midha V, Sood N, Avasthi G, Sehgal A. Prevalence of CD among school children in Punjab, North India. J Gastroenterol Hepatol. 2006;21:1622–1625.
- Catassi C, Cobellis G. Coeliac disease epidemiology is alive and kicking, especially in the developing world. Dig Liver Dis. 2007;39:908–910.
- Catassi C, Rätsch IM, Gandolfi L, Why is coeliac disease endemic in the people of Sahara? Lancet. 1999;354:647–648.
- Catassi C, Doloretta Macis M, Rätsch IM, De Virgilis S, Cucca F. The distribution of DQ genes in the Saharawi population provides only a partial explanation for the high CD prevalence. Tissue Antigens. 2001;58:402–406.
- Lohi S, Mustalahti K, Kaukinen K, Increasing prevalence of coeliac disease over time. Aliment Pharmacol Ther. 2007;26:1217–1225.
- Catassi C, Kryszak D, Bhatti B, Natural history of celiac disease autoimmunity in a USA cohort followed since 1974. Ann Med. 2010;42:530–538.
- Bach JF. The effect of infections on susceptibility to autoimmune and allergic diseases. N Engl J Med. 2002;347:911–920.
- Ivarsson A, Persson LA, Nyströ ML, Epidemic of coeliac disease in Swedish children. Acta Paediatr. 2000;89:165–171.
- Gobbetti M, Rizzello GM, Di Cagno R, De Angelis M. Sourdough lactobacilli and celiac disease. Food Microbiol. 2007;24:187–196.
- Zanoni G, Navone R, Lunari C, In celiac disease, a subset of autoantibodies against transglutaminase binds toll-like receptor 4 and induces activation of monocytes. PLoS Med. 2006;3:e358.
- Fasano A. Zonulin and its regulation of intestinal barrier function: the biological door to inflammation, autoimmunity, and cancer. Physiol Rev. 2011;91:151–175.
- Clemente MG, De Virgiliis S, Kang JS, Early effects of gliadin on enterocyte intracellular signalling involved in intestinal barrier function. Gut. 2003;52:218–223.
- Drago S, El Asmar R, Di Pierro M, Gliadin, zonulin and gut permeability: effects on celiac and non-celiac intestinal mucosa and intestinal cell lines. Scand J Gastroenterol. 2006;41:408–419.
- Schumann M, Richter JF, Wedell I, Mechanisms of epithelial translocation of the alpha(2)-gliadin-33mer in coeliac sprue. Gut. 2008;57:747–754.
- Matysiak-Budnik T, Moura IC, Arcos-Fajardo M, Secretory IgA mediates retrotranscytosis of intact gliadin peptides via the transferrin receptor in CD. J Exp Med. 2008;205:143–154.
- Tjon JM, van Bergen J, Koning F. CD: How complicated can it get? Immunogenetics. 2010;62: 641–645.
- Molberg O, McAdam SN, Korner R, Tissue transglutaminase selectively modifies gliadin peptides that are recognized by gut-derived T cells in celiac disease. Nat Med. 1998;4:713–717.
- Vader LW, De RA, van der Wal Y, Specificity of tissue transglutaminase explains cereal toxicity in celiac disease. J Exp Med. 2002;195:643–649.
- Nilsen EM, Jahnsen FL, Lundin KE, Gluten induces an intestinal cytokine response strongly dominated by interferon gamma in patients with CD. Gastroenterology. 1998;115:551–563.
- Schuppan D, Junker Y, Barisani D. CD: From pathogenesis to novel therapies. Gastroenterology. 2009;137:1912–1933.
- Di Sabatino A, Corazza GR. Coeliac disease. Lancet. 2009;373:1480–1493.
- Meresse B, Curran SA, Ciszewski C, Reprogramming of CTLs into natural killer-like cells in CD. J Exp Med. 2006;203:1343–1355.
- Meresse B, Chen Z, Ciszewski C, Coordinated induction by IL15 of a TCR-independent NKG2D signaling pathway converts CTL into lymphokine-activated killer cells in CD. Immunity. 2004;21:357–366.
- Jabri B, Sollid LM. Tissue-mediated control of immunopathology in coeliac disease. Nat Rev Immunol. 2009;9:858–870.
- Maiuri L, Ciacci C, Ricciardelli I, Association between innate response to gliadin and activation of pathogenic T cells in coeliac disease. Lancet. 2003;362:30–37.
- Londei M, Ciacci C, Ricciardelli I, Gliadin as a stimulator of innate responses in celiac disease. Mol Immunol. 2005;42:913–918.
- Wierink CD, van Diermen DE, Aartman IH, Heymans HS. Dental enamel defects in children with coeliac disease. Int J Paediatr Dent. 2007;17:163–168.
- Bucci P, Carile F, Sangianantoni A, D'Angiò F, Santarelli A, Lo Muzio L. Oral aphthous ulcers and dental enamel defects in children with coeliac disease. Acta Paediatr. 2006;95:203–207.
- Rubio-Tapia A, Murray JA. The liver in celiac disease. Hepatology. 2007;46:1650–1658.
- Halfdanarson TR, Litzow MR, Murray JA. Hematologic manifestations of celiac disease. Blood. 2007;109:412–421.
- Hershko C, Patz J. Ironing out the mechanism of anemia in celiac disease. Haematologica. 2008;93:1761–1765.
- Nicolas ME, Krause PK, Gibson LE, Murray JA. Dermatitis herpetiformis. Int J Dermatol. 2003;42: 588–600.
- van Rijn JC, Grote FK, Oostdijk W, Wit JM. Short stature and the probability of coeliac disease, in the absence of gastrointestinal symptoms. Arch Dis Child. 2004;89:882–883.
- Fasano A, Counts D. Commentary on “anti-pituitary antibodies in children with newly diagnosed celiac disease: a novel finding contributing to linear growth”. Am J Gastroenterol. 2010;105:697–698.
- Delvecchio M, De Bellis A, Francavilla R, Anti-pituitary antibodies in children with newly diagnosed celiac disease: a novel finding contributing to linear-growth impairment. Am J Gastroenterol. 2010;105:691–696.
- Gadewar S, Fasano A. Celiac disease: is the atypical really typical? Summary of the recent National Institutes of Health Consensus Conference and latest advances. Curr Gastroenterol Rep. 2005;7:455–461.
- Mora S. Celiac disease in children: impact on bone health. Rev Endocr Metab Disord. 2008;9:123–130.
- Stenson WF, Newberry R, Lorenz R, Baldus C, Civitelli R. Increased prevalence of celiac disease and need for routine screening among patients with osteoporosis. Arch Intern Med. 2005;165:393–399.
- Fasano A, Catassi C. Coeliac disease in children. Best Pract Res Clin Gastroenterol. 2005;19:467–468.
- Bushara KO. Neurologic presentation of CD. Gastroenterology. 2005;128:S92–S97.
- Lionetti E, Francavilla R, Pavone P, The neurology of coeliac disease in childhood: what is the evidence? A systematic review and meta-analysis. Dev Med Child Neurol. 2010;52:700–707.
- Holmes GKT. Screening for coeliac disease in type 1 diabetes. Arch Dis Child. 2002; 87:495–499.
- Larizza D, Calcaterra V, De Giacomo C, Coeliac disease in children with autoimmune thyroid disease. J Pediatr. 2001;139:738–740.
- Book L, Hart A, Black J, Prevalence and clinical characteristics of CD in Down's syndrome in a U.S. study. Am J Med Gen. 2001;98:70–74.
- Haboubi NY, Taylor S, Jones S. Coeliac disease and oats: a systematic review. Postgrad Med J. 2006;82:672–678.
- Hopman EG, Le Cessie S, von Blomberg BM, Mearin ML. Nutritional management of the gluten-free diet in young people with CD in the Netherlands. J Pediatr Gastroenterol Nutr. 2006;43:102–108.
- Catassi C, Fabiani E, Iacono G, A prospective, double-blind, placebo-controlled trial to establish a safe gluten threshold for patients with celiac disease. Am J Clin Nutr. 2007;85:160–166.
- Tack GJ, Verbeek WH, Schreurs MW, Mulder CJ. The spectrum of celiac disease: epidemiology, clinical aspects and treatment. Nat Rev Gastroenterol Hepatol. 2010;7:204–213.
- Schuppan D, Junker Y, Barisani D. Celiac disease: from pathogenesis to novel therapies. Gastroenterology. 2009;137:1912–1933.
- Lerner A. New therapeutic strategies for celiac disease. Autoimmun Rev. 2010;9:144–147.
- Frisoni M, Corazza GR, Lafiandra D, Wheat deficient in gliadins: Promising tool for treatment of coeliac disease. Gut. 1995;36:375–378.
- Spaenij-Dekking L, Kooy-Winkelaar Y, van Veelen P, Natural variation in toxicity of wheat: potential for selection of nontoxic varieties for celiac disease patients. Gastroenterology. 2005;129:797–806.
- Pizzuti D, Buda A, D'Odorico A, Lack of intestinal mucosal toxicity of Triticum monococcum in celiac disease patients. Scand J Gastroenterol. 2006;41:1305–1311.
- Gianfrani C, Siciliano RA, Facchiano AM, Transamidation of wheat flour inhibits the response to gliadin of intestinal T cells in celiac disease. Gastroenterology. 2007;133:780–789.
- Di Cagno R, De Angelis M, Auricchio S, Sourdough bread made from wheat and nontoxic flours and started with selected lactobacilli is tolerated in celiac sprue patients. Appl Environ Microbiol. 2004;70:1088–1096.
- Stepniak D, Spaenij-Dekking L, Mitea C, Highly efficient gluten degradation with a newly identified prolyl endoprotease: implications for celiac disease. Am J Physiol Gastrointest Liver Physiol. 2006;291:621–629.
- Mitea C, Havenaar R, Drijfhout JW, Efficient degradation of gluten by a prolyl endoprotease in a gastrointestinal model: implications for coeliac disease. Gut. 2008;57:25–32.
- Gass J, Bethune MT, Siegel M, Combination enzyme therapy for gastric digestion of dietary gluten in patients with celiac sprue. Gastroenterology. 2007;133:472–480.
- Pinier M, Verdu EF, Nasser-Eddine M, Polymeric binders suppress gliadin-induced toxicity in the intestinal epithelium. Gastroenterology. 2009;136:288–298.
- Kelly CP, Green PH, Murray JA, Intestinal permeability of larazotide acetate in celiac disease: results of a phase IIB 6-week gluten-challenge clinical trial [abstract]. Gastroenterology. 2009;136(Suppl 1):A474.
- Molberg O, McAdam S, Lundin KE, T cells from celiac disease lesions recognize gliadin epitopes deamidated in situ by endogenous tissue transglutaminase. Eur J Immunol. 2001;31:1317–1323.
- Maiuri L, Ciacci C, Ricciardelli I, Unexpected role of surface transglutaminase type II in celiac disease. Gastroenterology. 2005;129:1400–1413.
- Kapoerchan VV, Wiesner M, Overhand M, Design of azidoproline containing gluten peptides to suppress CD4+ T-cell responses associated with celiac disease. Bioorg Med Chem. 2008;16:2053–2062.
- Xia J, Siegel M, Bergseng E, Inhibition of HLA-DQ2-mediated antigen presentation by analogues of a high affinity 33-residue peptide from alpha2-gliadin. J Am Chem Soc. 2006;128:1859–1867.
- Siegel M, Xia J, Khosla C. Structure-based design of alphaamido aldehyde containing gluten peptide analogues as modulators of HLA-DQ2 and transglutaminase 2. Bioorg Med Chem. 2007;15: 6253–6261.
- Xia J, Bergseng E, Fleckenstein B, Cyclic and dimeric gluten peptide analogues inhibiting DQ2-mediated antigen presentation in celiac disease. Bioorg Med Chem. 2007;15:6565–6573.
- Keech CL, Dromey J, Chen Z, Immune tolerance induced by peptide immunotherapy in an HLA-DQ2-dependent mouse model of gluten immunity. Gastroenterology. 2009;136:A355.
- Baslund B, Tvede N, Danneskiold-Samsoe B, Targeting interleukin-15 in patients with rheumatoid arthritis: a proof-of-concept study. Arthritis Rheum. 2005;52:2686–2692.
- Hommes DW, Mikhajlova TL, Stoinov S, Fontolizumab, a humanised anti-interferon gamma antibody, demonstrates safety and clinical activity in patients with moderate to severe Crohn's disease. Gut. 2006;55(8):1131–1137.
- Costantino G, della Torre A, Lo Presti MA, Treatment of life-threatening type I refractory coeliac disease with long-term infliximab. Dig Liver Dis. 2008;40:74–77.
- Vivas S, Ruiz de Morales JM, Ramos F, Alemtuzumab for refractory celiac disease in a patient at risk for enteropathy-associated T-cell lymphoma. N Engl J Med. 2006;354:2514–2515.
- Salvati VM, Mazzarella G, Gianfrani C, Recombinant human interleukin 10 suppresses gliadin dependent T cell activation in ex vivo cultured coeliac intestinal mucosa. Gut. 2005;54:46–53.
- Silano M, Agostoni C, Guandalini S. Effect of the timing of gluten introduction on the development of celiac disease. World J Gastroenterol. 2010;16:1939–1942.