Abstract
Most of the cases of non-fulminant hepatitis A carry good renal and overall prognosis. Here we present a case of acute renal failure associated with acute non-fulminant hepatitis A, for which kidney biopsy showed interstitial nephritis and tubular necrosis. We also review the literature and possible pathogenesis.
INTRODUCTION
Renal involvement is well known in association with viral hepatitis B and C infection, but rarely recognized with hepatitis A virus (HAV). In 1978, S. P. Wilkinson first described acute renal failure associated with non-fulminant hepatitis A.Citation[1] Since then, more and more cases of non-fulminant hepatitis A with renal involvement have been reported. Usually identified as a mild, self-limited disease lacking extrahepatic involvement, HAV has recently been shown to have the potential to cause a broad spectrum of systemic complications, ranging from arthritis, vasculitis, and cryoglobulinemia to acute renal failure, fulminant hepatic failure, and death. Described here is a case of acute non-fulminant hepatitis A complicated by acute renal failure.
CASE REPORT
A 36-year-old previously healthy male presented to a local hospital in the Philippines with intermittent fever up to 41°C for four days. The fever subsided after he was admitted to the hospital. For the next four days, he experienced progressive icteric skin, mild right upper quadrant dull pain, anorexia, nausea, vomiting, and general weakness. He denied headache, neck stiffness, sore throat, cough, chest pain, diarrhea, dysuria, gross hematuria, decreased urine output, flank pain, retroorbital pain, myalgia, arthralgia, or skin rash. He is Taiwanese, and has been living in Subic Bay in the Philippines for three to four years. He had eaten local seafood several days prior to admission and denied intravenous drug use, previous blood transfusions, tattooing, homosexual activity, exposure to non-steroidal anti-inflammatory drugs or aminoglycosides or contrast medium, or insect bites. Dengue fever was suspected, and he was then referred to Chang Gung Memorial Hospital.
Upon admission, his temperature was 35.9°C, pulse rate 70 bpm, respiratory rate 15/min, and blood pressure 149/98 mmHg. A physical examination revealed icteric skin and sclera, bilateral lung basal crackles, and bilateral pretibial grade I pitting edema. Other physical findings were unremarkable. The white blood cell count was 6,900/uL, hemoglobin 13.2 g/dL, and platelet count 234,000/uL. Biochemistry tests showed blood urine nitrogen (BUN) 77 mg/dL, creatinine (Cr) 17.98 mg/dL, serum sodium 136 meq/L, serum potassium 4.5 meq/L, total bilirubin 8.3 mg/dL, direct bilirubin 5.6 mg/dL, aspartate aminotransferase (AST) 112 U/L, alanine aminotransferase (ALT) 990 U/L, alkaline phosphatase (ALK-P) 23 U/L, and serum albumin/globulin 2.7/5.0 g/dL. The prothrombin time was mildly prolonged (13.5 s, INR 1.3). Arterial blood gas showed metabolic acidosis with respiratory compensation (pH 7.379, PCO2 32.5, PO2 96.6, HCO3 18.7, Sat 97.3%). Urinary analysis showed microscopic hematuria (blood 2+, RBC 24/uL), pyuria (WBC 38/uL), but no proteinuria. The urine sodium was 88 meq/L. Serologic tests were positive for anti-HAV IgM (4.72/1.2/ABBOTT) and negative for HBsAg, anti-HBs Ab, anti-HBc IgM, and anti-HCV Ab. Serum myoglobin and creatine phosphokinase were within normal limits. Elevated serum n IgG 2150 mL/dL, IgA 437 mL/dL, and IgE 373 IU/mL were found. Serum protein electrophoresis showed a faint band of IgM-kappa. Serum C3 and C4 were within normal limits. Cryoglobulin was positive (IgG 1+, IgA1+, IgM 1+). P-ANCA and C-ANCA, ANA, and RF were within normal ranges, and the blood culture was negative. Abdominal sonography showed one gallstone, without biliary tract dilatation; normal liver texture; and neither ascites nor splenomegaly. Kidney sonography showed bilateral enlarged and swollen kidneys (kidney length left 12.7 cm, right 12.5 cm), increased cortical echogenicity, and prominent papillae, consistent with acute parenchymal disease.
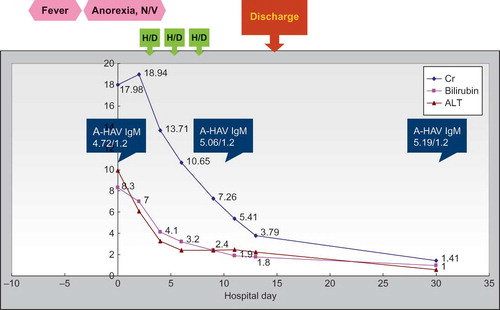
Under the suspicion of leptospirosis or tick borne disease, doxycycline 100 mg BID was given empirically from the first day of admission. In view of progressive azotemia (Cr up to 18.94 mg/dL) with uremic symptoms, hemodialysis was initiated from the third day of admission. Leptospirosis, scrub typhus, and murine typhus antibodies were confirmed negative. Doxycycline was discontinued after a one-week course. The azotemia and hyperbilirubinemia gradually improved. Because there were no further uremic symptoms, hemodialysis was ceased on the seventh day of admission after a total of three sessions. The amount of urine collected during the hospital course was around 1200–2500 ml per day.
He received a kidney biopsy on the tenth day of admission. An H&E stain (see A and 1B) showed interstitial nephritis, tubular necrosis with regeneration, glomerulosclerosis and arteriolar sclerosis. An immunofluorescence stain showed eight glomeruli with irregular faint IgM deposition, tubules with 1+ C3 deposition, and arteries with 1-2+ C3 deposition. By the time of discharge on the fourteenth day of admission, Cr had decreased to 3.79 mL/dL, and total bilirubin had decreased to 1.8 mL/dL. One month after initial presentation, Cr further improved to 1.41 mL/dL, and total bilirubin improved to 1.0 mL/dL (see ).
Figure 1. (a) H&E stain of kidney biopsy shows mononuclear cells infiltration of the interstitium, suggesting interstitial nephritis. (b) H&E stain of kidney biopsy shows tubular necrosis.
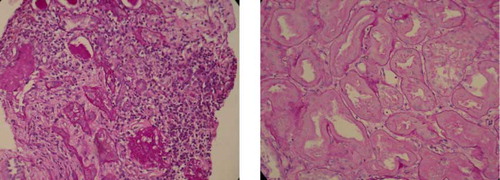
Figure 2. Clinical course of this patient. Abbreviations: H/D = hemodialysis, N/V = nausea/vomiting, Cr = creatinine (mg/dL), ALT = alanine aminotransferase (U/L). A-HAV IgM has a cutoff value of 1.2/ABBOTT. Bilirubin is measured in mg/dL.
DISCUSSION
Hepatitis B and C are both known to show extrahepatic involvement, including various nephropathies. The most notable renal manifestation of hepatitis B is membranous nephropathy. Other reported nephropathies associated with hepatitis B included membranoproliferative glomerulonephritis (MPGN), mesangial proliferative glomerulonephritis, IgA nephropathy, serum-sickness-like syndrome, and polyarteritis nodosa.Citation[2] Hepatitis C has been reported to be associated with cryoglobulinemia-related MPGN, immunotactoid glomerulonephritis (GN), fibrillary GN, amyloidosis, non-cryoglobulinemia-related MPGN, mesangial proliferative GN, membranous nephropathy, focal segmental sclerosis, and interstitial nephritis.Citation[3]
Although far less common, since 1978, hepatitis A complicated by nephropathies has drawn clinicians' attention. Some cases merely have mild proteinuria, microscopic hematuria, and mild urinary sediment abnormalities. A review of the literature revealed 36 case reports and two reviews related to hepatitis A and renal failure. In total, forty-seven patients were reported. summarizes the data of these forty-seven patients, plus the data for our case, making a total of forty-eight patients.
Table 1 Demographic and clinco-pathological features
Among the forty-eight patients, only one case developed fulminant hepatitis with hepatic coma,Citation[4] and the others had non-fulminant hepatitis. The ages ranged from 7 to 77 (see ) with a male predominance. ALT ranged from 259 to 6629 U/L, total bilirubin from 1.5 to 59.5 mg/dL, and serum creatinine level from 3.99 to 21 mg/dL. Most of the urinary analysis showed mild proteinuria and microscopic hematuria, while two patients had proteinuria of nephrotic range.Citation[5,6] The spot urine sodium concentration (UNa) ranged from 24 to 98 meq/L, except for two cases which had initial levels of 2 meq/L.Citation[1] However, both of these cases had subsequently high UNa.
Table 2 Clinical characteristics
Twenty-three patients received kidney biopsies (see ). The most common pathology was acute tubular necrosis. There were also MPGN, interstitial nephritis, or mixed presentations, although notably two of the biopsies were normal. Thirty-four patients received renal replacement therapy (see ), and ten only received conservative supportive treatment.
Table 3 Kidney biopsy (23 out of 48)
Table 4 Renal replacement therapy
Regarding the outcomes (see ), most of the patients had spontaneous and full renal and liver recovery without sequelae, mostly within 2–3 months. However four of the patients died,Citation[1,4,7,8] and one patient with MPGN had permanent renal insufficiency.Citation[6] Other comorbidities included generalized tonic-clonic seizure,Citation[9] disseminated intravascular coagulation or coagulopathy,Citation[6,10–12] type 2 diabetes mellitus,Citation[13] and pneumococcal peritonitis.Citation[14] In the case with generalized tonic-clonic seizure, the electroencephalography did not show the typical findings of hepatic encephalopathy, and the serum ammonia level was normal. The seizures subsided after hemodialysis.
Table 5 Outcome
Although there have been plenty of cases reported over the past few decades, the pathogenesis of hepatitis B and C related nephropathies have not been clearly defined. The most accepted mechanism of hepatitis B-related nephropathies is immune complex mediated injury,Citation[2] while that of hepatitis C related nephropathies, cryoglobulinemia, and immune complex mediated injury.Citation[3] Meanwhile, other mechanisms, such as direct cytopathic effects of the virus and indirect effects of virus-induced cytokines, virus-induced immunological effectors (T lymphocytes or antibodies), and virus-induced endothelial injury, have been proposed.Citation[2,3] Actually, the causative role of the hepatitis virus remains controversial, and the diagnosis is mainly supported by clinical time course and by exclusion.
Similarly, whether hepatitis A has a causative effect on nephropathies remains a key question. Currently, we favor hepatitis A as a cause of acute renal failure rather than just a coincidence, based on the following reasons. Acute renal failure usually has an average onset of within ten days after jaundice development, and achieves remission during the convalescent phase of hepatitis A infection. In these cases, there are no historical, clinical, or lab evidence of antecedent renal disease. There is neither systemic disease nor offending agent to explain the acute kidney injury.
Several hypotheses of the pathogenesis of hepatitis A‐related nephropathy have been postulated. First, acute hepatitis-related anorexia, vomiting, and diarrhea can cause intravascular volume depletion and activation of the rennin-angiotensin-aldosterone system (RAAS), both of which can compromise effective renal perfusion. However, except for Wilkinson's cases with low UNa, most patients have high UNa without hypotension or other signs of volume depletion.Citation[1] Therefore, pre-renal factors do not seem to be a good explanation. Second, hyperbilirubinemia secondary to hepatic dysfunction is believed to cause systemic vascular resistance reduction, predisposing renal vasoconstriction, and left ventricular contractility impairment, all of which in turn can compromise effective renal blood flow.Citation[15] On the other hand, elevated bile salt levels secondary to hepatic dysfunction cause direct renal tubular injury by non-specific detergent effects.Citation[16] To support the hypothesis of hyperbilirubinemia and bile salts inducing acute renal failure, most cases have a parallel relationship between elevated bilirubin and creatinine, except for Lin's case.Citation[17] In addition, some cases have low levels of bilirubin. Therefore, hyperbilirubinemia may not fully explain the mechanism. Third, kidney biopsies in two cases showed normal findings, implicating the possibility of hepatorenal syndrome.Citation[14,18] However, except for one case,Citation[4] almost all cases have non-fulminant hepatitis without hepatic failure, and most cases have high UNa. Therefore, the hypothesis of hepatorenal syndrome is not favored. It is possible that the normal kidney biopsy results may be false-negative due to there being un-sampled focal lesions or technical limitations. Fourth, immune complex mediated renal injury has been postulated. In 1978, Mathiesen et al. injected hepatitis A antigen isolated from acutely infected patients into marmosets intravenously, and the deposition of hepatitis A antigens at glomeruli basement membranes was detected in one of the eight marmosets.Citation[19] In 1981, Morita et al. produced proliferative glomerulonephritis and renal vasculitis in seven of eight marmosets by intravenous inoculation of hepatitis A virus from acutely infected patients.Citation[20] Immunofluorescence microscopy revealed mesangial deposits of IgG and IgM and capillary loop deposits of IgA and C3, although there was no antigen identified. In some of the reported cases, kidney biopsies have revealed various immune complex deposition, including IgG, IgA, IgM, C3, and Clq.Citation[6,21–23] This evidence suggests an important role of immune complexes. Fifth, endotoxinemia secondary to hepatic Kupffer cell dysfunction has been proposed to cause systemic vasodilatation, renal vasoconstriction, and release of cytokines, including some vasoactive mediators, which in turn compromise effective renal perfusion.Citation[16] On the other hand, endotoxinemia also induces platelet aggregation and release of nitrite oxide, with resultant intrarenal thrombosis and disseminated intravascular coagulation.Citation[16] In some cases, the endotoxin levels and anti-endotoxin antibody titers were elevated. Elevated FDP levels, prolonged PT, and intraglomerular deposition of fibrinogen were observed in Kamura's case.Citation[11] In Watanabe's case, renal function did not respond to hemodialysis, but improved dramatically after plasmapheresis.Citation[24] In Suga and Corpechot's cases, the renal function also recovered after plasmapheresis. These cases implicate the possible role of immune complexes or endotoxinemia.Citation[25,26] Sixth, like hepatitis B and C, a direct cytopathic effect of hepatitis A virus has been postulated.Citation[27]
In our case, the high UNa and lack of clinical evidence of dehydration precluded pre-renal azotemia. The high UNa and non-fulminant course also excluded hepatorenal syndrome. Because renal function improved parallel to liver function, the role of hyperbilirubinemia cannot be excluded. The presence of serum circulating IgG, IgM, and IgA and renal deposition of IgM and C3 strongly implicates the role of immune complexes. Unfortunately, we did not have the clinical tools to detect the presence of hepatitis A antigen in the kidneys.
The pathogenesis of hepatitis A related acute renal failure needs further studies, and may be multifactorial. The variability of nephropathies may be associated with host immune response differences and host genetic factors.
CONCLUSION
In conclusion, as is already known with hepatitis B and C, hepatitis A is potentially associated with various extrahepatic renal manifestations. A high suspicion of viral hepatitis, including acute hepatitis A, should arise when a patient presents with concomitant acute hepatitis and renal failure. The prognosis is mostly favorable, though not absolutely so. Further studies are needed to clarify the pathogenesis.
REFERENCES
- Wilkinson SP, Blendis LM, Williams R. Frequency and type of renal and electrolyte disorders in fulminant hepatic failure. Br Med J. 1974:186–189.
- Bhimma R, Coovadia HM. Hepatitis B virus-associated nephropathy. Am J Nephrol. 2004;24:198–211.
- Barsoum RS. Hepatitis C virus: From entry to renal injury-facts and potentials. Nephrol Dial Transplant. 2007;22:1840–1848.
- Mizuiri K, Kameyama M, Sagawa Y. A report of a case with fulminant hepatitis A associated with acute renal failure. Gastroenterology. 1985;20:470–475.
- Chio F, Bakir A. Acute renal failure in hepatitis A. Int J Artificial Organs. 1992;15:413–416.
- Zikos D, Grewal K, Craig K. Nephrotic syndrome and acute renal failure associated with hepatitis A virus infection. Am J Gastroenterol. 1995;90:295–298.
- Ogawa M, Hori J, Ueda S, Ohto M, Hirasawa H, Odaka M. A fatal case of acute renal failure associated with nonfulminant hepatitis A. Clin Nephrol. 1994;42:205–206.
- Beyazit Y, Guven GS, Kekilli M, Koklu S, Yolcu OF, Shorbagi A. Acute pericarditis and renal failure complicating acute hepatitis A infection. South Med J. 2006;99(1):82–84.
- Okushin H, Yamada G, Nishihara T. A case of sporadic type A hepatitis with acute renal failure. Liver. 1981;22:1299–1305.
- Schmidli RS, Lynn KL. Acute renal failure complicating non-fulminant hepatitis A infection: A case report. N Z Med J. 1990;103:375.
- Kamura M, Nagashima T, Imai T. A case of acute hepatitis A with renal failure caused by fibrinogen deposits. Jpn J Gastroenterol. 1993;90:2936–2939.
- Amaki S, Moriyama M, Motohashi T. Four cases of hepatitis A with acute renal failure. J Nihon Univ Med Assoc. 1989;48:343–349.
- Vesely DL, Dilley RW, Duckworth WC, Paustian FF. Hepatitis A-induced diabetes mellitus, acute renal failure, and liver failure. Am J Med Sci. 1999;317(6):419–424.
- Phillips AO, Thomas DM, Coles GA. Acute renal failure associated with non-fulminant hepatitis A. Clin Nephrol. 1993;39:156–157.
- Green J, Beyar R, Bomzon L, Finbery JP, Better OS. Jaundice, the circulation and the kidney. Nephron. 1984;37:145–152.
- Green J, Better OS. Systemic hypotension and renal failure in obstructive jaundice-mechanistic and therapeutic aspects. J Am Soc Nephrol. 1995;5:1853–1871.
- Lin CC, Chang CH, Lee SH, Chiang SS, Yang AH. Acute renal failure in non-fulminant hepatitis A. Nephrol Dial Transplant. 1996;11(10):2061–2066.
- Nachbaur K, Konig P, Rumpelt HJ, Schobel B, Lhotta K, Vogel W. Acute renal failure complicating non-fulminant hepatitis A. Clin. Nephrol. 1996;45(6):398–400.
- Mathiesen LR, Drucker J, Lorenz D, Localization of hepatitis A antigen in marmoset organs during acute infection with hepatitis A virus. J Infect Dis. 1987;138:369–377.
- Morita M, Kitajima K, Yoshizawa H, Glomerulonephritis associated with arteritis in marmosets infected with hepatitis A virus. Br J Exp Pathol. 1981;62:103–112.
- Takeshita S, Yamakado M, Nagano M, Umezu M, Tagawa H. A case of sporadic acute type A hepatitis associated with acute renal failure. Nippon Jinzo Gakkai Shi. 1994;36:871–875.
- Mccann UGII, Rabito F, Shah M, Nolan CRIII, Lee M. Acute renal failure complicating nonfulminant hepatitis A. West J Med. 1996;165(5):308–310.
- Imatake M, Motohashi T, Amaki S. A case of hepatitis A associated with thrombocytopenia, leukopenia, and acute renal failure. Jpn J Gastroenterol. 1990;87:1706–1709.
- Watanabe S, Nomoto H, Matsuda M. A case of acute renal failure associated with type A acute hepatitis responds dramatically to plasmapheresis. Tokai J Exp Clin Med. 1986;11:1–4.
- Corpechot C, Cadranel JF, Hoang C. Cholestatic hepatitis A in adults—clinical, biological, and histopathological study of nine cases. Gastroenterol Clin Biol (Paris). 1994;18:743–750.
- Suga M, Shibata K, Akahonai Y. A case of type A fulminant hepatitis with renal failure cured with plasma exchange. Acta Hepatol. 1984;25:241–245.
- Kawai K, Tomita E, Sugihara J. A case of acute hepatitis, type A, with acute renal failure: Report of a case with renal biopsy. Jpn J Gastroenterol. 1983;80:1345–1348.
- Yuzo W. A case report of sporadic acute hepatitis, type A associated with acute renal failure. Kidney Dial. 1983;15:71–75.
- Kramer MR, Hershko C, Slotki IN. Acute renal failure associated with non-fulminant type A viral hepatitis. Clin Nephrol. 1986;25:219.
- Garel D, Vasmant D, Mougenot B, Bennsman A. Glomerular nephropathy with mesangial proliferation and acute hepatitis A virus infection: Two cases. Ann Pediatr (Paris). 1986;33:185–188.
- Eng C, Chopra S. Acute renal failure in non-fulminant hepatitis A. J Clin Gastroenterol. 1990;12:717–718.
- Tsuru T, Ishibashi H, Matsuishi E. Acute renal failure associated with acute type A hepatitis with a mild liver damage. Fukuoka Acta Med. 1990;81:337–341.
- Blum A, Ben-Yehuda A. Acute reversible renal failure in acute hepatitis A. Harefuah. 1990;118:388.
- Mattoo TK, Mahmood MA, al-Sowailem AM. Acute renal failure in non-fulminant hepatitis A infection. Ann Trop Pediatr. 1991;11:213–215.
- Ilan Y, Galun E. Glomerulonephritis associated with acute HAV infection. J Clin Gastroenterol. 1992;15:85.
- Geltner D, Naot Y, Zimhoni O. Acute oliguric renal failure complicating type A nonfulminant viral hepatitis. J Clin Gastroenterol. 1992;14:160–162.
- Konishi N, Takeshita K, Yasui H, Hatta I. A case of acute hepatitis A associated with acute renal failure from the onset. Jpn J Nephrol. 1993;35(9):1103–1106.
- Malbrain MLNG, Lambrecht GLY, Brans B, Lins RL, Daelemans R. Acute renal failure in non fulminant hepatitis A. Clin Nephrol. 1994;41:180–181.
- Faust RL, Pimstone N. Acute renal failure associated with nonfulminant hepatitis A viral infection. Am J Gastroenterol. 1996;91:369–372.
- Jamil SM, Massry SG. Acute anuric renal failure in nonfulminant hepatitis A infection. Am J Nephrol. 2009;18(4):329–332.
- Akcay A, Altun B, Usalan C, Ulusoy S. Hepatitis A viral infection-related acute renal failure. Nephron. 1999;83:191.
- Adas M, Tanakol R, Yarman S, Boztepe H, Ecder T, Alagol F. Acute renal failure associated with nonfulminant hepatitis A virus infection. Ren Fail. 2002;24(1):97–102.
- Vaboe AL, Leh S, Forslund T. Interstitial nephritis, acute renal failure in a patient with non-fulminant hepatitis A infection. Clin Nephrol. 2002;57(2):149–153.
- Kim SE, Kim SJ, Kim HS, Kim HS, Nam ES, Lee SK, Two cases of acute renal failure associated with non-fulminant acute hepatitis A. Korean J Gastroenterol. 2009;48(6):421–426.