Abstract
Thomas syndrome is a rare syndrome including Potter sequence, renal anomalies, heart defects, cleft palate with other oropharyngeal anomalies. Here, we report a newborn with Potter sequence, bilateral renal hypoplasia and cystic dysplasia, multiple cardiovascular malformations, long large ears, frontal bossing, small lips, partial simple toe syndactyly, and cleft palate. To our best knowledge, this patient may be considered as a new variant of Thomas syndrome or a new syndrome.
INTRODUCTION
“Potter’s sequence” or “oligohydramnios sequence” is a term used to describe the typical physical appearance of a fetus or neonate due to a dramatically decreased amniotic fluid volume (oligohydramnios), or absent amniotic fluid (anhydramnios), secondary to renal diseases such as bilateral renal agenesis. Other causes of Potter syndrome can be obstruction of the urinary tract, multicystic kidney disease, renal hypoplasia, and rupture of the amniotic sac.Citation1–3
Thomas syndrome is a lethal multiple congenital anomalies syndrome consisting of small kidneys, bilateral cleft lip/cleft palate, complex heart defects, bilateral renal agenesis/hypoplasia, and other malformations and it is probably inherited as an autosomal recessive trait.Citation4–6
Here, we report a newborn with Potter sequence, cleft palate, bilateral renal hypoplasia and cystic dysplasia, and cardiac malformations suggesting a variant of Thomas syndrome or a new syndrome.
CASE PRESENTATION
A newborn baby was referred to our neonatal intensive care unit with severe respiratory problem. The infant weighed 2000 g and in the range of small for gestational age (third percentile) measured 47 cm in length (third percentile) and 31 cm in head circumference (third percentile). In her prenatal history intrauterine growth failure, oligohydramnios and reduced renal size was complicated to the pregnancy since 16th week.
Our patient was delivered vaginally at 38 weeks of gestation and was the only living child of a 27-year-old mother. The mother had three pregnancies. The first pregnancy was complicated by mole hydatiform. The second baby was born at 26 gestation weeks with a birthweight of 825 g and died immediately after birth. Autopsy had not been performed.
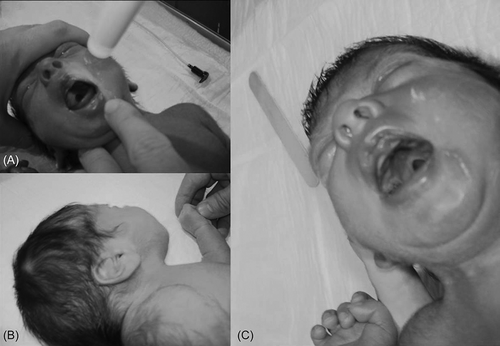
On physical examination of our patient pulse rate was 160/min; respiratory rate, 82/min; blood pressure, 60/35 mmHg; body temperature, 36°C. Some anomalies were noted at birth including Potter face, long large ears, frontal bossing, small lips, partial simple toe syndactyly 2–3, and cleft palate (). Respiratory system examination revealed tachypnea with thoracal retractions, and cardiovascular system examination revealed heart murmur.
Figure 1. (A) Potter face; (B) large, malformed ears; and (C) cleft palate.
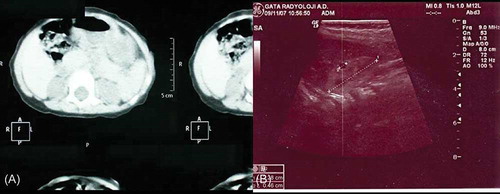
Laboratory studies yielded the following values (normal ranges are given in parentheses): white blood cell count, 12,500/mm3 (6000–17,500) with a normal blood differential count; blood urea nitrogen (BUN), 200 mg/dL (5.7–20.1); creatinine, 2.6 mg/dL (0.1–0.9); sodium, 134 mmol/L (139–146); potassium, 4.1 mmol/L (3.5–6.0); phosphorus, 3.3 mg/dL (4.9–7.9); and glucose, 70 mg/dL (60–100). Arterial blood gas values showed a pH of 7.03 (7.35–7.45) and bicarbonate: 2.3 mmol/L (21–28). Urine and serum ketone bodies were positive. Blood chemistry tests and urinary amino acids, mucopolysaccharides, and serum hormone levels were within normal limits for her age. TORCH and viral serologies including human papillomavirus serology were negative. Chest X-ray showed mild lung hypoplasia. Since the BUN and creatinine levels were high and the urine outflow was decreased, we diagnosed acute renal failure. Bilateral renal hypoplasia and cystic dysplasia were documented by ultrasonography (USG) and CT (). Right kidney was 1.5 cm in length (normal values 2.5–4.8 cm), 0.8 cm in depth (normal values 1.3–2.7 cm). Patent ductus arteriosus, ventricular septal defect, and atrial septal defect were demonstrated on echocardiographic examination. Chromosome analysis was normal on peripheral blood (46, XX).
Figure 2. (A) CT image of bilateral renal hypoplasia. (B) Ultrasonographic image showing renal hypoplasia and cysts. Renal size for right kidney was 1.5 cm in length (normal values for neonates 2.5–4.8 cm) and 0.8 cm in depth (normal values for neonates 1.3–2.7 cm).
DISCUSSION
Thomas syndrome is a lethal multiple congenital anomalies syndrome (Potter sequence, bilateral renal agenesis/hypoplasia, bilateral cleft lip/cleft palate, complex heart defects, and other malformations). It is probably inherited as an autosomal recessive trait.Citation4–7
Our patient was born with severe growth retardation. She had Potter face and cleft palate and soon after birth she had severe respiratory failure. To evaluate the reason for respiratory problems, chest X-ray and cardiac echocardiography were done. The chest X-ray showed mild lung hypoplasia. Echocardiography was done for the differential diagnosis of heart murmur detected on physical examination. Patent ductus arteriosus, ventricular septal defect, and atrial septal defect were detected. She was treated by mechanical ventilation for 2 weeks of duration. Since the urine outflow was decreased and blood chemical analyses were consistent with acute renal failure, renal USG was done to rule out renal aplasia or hypoplasia. Bilateral renal hypoplasia was detected on USG. Right kidney was 1.5 cm in length (normal values 2.5–4.8 cm), 0.8 cm in depth (normal values 1.3–2.7 cm). The measurements were consistent with bilateral renal hypoplasia.Citation8
Other congenital anomalies were long large ears, small lips, frontal bossing, and partial simple toe syndactyly 2–3. Some of these anomalies had not been reported in Thomas syndrome by other authors before. These findings let us to consider that our patient may be a new variant of Thomas syndrome or a new syndrome ().
Table 1. Thomas Syndrome—clinical variability.
PotterCitation1,2 originally described the association of bilateral renal agenesis with oligohydramnios and compression deformities of the face and the limbs together with pulmonary hypoplasia. Potter sequence is known to be causally heterogeneous and pathogenetically variable. Potter sequence is often related with other malformations such as cardiac malformations or cleft lip/cleft palate. Almost all of the patients are nonsyndromic, some are of syndromic origin, chromosomal, monogenic, teratogenic, disruptive, or unknown cause. Potter sequence may occur with associated congenital abnormalities (syndromic patients), or without them (nonsyndromic patients).Citation3,9
Holzgreve et al.Citation10 reported a fetus with Potter sequence, heart defect, polydactyly, and cleft palate together with additional malformations, in particular adhesion of the tongue to the posterior cleft palate, and skeletal defects. Leguis et al.Citation11 made the second observation for Holzgreve syndrome. Thomas et al.Citation4 reported two sibs with cardiac and renal abnormalities. The first baby had hypoplastic left heart and renal hypoplasia; the second baby had complex congenital heart defect, renal agenesis, and cleft lip and palate. Thomas suggested that these patients represent the first familial examples of the Holzgreve syndrome. Thomas et al.Citation4 emphasized that Holzgreve and Thomas syndromes are different entities. Briscioli et al.Citation6 proposed that the two sibs reported by Thomas et al.,Citation4 the patient reported by themselves, the four sibs reported by Zlotogora et al.Citation7 did not have polydactyly and intrabuccal bands as did the patients described by Holzgreve et al.Citation10 and Legius et al.Citation11
On the basis of history, clinical, and laboratory findings, our patient described in the present report is very similar to those reported by Cury et al.,Citation3 Thomas et al.,Citation4 Zlotogora et al.,Citation7 and Briscioli et al.Citation6
Bonnet et al.Citation5 and Leguis et al.Citation11 each reported an isolated patient with the similar combination of Potter sequence, heart defect, polydactyly, and cleft palate. The patient reported by Leguis et al.Citation11 also had intrabuccal bands. Holzgreve syndrome includes both intrabuccal bands and polydactyly in addition to the clinical findings seen in Thomas syndrome. Since our patient does not have polydactyly and intrabuccal bands, Holzgreve syndrome may be excluded in differential diagnosis.
The molecular genetic basis of Thomas and Holzgreve syndromes has not been understood yet, the diagnosis can only be made by clinical findings. In most of the reports, Thomas syndrome was diagnosed during the neonatal period. Pediatricians should be alert for congenital abnormalities in patients with Potter sequence as this may indicate the presence of a rare disorder. A comprehensive medical history and a physical evaluation are essential to rule out other abnormalities. Genetic consultation should be offered to the family for future planning.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
REFERENCES
- Potter EL. Bilateral renal agenesis. J Pediatr. 1946;29:68–76.
- Potter EL. Bilateral absence of ureters and kidneys. Am J Obstet Gynecol. 1965;25:3–12.
- Curry CJR, Jensen K, Holland J, Miller L, Hall ED. The Potter sequence. Am J Med Genet. 1984;19:679–702.
- Thomas IT, Honore GM, Jewett H, Velvis H, Garber P, Ruiz C. Holzgreve syndrome: Recurrence in sibs. Am J Med Genet. 1993;45:767–769.
- Bonnet J, Cordier MP, Ollagnon E, . Hypoplasierknale, polydactylie, cardiopathie: Un nouveau syndrome. J Genet Hum. 1987;35:279–289.
- Briscioli V, Lalatta F, Rizzuti T, Fesslova V. Thomas syndrome: Clinical variability and autosomal recessive inheritance. Am J Med Genet. 1997;71:373–374.
- Zlotogora J, Ariel J, Ornoy A, Yagel S, Eidelman AI. Thomas syndrome: Potter sequence with cleft lip/palate and cardiac anomalies. Am J Med Genet. 1996;2:224–226.
- Aamir D, Ashraf A, Habib R. A comparative study of renal size in newborn babies. Gomal J Med Sci. 2006;4:65–69.
- Al Saadi AA, Yoshimoto M, Bree R, . A family study of renal dysplasia. Am J Med Genet. 1984;19:669–677.
- Holzgreve W, Wagner H, Rehder H. Bilateral renal agenesis with Potter phenotype, cleft palate, anomalies of the cardiovascular system, skeletal anomalies including hexadactyly and bifid metacarpal: A new syndrome? Am J Med Genet. 1984;18:177–184.
- Leguis E, Moerman P, Fryns JP, Vandenberghe K, Eggermont E. Holzgreve-Wagner-Rehder syndrome. Potter sequence associated with persistent buccopharyngeal membrane: A second observation. Am J Med Genet. 1988;31:269–272.