Abstract
Fabry disease is a rare X-linked recessive glycosphingolipid storage disease that is caused by a deficiency of the lysosomal α-galactosidase A (GLA) enzyme, encoded by the GLA gene. This deficiency leads to the accumulation of glycosphingolipids throughout the body, which, in turn, causes multisystem diseases associated with renal, cardiovascular, and cerebrovascular complications. Recent molecular studies of GLA have demonstrated the existence of atypical variants in Fabry disease, suggesting significant genotype–phenotype correlations. In this study, we describe a renal variant of Fabry disease caused by a novel small insertion mutation in the GLA gene.
INTRODUCTION
Fabry disease is a rare X-linked recessive glycosphingolipid storage disease caused by mutations in the α-galactosidase A (GLA) gene.Citation1 Defects in the lysosomal GLA enzyme encoded by GLA gene result in the systemic accumulation of partially metabolized glycosphingolipids. This leads to the development of multisystem diseases associated with renal, cardiovascular, and cerebrovascular complications. Fabry disease comprises classical and variant phenotypes. The classical phenotype produces a number of clinical manifestations, including angiokeratoma, acroparesthesia, hypohidrosis, renal failure, and vasculopathy of the heart and brain. On the other hand, the variant phenotype manifests only one or two symptoms and is termed “oligosymptomatic.”
The genomic sequence and full-length cDNA of the human GLA gene, which is located on chromosome Xq22, have previously been characterized.Citation2 While most mutations result in classical phenotypes, some mutations generate variant phenotypes with manifestations confined to the heart or the kidney. However, the data available from previous studies are quite limited,Citation3 and therefore, further research to determine the genotype–phenotype correlations in Fabry disease is warranted.
In this report, we describe a case of renal variant of Fabry disease caused by a small insertion in the GLA gene. To our knowledge, this is the first description of this kind of a mutation, and not many cases of renal variant of Fabry disease have been reported.
CASE REPORT
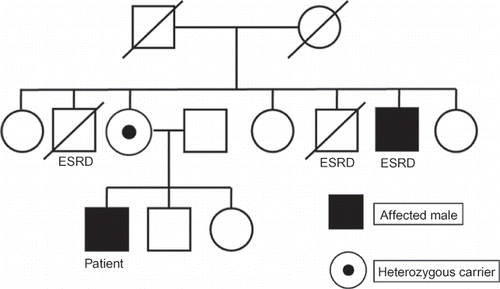
We evaluated a case of proteinuria observed for 8 years in a 34-year-old man. As shown in , the family pedigree revealed that the older and younger brothers of the patient’s mother had died of end-stage renal disease (ESRD), and another younger brother also suffered from ESRD. There was no medical history of hypertension or diabetes mellitus, the patient did not complain of acroparesthesia, and hypohidrosis and angiokeratoma were not present.
Figure 1. The family pedigree. ESRD, end-stage renal disease.
Urinalysis revealed normal microscopic findings and proteinuria; the 24 h protein excretion was 210 mg/day. In addition, we obtained the following values: 7.2 mg/dL, blood urea nitrogen; 6.8 g/dL, total serum protein; 4.5 g/dL, serum albumin; 0.7 mg/dL, serum creatinine; and 73.3 mL/min, creatinine clearance. Kidney ultrasonography revealed cortical thinning and poor differentiation of the corticomedullary junction. Electrocardiogram showed normal sinus rhythm and echocardiography showed normal chamber size and thickness, with normal ejection fraction. Ophthalmic examination did not indicate corneal opacity.
Percutaneous ultrasound-guided renal biopsy was performed by a nephrologist. Light microscopy examination revealed markedly enlarged podocytes with a foamy cytoplasm. Furthermore, foamy vacuoles were also detected in tubular epithelial cells. While the glomeruli were normocellular, some glomeruli showed global glomerulosclerosis. Electron microscopy identified whorled myelin figures—composed of concentrically arranged lamellae of electron-dense material—in the cytoplasm of both podocytes and tubular epithelial cells ().
Figure 2. Renal biopsy findings. (A) Hematoxylin and eosin (H&E) staining. Podocytes of the glomeruli display a foamy appearance and an expanded cytoplasm (×400). (B) H&E staining. Foamy tubular epithelial cells are shown (×400). (C) Toluidine blue staining. A semi-thin section stained with toluidine blue for electron microscopy shows inclusions within the cytoplasm of the podocytes (×200). (D) Electron microscopy. Diffuse, whorled myelin figures in the cytoplasm of the podocytes are shown (×3000).
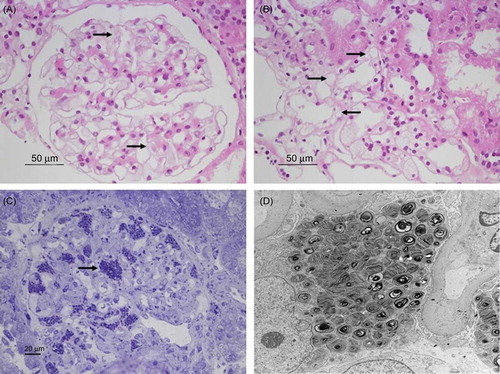
Figure 3. Nucleotide sequencing of the GLA gene by using genomic DNA. (A) Control. (B) Patient DNA sequence reveals a homozygous small insertion mutation, that is, c.1030_1031insT (p.Leu344fsX31). (C) Patient’s mother DNA sequence shows that the mother is a heterozygous carrier for c.1030_1031insT (p.Leu344fsX31).
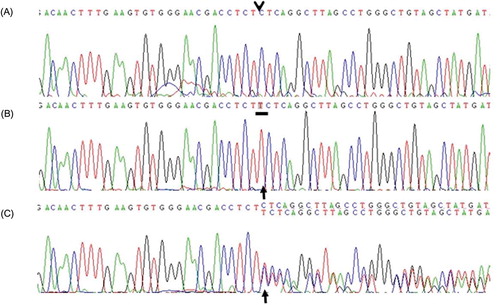
Leukocyte α-galactosidase activity was significantly reduced to <0.01 nmol/h/mg (normal range 25–126). For mutational analysis, genomic DNA was isolated from peripheral blood leukocytes by using the Quick Gene DNA isolation kit (Fujifilm, Tokyo, Japan). Polymerase chain reaction amplification of exon 7 of the GLA gene was performed using the following primers: sense 5′-gaatgccaaactaacagggc-3′ and antisense 5′-gccacctagccttgagcttt-3′. DNA sequencing was carried out with the Big Dye Terminator v3.1 Cycle Sequencing Ready Reaction Kit (Applied Biosystems, Foster City, CA, USA), according to the manufacturer’s instructions. Sequence analysis of the patient’s GLA gene identified an insertion mutation; a T nucleotide was inserted between the T and C nucleotides at positions 1030 and 1031, respectively (c.1030_1031insT), which was considered responsible for a shift in the reading frame of the leucine residue at position 344 and the formation of a premature stop codon ().
Once the diagnosis of Fabry disease was confirmed, the leukocyte α-galactosidase activity was measured in the other family members and their DNA was sequenced. The α-galactosidase activity in the patient’s mother had decreased to 4.06 nmol/h/mg, and sequence analysis of the GLA gene revealed that she was a heterozygous carrier for c.1030_1031insT. We also confirmed Fabry disease in the mother’s younger brother by measurement of leukocyte α-galactosidase enzyme activity and DNA sequencing.
DISCUSSION
In this study, we identified a novel mutation of the GLA gene that leads to a renal variant of Fabry disease. In the affected male patients, the first symptoms of Fabry disease—angiokeratoma, acroparesthesia, and ocular changes—appear in childhood or adolescence. Progressive vascular accumulation of glycosphingolipids subsequently leads to renal failure as well as cardiac and cerebrovascular diseases. However, the patient considered in our study displayed renal manifestations without typical Fabry complications. Creatinine clearance declined less when compared with a matched age group, and kidney ultrasonography revealed atrophic changes. Moreover, microscopic examination showed diffuse sclerotic change in some glomeruli.
DNA sequencing revealed a novel small insertion in the GLA gene of the patient (c.1030_1031insT). This insertion resulted in a shift in the reading frame of Leu344 and the formation of a premature stop codon; this may cause the production of a nonfunctional protein or the absence of the gene product.
Patients suffering from Fabry disease who display only one or two manifestations are referred to as oligosymptomatic. Currently, the cardiac variant is the most common variant of Fabry disease.Citation4 However, analysis of genotype–phenotype correlations in Fabry disease is complicated by numerous factors, including the high proportion of private mutationsCitation5 and the large phenotypic heterogeneity associated with individual mutations. Despite these limitations, some studies have demonstrated the existence of significant genotype–phenotype correlations.Citation3
The level of GLA enzyme activity influences the extent of the disease; in particular, a late-onset variant of Fabry disease is associated with residual enzyme activity. However, in this study, we could not detect enzymatic activity in the patient. Enzyme replacement therapy using GLA is highly specific for Fabry disease. Therefore, we initiated this therapy in the patient with an initial fortnightly dose of 1 mg/kg agalsidase-β (Fabrazyme®; Genzyme Co., MA, USA).
Interestingly, in 1995, the patient’s mother donated her left kidney to her older brother, who suffered from ESRD. Unfortunately, in his case, hemodialysis was resumed 3 years after kidney transplantation and he died in 2009. We do not know exactly why graft failure occurred so early, because we did not access his medical record. However, it is possible that transplantation of a kidney graft obtained from a carrier of Fabry disease affects graft function. Indeed, renal transplantation from a donor who is a heterozygote female relative to a recipient with Fabry disease is generally considered unsafe, as glycosphingolipid accumulation may already be present in the donor in the absence of clinical symptoms.Citation6 Moreover, renal function in the heterozygote female donor may decrease following kidney donation.Citation7
In conclusion, we reported a novel small insertion mutation in the GLA gene (c.1030_1031insT), which abrogates the activity of GLA and causes a renal variant of Fabry disease. Our study illustrates that Fabry disease can present manifestations exclusive to the renal system, despite complete lack of enzyme activity, and it also suggests the presence of a genotype–phenotype correlation for the c.1030_1031insT mutation.
ACKNOWLEDGMENT
This research was supported by the Korea Science and Engineering Foundation through the Medical Research Center for Gene Regulation grant (2011-0030732) at Chonnam National University.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
REFERENCES
- Brady RO, Gal AE, Bradley RM, Martensson E, Warshaw AL, Laster L. Enzymatic defect in Fabry’s disease. Ceramide trihexosidase deficiency. N Engl J Med. 1967;276:1163–1167.
- Shabbeer J, Yasuda M, Luca E, Desnick RJ. Fabry disease: 45 novel mutations in the alpha-galactosidase A gene causing the classical phenotype. Mol Genet Metab. 2002;76:23–30.
- Altarescu GM, Goldfarb LG, Park KY, . Identification of fifteen novel mutations and genotype-phenotype relationship in Fabry disease. Clin Genet. 2001;60:46–51.
- von Scheidt W, Eng CM, Fitzmaurice TF, . An atypical variant of Fabry’s disease with manifestations confined to the myocardium. N Engl J Med. 1991;324:395–399.
- Schafer E, Baron K, Widmer U, . Thirty-four novel mutations of the GLA gene in 121 patients with Fabry disease. Hum Mutat. 2005;25:412.
- Niaudet P. Living donor kidney transplantation in patients with hereditary nephropathies. Nat Rev Nephrol. 2010;6:736–743.
- Popli S, Molnar ZV, Leehey DJ, . Involvement of renal allograft by Fabry’s disease. Am J Nephrol. 1987;7:316–318.