Abstract
Joubert syndrome (JBTS) is a rare autosomal recessive disorder with an underestimated prevalence due to lack of recognition of clinical signs or failure to diagnose this pathology. JBTS is clinically heterogeneous, and it is characterized by a multiple organ involvement predominantly due to the requirement for Joubert gene function in several tissues. Renal disease affects approximately 30% of patients with JBTS, presenting itself in most cases as nephronophthisis (NPHP), a structural tubulo-interstitial disorder characterized by thickened basal membrane of the tubular epithelium and progressive interstitial fibrosis, associated with cysts at the cortico-medullary junction. We propose three cases concerning three patients with JBTS having different years of illness and degrees of renal impairment, evaluating the parameters of renal function at the time of genetic diagnosis and seen after a follow-up of 7 years. We measured neutrophil gelatinase-associated lipocalin (NGAL), considered as an excellent predictor of kidney injury, to evaluate whether this biomarker might be an early biomarker for JBTS-related kidney disease. NGAL was high in all three cases, but with different levels, indicating a tubular suffering typical of this syndrome, with dissimilar severity in the analyzed subjects. NGAL could represent an early indicator of renal damage useful to start an intensive nephrologic follow-up.
INTRODUCTION
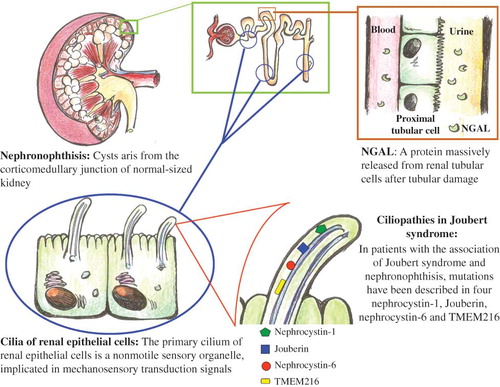
Joubert syndrome (JBTS) is a rare and underestimated autosomal recessive disorder with prevalence in the United States of 1 in 100,000 with renal disease affecting approximately 30% of patients with JBTS.Citation1 JBTS is clinically heterogeneous and is characterized by a multiple organ involvement, nephronophthisis (NPHP), or cystic dysplasia, in association with retinal degeneration, aplasia/hypoplasia of the cerebellar vermis causing ataxia, facultative symptoms of psychomotor retardation, polydactyly. Some of these findings are not apparent at birth. The pathognomonic feature is a complex midbrain–hindbrain malformation evident on brain magnetic resonance imaging (MRI), which is characterized by hypo-/dysplasia of the cerebellar vermis, elongated, thickened, and misoriented superior cerebellar peduncles and a deep interpeduncular fossa “molar tooth sign.”Citation2 This clinical pleiotropism can probably be explained by the genetic basis of these syndromes. The multiorgan involvement in JBTS is predominantly due to the requirement for Joubert gene function in multiple tissues. Ten causative genes have been identified to date: INPP5E, TMEM216, AHI1, NPHP1, CEP290, TMEM67, RPGRIP1L, ARL13B, CC2D2A, and OFD1. All these genes encode for proteins of the primary cilium, including JBTS in the group of “ciliopathies.” This is a structural tubulo-interstitial disorder characterized by irregular, thickened basal membrane of the tubular epithelium, and progressive interstitial fibrosis, associated with small cysts at the cortico-medullary junction. Glomeruli are often normal, although some of them may be completely sclerosed and others may show periglomerular fibrosis. In patients with the association of JBTS and NPHP, mutations have been described in four different genes: NPHP1 (encodes nephrocystin-1), AHI1 (encodes Jouberin), NPHP6 (encodes nephrocystin-6), and TMEM216.Citation3–6 In particular, nephrocystin-1 has been localized in renal cilium and it is expressed in renal collecting ducts.Citation7 Jouberin is localized in the renal collecting duct and interacts with nephrocystin-1.Citation8 Nephrocystin-6 is a centrosomal protein.Citation9 TMEM216, localized at the base of primary cilia, encodes an uncharacterized tetraspan transmembrane protein.Citation6 Clinically, NPHP usually presents with urine-concentrating defects (salt-losing renal insufficiency) in the first or second decade of life: it manifests by polydipsia, polyuria, anemia, and growth failure, with progression to end-stage renal disease approximately by 13 years of age.Citation10 Early ultrasound changes during the disease include increased renal echogenicity, cysts, loss of cortico-medullary differentiation, with small, scarred kidneys only observed after the progression of the disease. It is important to note that NPHP does not recur in transplanted kidneys. Prognostic information in literature is limited by small numbers of patients, diverse ascertainment strategies, and short duration of follow-up and lack of standardized assessments. Prognosis depends mostly on renal and hepatic complications that, if not timely diagnosed and managed, represent the major causes of death in JBTS patients. Neutrophil gelatinase-associated lipocalin (NGAL) is a 25-kDa protein massively released from renal tubular cells after various injuring stimuli: its levels, both in plasma and in urine, rise before any increase occurs in creatinine levels, thus facilitating a more reliable prediction of renal damage.Citation11 The most diagnostic use of this protein is in the field of acute kidney injury. Furthermore, chronic renal damage could influence the physiological balance of this protein in a way similar to that observed for acute injury conditions. In this view, chronically damaged tubular cells would produce a great quantity of NGAL; thus, the increased NGAL levels subsequently observed would not be the consequence of a decrease in renal protein clearance capacity (because of tubular impairment), but rather of active chronic stress-induced production of this protein by the same injured cells.Citation12 There are no data on the levels of this biomarker in patients with JBTS. The aim of our case report was to evaluate serum and urinary NGAL in three siblings with this syndrome with different degrees of renal impairment. We evaluated three cases concerning three members of a family with JBTS, due to TMEM216 gene mutation, having different years of illness and different degrees of renal impairment. At the time of diagnosis and after a follow-up period, in all three cases, renal ultrasonography underlined normal-sized kidneys with increased cortical echogenicity, maintenance of cortico-medullary differentiation with a diagnosis of NPHP. We evaluated the parameters of renal function observed at the time of genetic diagnosis of the syndrome and those seen after a follow-up of 7 years. We also evaluated serum (s) and urine (u) levels of NGAL in these subjects in order to evaluate its diagnostic power of impaired renal function, comparing it with laboratory data and with data obtained by renal scintigraphy. Even considering that our observation consist of only three cases of JTBS, in order to have a normal range of serum and urinary NGAL values, we determined the levels of this marker in 10 healthy subjects well matched for age and sex. Electrolytes, calcium, and phosphorus were normal. Proteinuria and microalbuminuria were absent except at the later time point in patient P.E. In all cases we found low values of urinary osmolality and urine gravity, indicating a urinary concentration defect ().
Table 1. Laboratory and instrumental assessment of renal function.
Case 1: P.A. (14-year-old boy): After birth, nystagmus, movement disorders, and developmental language disorders were detected. There was also a delay of growth for the first 8 months of life with repeated episodes of infection at the level of the upper respiratory tract. JBTS diagnosis was performed at the age of 2 years with the molar tooth sign on brain MRI.
Case 2: P.S. (18-year-old girl): At birth, tremors and difficulty in sucking with a nystagmus, movement disorder, and developmental language disorders were detected. JBTS diagnosis was performed at the age of 11 years with the molar tooth sign on brain MRI.
Case 3: P.E. (23-year-old girl): At 3 months of life for hypotonia, the girl was subjected to brain CT scan that showed cerebellar atrophy. She also had nystagmus, movement disorder, and developmental language disorders. JBTS diagnosis was performed at the age of 6 years with the molar tooth sign on brain MRI.
DISCUSSION
The diagnosis of JBTS is difficult because of the current absence of a specific and simple laboratory test. In fact, the certainty of the diagnosis is given by genetic analysis that is always late with respect to clinical and radiological data, which are relatively specific to the disease through the molar tooth sign on MRI. Renal involvement changes the nature of JBTS and also it can develop itself over the age of 10 years, so it must be diagnosed as early as possible. Renal ultrasonography is useful in evaluating the presence of cysts in the medulla cortico-medullary junction but this is not enough when the function is being investigated. Low values of urinary osmolality and urine gravity indicate a urinary concentration defect. Renal cortical scintigraphy with technetium-labeled diethylene-triamine-pentaacetate (99mTc-DTPA) is usually used for evaluating the functioning renal tissue and it is an excellent agent for the visualization of renal parenchyma. In our cases, renal involvement of JBTS was clearly diagnosed with bilaterally decreased uptake of 99mTc-DTPA in the kidneys in renal cortical scintigraphy. Visual demonstration of poor radiopharmaceutical uptake, increased background activity, and bladder visualization suggest that there is a possible tubular function defect in NPHP in JBTS and this is the cause of the failure in the uptake of the radioisotope by the tubule. A tubular agent that shows the functioning tubular mass like NGAL can shed light into the presence of a functional abnormality. In this case report we have shown that the levels of sNGAL and uNGAL were higher when comparing with the physiological values found in healthy subjects. In all patients we excluded the presence of urinary tract infection and leukocyturia, responsible for altering NGAL sensibility and specificity. The three patients studied have a different renal involvement. sNGAL and uNGAL were high in all three cases, but with different levels, indicating a tubular suffering typical of this syndrome, with dissimilar severity in subjects analyzed. In fact, the values of this biomarker were parallel to the severity of renal function, presenting the highest values the child P.E., suffering from a more severe kidney disease. It is interesting to note that P.S., with a normal creatinine, azotemia, and scintigraphic data, presents NGAL values also higher than healthy subjects, expression of “subclinical” involvement of renal tubules damaged in a manner which does not affect the total function of the kidneys, organs with a note “functional reserve” (). This shows how the NGAL is also a very early marker of renal damage (). In addition, proteinuria and microalbuminuria were, at least in our case, not very useful in monitoring the glomerular and tubular damages, with their values that have always been found in the normal range.
Figure 1. sNGAL, uNGAL, and GFR in patients studied. It can be seen that there is a different renal impairment, given the three different values of GFR obtained using renal scintigraphy. The values of the NGAL present a different trend in the three cases and reflect, in inverse way, the renal injury. In fact, P.E. has the highest values of NGAL, due to the most severe renal impairment evidenced by the lower value of GFR.
Note: NGAL, neutrophil gelatinase-associated lipocalin; GFR, glomerular filtration rate.
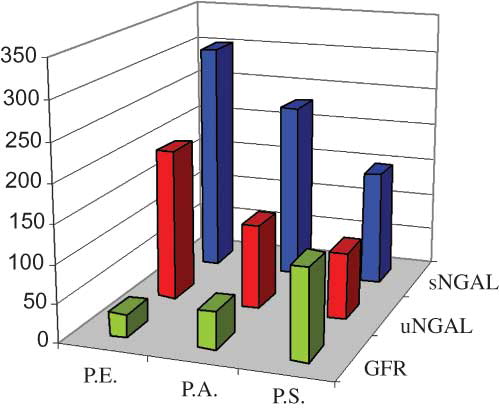
Figure 2. JBTS, tubular damage, and NGAL. The gene products associated with Joubert syndrome are known to localize to primary cilium. Its disruption leads to cystic kidney disease. The increase in NGAL values may express the degree of subclinical tubular impairment, thus representing an earlier measurable index of renal suffering.
Note: JBTS, Joubert syndrome; NGAL, neutrophil gelatinase-associated lipocalin.
Early detection of kidney impairment is a fundamental objective in managing these patients. Assuming that the data obtained relate only to three patients with this syndrome, however, the NGAL could represent an early indicator of renal damage on which to base an intensive nephrologic follow-up and using it as an indicator of therapeutic response.
ACKNOWLEDGMENTS
This work has never been published or submitted for publication elsewhere. The final manuscript has been reviewed and approved by all authors. The authors deny any commercial affiliation or other arrangements that could be considered to pose a conflict of interest regarding the submitted article.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
REFERENCES
- Parisi MA, Doherty D, Chance PF, Glass IA. Joubert syndrome (and related disorders). Eur J Hum Genet. 2007;15:511–521.
- Maria BL, Quisling RG, Rosainz LC, . Molar tooth sign in Joubert syndrome: Clinical, radiologic, and pathologic significance. J Child Neurol. 1999;14:368–376.
- Castori M, Valente EM, Donati MA, NPHP1 gene deletion is a rare cause of Joubert syndrome related disorders. J Med Genet. 2005;42:e9.
- Utsch B, Sayer JA, Attanasio M, . Identification of the first AHI1 gene mutations in nephronophthisis-associated Joubert syndrome. Pediatr Nephrol. 2006;21:32–35.
- Sayer JA, Otto EA, O’Toole JF, . The centrosomal protein nephrocystin-6 is mutated in Joubert syndrome and activates transcription factor ATF4. Nat Genet. 2006;38:674–681.
- Valente EM, Logan CV, Mougou-Zerelli S, . Mutations in TMEM216 perturb ciliogenesis and cause Joubert, Meckel and related syndromes. Nat Genet. 2010;42:619–625.
- Eley L, Moochhala SH, Simms R, Hildebrandt F, Sayer JA. Nephrocystin-1 interacts directly with Ack1 and is expressed in human collecting duct. Biochem Biophys Res Commun. 2008;371:877–882.
- Eley L, Gabrielides C, Adams M, Johnson CA, Hildebrandt F, Sayer JA. Jouberin localizes to collecting ducts and interacts with nephrocystin-1. Kidney Int. 2008;74:1139–1149.
- Helou J, Otto EA, Attanasio M, . Mutation analysis of NPHP6/ CEP290 in patients with Joubert syndrome and Senior-Loken syndrome. J Med Genet. 2007;44:657–663.
- Saunier S, Salomon R, Antignac C. Nephronophthisis. Curr Opin Genet Dev. 2005;15:324–331.
- Bolignano D, Coppolino G, Lacquaniti A, . From kidney to cardiovascular diseases: NGAL as a biomarker beyond the confines of nephrology. Eur J Clin Invest. 2010;40:273–276.
- Bolignano D, Donato V, Coppolino G, . Neutrophil gelatinase-associated lipocalin (NGAL) as a marker of kidney damage. Am J Kidney Dis. 2008;52:595–605.