Abstract
Tissue hypoxia is a pathologic feature of many human diseases like cancer, myocardial infarction, stroke, and kidney disease. Convincing data from clinical studies in patients with chronic renal failure point to chronic hypoxia of kidneys as the end result of multiple processes and mechanisms. In acute as well as chronic diseases, tissue hypoxia not only implies a risk of energy deprivation but also induces regulatory mechanisms with profound influence on gene expression. Moreover, once established, accumulating evidence points to this chronic hypoxia as the central player along with final common pathway to end-stage renal disease (ESRD). An evolutionarily preserved oxygen-sensing mechanism enables cells to adapt and maintain homeostasis under hypoxic conditions by transcriptional activation of a host of genes mediating metabolic adaptation, angiogenesis, energy conservation, erythropoiesis, in addition to cell survival. The endogenous oxygen-sensing mechanism incorporates hypoxia-inducible factors (HIFs) that hub cellular response to hypoxia and comprises a family of oxygen-sensitive basic helix–loop–helix proteins that control the cellular transcriptional response to hypoxia. Hypoxia-inducible factor 1 (HIF-1) is thus a significant mediator of physiological responses to acute and chronic hypoxia. Since HIF is activated to suboptimal levels in pathogenic renal states, therapeutic activation holds a promising novel and effective approach to the treatment of ESRD. Current insights into the regulation of HIF may augment the understanding of the role of hypoxia in renal failure progression and may unbolt new options to improve hypoxia tolerance and induce nephroprotection.
OXYGENATION OF THE KIDNEY
Oxygenation of the kidney is characterized by a remarkable paradox in light of some facts: in relation to their weight, kidneys are highly perfused organs of an organism receiving an overall oxygen supply of more than 80 mL/min × 100 g weight, of which less than 10% is only consumed during its passage through the kidneys.Citation1 Moreover, tissue oxygen tensions in the renal parenchyma are also lower than in most other organs.Citation2 The uniqueness of the architecture of the renal vasculature explains the discrepancy between high oxygen supply and low tissue oxygen tensions of the kidney. In renal cortex as well as medulla, branches of the renal arteries and veins run stringently parallel and in close contact with each other over long distances which allows oxygen to diffuse from the arterial system into the venous system before entering the capillary bed,Citation3,4 thus subjecting oxygen to a countercurrent exchange comparable with urea, and this mechanism is particularly relevant in the renal medulla, where it leads to oxygen tensions below 10 mmHg.Citation2 However, preglomerular arteries of the renal cortex also undergo the countercurrent exchange of oxygen, where oxygen tensions are about 30 mmHg with marked variability.Citation2,4 Moreover, most tubular segments have a very limited capacity for anaerobic energy generation and are dependent on oxygen to maintain active transtubular reabsorption of solutes. Thus, the combination of limited tissue oxygen supply and high oxygen demand may be considered as the main reason for the susceptibility of the kidney to acute ischemic injury.Citation5
HYPOXIA-INDUCIBLE FACTORS
Hypoxia-inducible factors (HIFs) belong to a group of transcription factors comprising the basic helix–loop–helix–PAS [per/aryl-hydrocarbon-receptor nuclear translocator (ARNT)/Sim]Citation6 family consisting of a labile α subunit (HIF-1 α, HIF-2 α, or HIF-3 α) and a common stable β (ARNT) subunit that heterodimerize to form a functional transcriptional complex. The heterodimer can translocate to the nucleus and transactivate a myriad of target genes in conjunction with a number of cofactors. The key mechanism of regulation of HIF activity is through oxygen-dependent proteasome degradation of the HIF-α subunit (). Binding of HIF to hypoxia response elements leads to direct transcriptional activation of hypoxia-inducible genes consensus sequences presenting in the regulatory regions.Citation7,8 HIF is considered as a master regulator of gene expression in response to hypoxia. Its target genes include erythropoietin (EPO), angiogenic growth factors, heme oxygenase-1, glucose transporter-1, and almost all glycolytic enzymes.Citation9 In addition, HIF target genes are also involved in cell survival decisions, including apoptosis, and both pro- and antiapoptotic effects have been described.Citation10,11
Figure 1. Hypoxia-inducible factor (HIF) regulation.
Notes: Under normoxic conditions, HIF-α is hydroxylated in the oxygen-dependent destruction domain (Pro402 or 564 of HIF-1 α) and at an asparagine residue in the C-terminal transactivation domain (Asn803 of HIF-1 α). Prolyl hydroxylation by prolyl hydroxylase domain (PHD) enzyme leads to binding of HIF-1 α to the pVHL–E3–ubiquitin ligase complex. HIF-α is consequently polyubiquitinated and degraded by the proteasome. Acetylation of HIF-α by arrest-defective-1 (ARD1) enhances binding to pVHL and subsequent ubiquitination. During hypoxia, prolyl hydroxylases are rendered inactive and HIF-α is stabilized and translocates to the nucleus, where it is SUMOylated by small ubiquitin-like modifier (SUMO) conjugases. Failure to deSUMOylate HIF-1 α [e.g., the absence of a sentrin/SUMO-specific protease 1 (SENP1)] targets HIF-1α for VHL/proteasome-dependent degradation by an alternate signal (prolyl hydroxylation independent) for pVHL binding. DeSUMOylated HIF-1 α escapes degradation, heterodimerizes with HIF-1β, and increases transcription of HIF target genes.
![Figure 1. Hypoxia-inducible factor (HIF) regulation.Notes: Under normoxic conditions, HIF-α is hydroxylated in the oxygen-dependent destruction domain (Pro402 or 564 of HIF-1 α) and at an asparagine residue in the C-terminal transactivation domain (Asn803 of HIF-1 α). Prolyl hydroxylation by prolyl hydroxylase domain (PHD) enzyme leads to binding of HIF-1 α to the pVHL–E3–ubiquitin ligase complex. HIF-α is consequently polyubiquitinated and degraded by the proteasome. Acetylation of HIF-α by arrest-defective-1 (ARD1) enhances binding to pVHL and subsequent ubiquitination. During hypoxia, prolyl hydroxylases are rendered inactive and HIF-α is stabilized and translocates to the nucleus, where it is SUMOylated by small ubiquitin-like modifier (SUMO) conjugases. Failure to deSUMOylate HIF-1 α [e.g., the absence of a sentrin/SUMO-specific protease 1 (SENP1)] targets HIF-1α for VHL/proteasome-dependent degradation by an alternate signal (prolyl hydroxylation independent) for pVHL binding. DeSUMOylated HIF-1 α escapes degradation, heterodimerizes with HIF-1β, and increases transcription of HIF target genes.](/cms/asset/457a982d-7740-443f-b13c-d98558d7b498/irnf_a_653754_f0001_b.jpg)
Under normoxic conditions, members of the prolyl hydroxylase domain (PHD) family of proteins (PHD1, PHD2, and PHD3) in the presence of oxygen, , and 2-oxoglutarate hydroxylate HIF-α at one or both conserved prolyl residues in HIF-α (proline 402 and 564 in HIF-1 α) (). VHL E3 ubiquitin ligase [composed of the von Hippel-Lindau tumor suppressor protein (pVHL), Cullin 2, elongin C, and Rbx1] recognizes hydroxylated HIF-α and polyubiquitinates it and grades it for destruction by the 26S proteasome. Under hypoxic condition, HIF-α escapes degradation, heterodimerizes with HIF-β (ARNT), and binds the transcriptional coactivator, CBP/p300, to form the active transcriptional complex that translocates to the nucleus mediating transactivation of hypoxia response element-containing genes.Citation12–15 Since the intracellular partial pressure of oxygen is at most times below the KM (of PHDs) for oxygen, the oxygen availability is a determinant of PHD activity.Citation16,17 Furthermore, PHD activity can also be modulated by a number of metabolic intermediates or PHD cofactors, including reactive oxygen species (ROS), ascorbate, succinate, fumarate, and nitric oxide (NO) ().Citation18
Figure 2. Regulation of hypoxia-inducible factor (HIF) prolyl hydroxylases.
Notes: HIF prolyl hydroxylase domains (PHDs) require oxygen, Fe2+, and 2-oxoglutarate for prolyl hydroxylation activity. Nitric oxide (NO), reactive oxygen species (ROS), the Krebs cycle metabolites succinate and fumarate, cobalt chloride (CoCl2), and Fe chelators such as desferroxamine may inhibit the PHD family of HIF prolyl hydroxylases during normoxia. A number of HIF prolyl hydroxylase inhibitors are being investigated for preclinical and clinical trials. NGF, nerve growth factor.
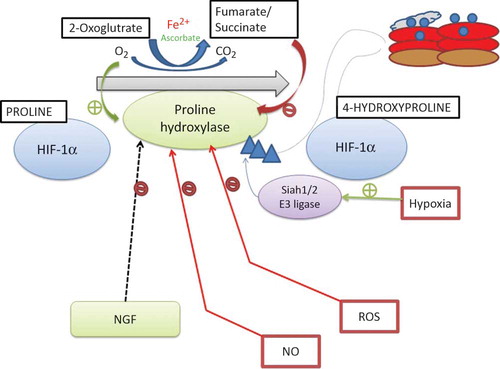
It has recently been revealed that HIF-α is regulated under hypoxic conditions in conjugation with small ubiquitin-like modifiers (SUMO) that regulate its transcriptional activity.Citation19–21 Hypoxia induces SUMOylation of HIF-1 α, through a proline hydroxylation-independent mechanism that promotes its binding to VHL protein, leading to its degradation. A study has demonstrated that mice lacking SENP1 (a sentrin/SUMO-specific protease 1) died from anemia secondary to EPO deficiency due to a defect in deSUMOylation which resulted in HIFs being targeted for degradation under hypoxic conditions.Citation22 The role of simulation in HIF regulation is still under intense investigation; alternative mechanisms have been proposed though. There are compelling data that show HIF-α regulation through mechanisms independent of oxygen content or VHL. For the case in point, Mekhail et al.Citation23,24 reported that a decrease in environmental pH (occurring early in tissue hypoxia before HIF induction by inactivation of PHD activity) resulted in sequestration of VHL in the nucleolus and neutralized VHL’s ability to facilitate degradation of HIF. In addition to hydroxylation at prolyl residues, factor-inhibiting HIF-1 (FIH-1) hydroxylates HIF at a conserved asparagine residue blocking the recruitment of transcriptional coactivators p300 and CBP, thus limiting HIF transactivation.Citation25–33 However, FIH-1 hydroxylates HIF only under normoxia and mild hypoxia but not under pronounced hypoxia, apparently to suppress the activity of HIF-α proteins that escape degradation under moderate hypoxia.Citation34,35 Thus, the complex regulatory pathways that govern HIF-α activity support the perception that inappropriate HIF activation can have far-reaching consequences on different aspects of cell growth, differentiation, and metabolism.
PROLYL HYDROXYLASE
The contribution of HIF prolyl hydroxylases (PHD1, PHD2, and PHD3) to the physiologic regulation of HIF remains uncertain. These respective isoforms have unique but overlapping patterns of tissue expression. Data from experiments using suppression by small interference RNA showed that each contributes in a non-redundant manner to the regulation of both HIF-1 α and HIF-2 α subunits and that the contribution of each PHD is strongly dependent on the abundance of the enzymeCitation36; PHD2 is substantially the most abundant and has dominant effect in most cells. Both PHD2 and PHD3 proteins are induced by hypoxia, the induction of PHD3 being particularly striking in certain cells and thus contributing greater than that of PHD2. PHD3 seems to contribute more substantially to the regulation of HIF-2 α. Many recent studies have been carried out with prolyl hydroxylase inhibitors to stabilize HIF and have been the focus as novel strategies in the treatment of many renovascular diseases.Citation37
Cobalt is now recognized as an inhibitor of PHD and thereby serves as a stimulator of HIF. A study has demonstrated that cobalt therapy leads to a significant erythropoietic response in association with improved appetite and greater tolerance for medications that are necessary to correct electrolyte abnormalities. A study has demonstrated the renoprotective effects of chemical preconditioning with cobaltous chloride in an ischemic model of renal injury.Citation38 In this study administration induced the upregulation of HIF-regulated genes, such as vascular endothelial growth factor (VEGF) and EPO, and protected the kidney against the tubulointerstitial damage induced by hypoxia. Cobalt treatment has also been shown to be effective when given after the initial insult in a chronic progressive glomerulonephritis model, a cyclosporine nephrotoxicity model, and a model of chronic renal failure with glomerular hypertension, demonstrating its therapeutic potential.Citation39–41 Long-term administration of cobalt in humans is hindered by various side effects, although effective results have been obtained in experimental animals. Thus, less toxic and more potent PHD inhibitors have to be sought and a variety of new candidates are now under development.Citation42 Apart from HIF prolyl hydroxylase, there is a single HIF asparaginyl hydroxylase, FIH that has been identified to date. Therapeutic potential of FIH inhibitors is also an interesting subject to be pursued.
RENAL DEVELOPMENT
A mismatch between oxygen supply and demand can result in regions of relative hypoxia and HIF activation during kidney development which is a rapid cell growth phase with congregation of the vasculature. HIF-1 α is expressed in nearly all tissues, whereas HIF-2 α is restricted to the endothelium, kidney, liver, and brain in terms of expression.Citation28 In developing kidney, HIF-1 α is expressed weakly in the outer cortex and strongly in some tubular, collecting duct epithelial cells and HIF-2 α is localized to glomeruli with dense staining in the nuclei of podocytes and microvascular endothelial cells. A study has shown that mice which were genetically deficient in HIF-1 α [embryonic day 9.5 (E9.5)] died at mid-gestation from vascular and neural tube defects.Citation43,44 Moreover, stem cells derived from HIF-1 α−/− mice failed to produce VEGF in response to hypoxia, and embryos displayed abnormalities in the yolk sac vasculature. Mice carrying germline deletions of HIF-2 α had varied phenotypes which were dependent on the genetic background.Citation45 Tian et al.Citation28 also observed bradycardia from defective catecholamine synthesis which led to embryonic lethality at mid-gestation, whereas study by Peng et al.Citation46 showed embryonic lethality at E9.5–E13.5 without any evidence of heart rate or catecholamine differences. Surprisingly, no defects in renal vascular development or nephrogenesis were observed in HIF-2 α null embryos in spite of no compensatory upregulation of HIF-1 α or HIF-3 α.Citation47 It has been possible to draw a few conclusions about the role of HIF genes in renal development using fetal organ culture.Citation48 Taken together, these data signify no identified role for HIF-2 α in renal vascular or glomerular development in mice. Moreover, VEGF is a critical mediator of vasculogenesis and an important downstream effector of HIF-α.Citation49,50 The specific role of VEGF in glomerular development was revealed by injecting VEGF-neutralizing antibodies into newborns during developmental stage of kidney which resulted in fewer nephrons and many abnormal glomeruli lacking glomerular tufts.Citation51 Additionally, glomerular selective deletion or overexpression of VEGF-A in mice may lead to severe and early glomerular disease.Citation52 Eremina and QuagginCitation53,54 demonstrated that tight regulation of VEGF-A signaling is essential for establishing and maintaining glomerular filtration barrier and thus strongly supports a pivotal role for VEGF-A in renal disease. The fundamental role of VEGF produced by podocytes is further highlighted by the development of thrombotic microangiopathy in humans treated with antibody-targeting VEGF and signifies its importance in the maintenance of healthy glomerular endothelial cells and a proper filtration barrier.Citation54
CHRONIC KIDNEY DISEASE
Renal biopsy specimens show that functional impairment of the kidney is better correlated with the degree of tubulointerstitial damage than with glomerular injury.Citation55–57 Fine et al.Citation58 projected that tubulointerstitial injury and loss of peritubular capillaries-induced chronic ischemic injury is the final common pathway to end-stage renal disease (ESRD). Hypoxia upregulates extracellular matrix production, suppresses turnover of collagen, and has been proposed to promote epithelial-to-mesenchymal transition which further promotes fibrosis, a characteristic of chronic renal disease. Thus, HIF induction may be considered as an important facet in the mechanistic role for progression of chronic renal diseases.Citation59,60 Studies from remnant kidney model revealed that tubular hypoxia, stabilization of HIF-1 α, and induction of hypoxia-responsive genes were observed before the histologic evidence of tubulointerstitial damage.Citation61 Tissue-specific ablation of HIF-1 α in mice using Cre-loxP-mediated gene targeting has shown to inhibit the development of tubulointerstitial disease, interstitial collagen deposition, epithelial-to-mesenchymal transition, and inflammation in response to unilateral ureteral obstruction.Citation62 Furthermore, tissue hypoxia (as detected by pimonidazole hydrochloride, which forms protein adducts at an oxygen tension of approximately 8 mmHg) is apparent in as early as 24 h and persists throughout the development of fibrosis, in contrast to tubulointerstitial fibrosis which appears approximately 4 days after ureteral ligation. Concomitantly, both HIF-1 α and HIF-2 α are detected as early as 1 day after ureteral ligation. This study revealed that the profibrotic effect of HIF-1 α was partially attributed to the HIF targets lysyl oxidases (Lox and LoxL2 genes), since pharmacologic blockade of these enzymes ameliorates fibrosis in vivo as well as hypoxia-induced cell migration in vitro.
Polycystic kidney disease is another important cause of chronic kidney disease (CKD) and HIFs have been recently implicated in its pathophysiology. Renal cysts are considered as a premalignant lesion in VHL-deficient clear cell renal cancer where loss of function of VHL leads to constitutive HIF activation suggesting a link between HIF and cystic disease. In support of this conception, a study has demonstrated that mice with conditional knockout of VHL in proximal tubular epithelial cells develop macroscopic and microscopic renal cysts.Citation63 Immunohistochemical analysis of kidneys amid autosomal dominant polycystic disease reveals that HIF-1 α is expressed in cystic epithelial cells and some infiltrating leukocytes, whereas HIF-2 α expression is limited to vascular endothelial cells, pericystic interstitial cells, and neutrophils. Despite the consequences, HIF accumulation in human as well as rat polycystic kidney disease is possibly responsible for increased EPO production and pericystic hypervascularity and may have an impact on progression of polycystic disease. The diverse role of HIFs in acute and chronic renal injuries suggests that the framework, cell type, and duration of HIF expression determine its effect on disease outcome. In addition, the specific roles of HIF-1 α and HIF-2 α and their respective downstream targets in chronic renal disease are unclear.Citation48,64,65
EFFECTS OF LOW OXYGEN TENSIONS ON RENAL CELL FUNCTION
In recent years, it has become clear that variations in oxygen tensions continuously regulate many facets of cellular functions most important being oxygen-dependent gene regulation mediated by the hypoxia-inducible transcription factors (HIF) (). Under normoxic conditions, these transcription factors are not detected in any region of the kidney but the unremittingly low oxygen tensions in the renal medulla.Citation66 Moreover, exposing animals to systemic hypoxia revealed a marked capacity of renal cells to induce a transcriptional response through HIF activation. HIF-1 α accumulated in tubular epithelial cells of most nephron segments, while the expression of HIF-2 α was found in peritubular endothelial cells and fibroblasts and in glomerular cells.Citation66 Ischemia/reperfusion,Citation66,67 renal infarction,Citation68or radiocontrast application Citation69 related acute renal hypoxia also induces HIF. However, data regarding HIF expression in chronic renal disease are quite limited. Tanaka and coworkers generated transgenic rats expressing a reporter gene driven by the HIF DNA-binding site, the “hypoxia response element,” as directly visualizing HIF, which rapidly degrades on reoxygenation is quite difficult. Subsequently, results from two different models, puromycin amino nucleoside nephropathy and the remnant kidney model, showed transgene activation and hence indirect evidence for induction of the HIF pathway.Citation67 Thus, the majority of cellular effects of HIF induction are likely to bestow adaptation and protection against hypoxic injury, and HIF induction is consequently a potential strategy for nephroprotection. Conversely, genes induced by hypoxia may also be maladaptive, promoting renal fibrosis under some circumstances.Citation59 Hypoxia also promotes the transdifferentiation of proximal tubular cells into myofibroblasts.Citation59,60
Conversely, hypoxia may indirectly promote renal injury by increasing the blood pressure. A study has demonstrated that exposure of rats to systemic hypoxia leads to a significant rise in blood pressure associated with microvascular endothelial changes, subtle tubulointerstitial injury, inflammation, and interstitial cell proliferation that persisted after return-to-normal ambient oxygen tensions.Citation70 Moreover, Johnson et al.Citation71 have postulated such microvascular changes as a uniform pathway for the development of salt-sensitive hypertension. Hypoxia modulates the phenotype of isolated papillary cellsCitation72 and plays a central role in stem cell responses in different organs.Citation73,74 The supplementary role of hypoxia is its effect on maintenance and recruitment of adult renal stem cells. A recent data indicate that the renal medulla serves as a forte for adult renal stem cells, which resume proliferation and are recruited into different zones of the kidney after tubular damage.Citation72
EVIDENCE FOR TISSUE HYPOXIA IN CKD
It is uncertain whether impaired renal perfusion affects tissue oxygenation. Since glomerular perfusion determines oxygen supply and oxygen consumption, the effect of changes in glomerular perfusion on the net balance of renal oxygen tensions is complex to predict. Renal artery stenosis has been shown to be an infrequent and mild stimulus for EPO formation, which is normally produced in inverse relationship to peritubular cortical oxygen tension.Citation75 Regrettably, limited data are available about the contours of oxygen tensions in kidneys with different forms of chronic disease. Several investigators have used immunostaining techniques with pimonidazole (bioreductive agent) which accumulates in hypoxic tissues and has been used as a radiosensitizer to treat malignant tumors.Citation76 Reports have revealed increased staining in an experimental glomerulonephritis model,Citation77 puromycin aminonucleoside nephropathy,67 a remnant kidney model,Citation61,67 as well as in an ischemia/reperfusion injury model several weeks after recovery.Citation78 The time course of changes in a progressive model of rat glomerulonephritis suggested that hypoperfusion of peritubular capillaries induces chronic hypoxia before progression of tubulointerstitial fibrosis.Citation77 On the contrary, single study showed higher oxygen tensions in the remnant kidney model using oxygen electrodes, rather than lower oxygen tensions in the diseased kidneys. This increase in renal oxygenation was explained due to an “inadequately” low EPO production despite moderate anemia, which occurs in this model.Citation79 Considering different results, it appears likely that changes in renal oxygen tensions are not uniform, but depend on the type of renal disease, time course of injury, and its progression. The supplementary recordings of tissue oxygen tensions to verify such influences would certainly be desirable in various models, in addition to information about oxygen tensions in human kidneys which would also be of significant interest. Recently, blood oxygen level-dependent (BOLD) magnetic resonance imaging (MRI) technique has been introduced for visualizing changes in renal oxygenation in humans.Citation80 BOLD MRI has also been used to demonstrate changes in oxygenation in an animal model of diabetic nephropathy,Citation80 but its systematic application in patients with CKD is not applied yet.
CHRONIC HYPOXIA IN THE KIDNEY IS MULTIFACTORIAL
Loss of Peritubular Capillaries and Fibrosis in Chronic Renal Disease
Chronic ischemia occurs through a number of mechanisms acting in concert. Histologic studies of human kidneys and various animal models have shown extensive tubulointerstitial injury which is associated with damage to renal arteries and arterioles and with distortion as well as loss of peritubular capillaries.Citation81–85 Thus, peritubular capillary blood supply and oxygenation to the corresponding region is absent in fibrotic kidneys with advanced renal disease. Even if the peritubular capillaries are intact, interstitial fibrosis still impairs the tubular oxygen supply because of the extended distance between the capillaries and tubular cells which reduces the efficiency of oxygen diffusion. Incidentally, hypoxia per se is a profibrogenic stimulus for tubular cells, interstitial fibroblasts, and renal microvascular endothelial cells. Under hypoxic conditions, tubular cells undergo epithelial–mesenchymal transdifferentiation to become myofibroblasts.Citation60 Moreover, hypoxia also activates fibroblasts and changes the extracellular matrix metabolism of resident renal cells.Citation59,86 On the whole, a fibrogenic response leads in turn to the destruction of peritubular capillaries. Additionally, renal tubular cells subjected to severe or long-lasting hypoxia develop mitochondria functional deficits leading to persistent energy deficits, subsequently causing apoptosis of cells.Citation87 Collectively, chronic hypoxia in this compartment leads to transdifferentiation or apoptosis (or both) of tubular cells, activation of resident fibroblasts, and further annihilation and loss of peritubular capillaries with progression of fibrosis which collectively set up a vicious cycle of regional hypoxia and progressive kidney failure in the late stages of disease.
Glomerular Damage and Hypoxia
Hypoxia plays a pathogenic role in the early stage of kidney disease before the development of structural tubulointerstitial injury. Impairment of the glomerular capillary bed, for example, in glomerulosclerosis automatically results in a decrease in peritubular perfusion (peritubular capillaries occur downstream of the glomerular efferent arterioles) and tubular oxygen supply. A study employing a model of accelerated glomerulosclerosis induced by repeated injection of anti-Thy1 antibody in uninephrectomized rats showed a decrease in blood flow in peritubular capillaries using intravital microscopy and physiologic lectin perfusion.Citation77 Thus, sluggish peritubular capillary blood flow is associated with hypoxia in the corresponding tubulointerstitium and precedes the development of histologic tubulointerstitial injury and peritubular capillary loss leading to CKD.
Hemodynamic Maladjustment in the Tubulointerstitium: Imbalance of Vasoactive Substances
An imbalance in vasoactive substances and associated intrarenal vasoconstriction causes chronic hypoxia in the kidney in the early stage of kidney disease even if glomeruli are intact. Futrakul et al.Citation88 from intrarenal hemodynamic studies in patients with severe glomerulonephritis using radioisotope techniques demonstrated that elevated efferent arteriolar resistance and decreased peritubular capillary flow were associated with reversible renal functional impairment and this reversible change in peritubular capillary flow may reflect an improvement in the imbalance of vasoactive substances in the kidney. Recently, these observations were extended to report a correlation between a decrease in peritubular capillary flow and tubular dysfunction in patients with type 2 diabetes and normoalbuminuria.Citation89 Moreover, these results support the conception that chronic hypoxia may induce tubulointerstitial injury eventually leading to ESRD in patients with a variety of kidney diseases. Local activation of renin-angiotensin system (RAS) is considered as the most important component among various vasoactive substances, since it leads to constriction of efferent arterioles, hypoperfusion of postglomerular peritubular capillaries, and subsequent hypoxia of the tubulointerstitium in the downstream compartment which further aggravates renal failure. To clarify the mechanism of these effects, a study employed remnant kidney model in rats induced by ligation of renal artery branches in which RAS was markedly activated. Computer-assisted morphologic analysis demonstrated narrowing and distortion of peritubular capillaries with decreased blood flow and hypoxia in a very early phase in this model before the development of structural kidney damage.Citation61 Besides, angiotensin II also damaged endothelial cells directly which was supported by the notion that administration of angiotensin II to rats causes the loss of peritubular capillaries and this effect was ameliorated by receptor blockade.Citation90,91 Another significant mechanism of angiotensin II-induced ischemia is inefficient cellular respiration and hypoxia via oxidative stress. Thus, angiotensin II induces chronic hypoxia via both hemodynamic and non-hemodynamic mechanisms.
Role of Anemia in Hypoxia of the Kidney
Third National Health and Nutrition Examination Survey and the National Kidney Foundation: Kidney Early Evaluation Program has shown that the risk for anemia significantly increases when glomerular filtration rate (GFR) falls below 60 mL/min per 1.73 m2 divulging the important role of anemia at a relatively early stage of renal dysfunction. The amount of O2 delivered to a specific organ is the product of blood flow and arterial O2 content. Anemia in kidney disease may accelerate the decline in renal function by inducing tubulointerstitial hypoxia.Citation92 A retrospective multivariate logistic analysis of 71,802 subjects performed by Iseki et al.Citation93 and an analysis of the data of the Reduction of Endpoints in non-insulin dependent diabetes mellitus (NIDDM) with the Angiotensin II Antagonist Losartan study of patients with type 2 diabetic nephropathy have confirmed anemia as an independent risk factor for ESRD.Citation94
Oxidative Stress and Inefficient Cellular Respiration
CKD is associated with oxidative stress (). Upregulation of angiotensin II in renal diseases promotes renal oxidative stress by stimulating nicotinamide adenine dinucleotide phosphate (NADPH) oxidase. Furthermore, renal anemia also contributes to oxidative stress as erythrocytes represent a major antioxidant component of the blood. Superoxide anion leads to decreased NO bioavailability through ONOO− formation. Adler and HuangCitation95 have shown that, since NO is a suppressor of mitochondrial respiration, depletion of NO by oxidative stress may stimulate mitochondrial respiration and uncouple it from chemical energy consumption, resulting in tissue hypoxia. A study employing kidneys of the spontaneously hypertensive rats (SHRs), which characteristically underwent oxidative stress, revealed enhanced oxygen usage relative to tubular sodium transport and lower intrarenal pO2.Citation96 Moreover, amelioration of oxidative stress has shown to improve renal oxygenation in a model of diabetic nephropathyCitation97 and in the angiotensin II continuous infusion model.Citation98 It is likely that the renal hypoxia in these models results from a decrease in NO bioavailability and subsequent uncoupling of mitochondrial respiration as a result of oxidative stress which eventually activates oxidative stress-related mechanism causing CKD.Citation99
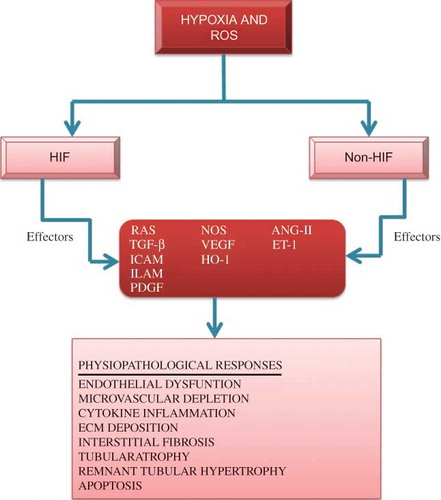
Figure 3. Representation for the association of hypoxia and reactive oxygen species (ROS) in hypoxia-driven renal parenchymal destructive processes and their related mediators.
Notes: HIF and non-HIF mechanisms, such as p53 or STAT3, are the key regulators of these processes. Some effectors may be protective as well as destructive and some with both potentially injurious and protective characteristics. ECM, extra cellular matrix; ICAM, intercellular adhesion molecule 1; ELAM, endothelial-leukocyte adhesion molecule; PDGF, platelet-derived growth factor; ET-1, endothelin-1; HO-1, hemeoxygenase-1; ANG-II, angiotensin-II; TGF-β, transforming growth factor-β; NOS, nitric oxide synthase; RAS, renin-angiotensin system.
Relative Hypoxia as a Result of Increased Metabolic Demand
Cells suffer from relative hypoxia even under the maintenance of otherwise normal blood flow in case of increased metabolic demand. Studies that have used the BOLD MRI technique have demonstrated that streptozotocin-induced diabetic kidneys suffer from tissue hypoxia at an early stage before the development of structural changes.Citation100 A possible explanation for this phenomenon may be the hyperfiltration that occurs early in diabetic nephropathy leading to the increased delivery of sodium to tubular cells, imposing an excessive tubular sodium reabsorption workload relative to oxygen supply, and subsequently resulting in tubular hypoxia. However, whether proteinuria causes functional hypoxia as a result of increased metabolic demand for reabsorption is a key query for future study.
ADMA, DDAH, NOS, AND ENDOTHELIAL DYSFUNCTION
Asymmetric dimethylarginine (ADMA) is an amino acid that has the property of inhibiting the activity of nitric oxide synthases (NOSs) with most potent effect on endogenous endothelial NOS (eNOS) inhibitor.Citation101 The kidney plays a main role in the elimination of ADMA, because of its double function of urinary excretion and enzymatic degradation of ADMA (due to the high dimethylarginine dimethylaminohydrolase (DDAH) content in renal tissue).Citation102,103 Compelling evidence shows that disruption of this enzyme, as in DDAH knockout model, impairs the degradation of ADMA and leads to inhibition of NO synthesis with known developments like hypertension as well as vascular dysfunction.Citation104 Data from experimental studies have revealed that ADMA reduces NO production by eNOS in vitro and in vivo in a dose-dependent mannerCitation105–107 and also that exogenous infusion of ADMA in humans increases systemic vascular resistance, reduces cardiac output, and impairs renal blood flow and sodium excretion.Citation108–110
ADMA, RENAL FUNCTION, AND PROGRESSION IN PATIENTS WITH CKD
Vallance et al.Citation111 have reported markedly increased dimethylarginine blood levels in patients with ESRD receiving hemodialysis therapy. Moreover, a result of several subsequent studies certainly has indicated that increased ADMA blood levels play an important role in the pathophysiology of cardiovascular disease in CKD.Citation101 A recent study has demonstrated that ADMA blood levels are elevated already early in the course of CKD, that is, even before true GFR (assessed by inulin clearance) is reducedCitation112 and hence it is hypothesized that ADMA as a potent NOS inhibitor could be involved in disease progression in CKD patients.Citation108 Additionally, many compelling evidences from clinical studies show that ADMA is indeed involved in the evolution of non-diabeticCitation113,114 and diabeticCitation115,116 progressive nephropathies. In these studies, high ADMA blood concentrations consistently predicted a more rapid decline in renal function. The supplementary support for the role of ADMA in CKD progression comes from recent studies in the rat remnant kidney model (5/6 nephrectomy)Citation117 and in an animal model of diabetic nephropathy.Citation118 Thus, lowering of ADMA blood levels by modulation of DDAH activity may consequently represent a novel therapeutic strategy for arresting progressive CKD. Although up to now results of studies on pharmacological intervention to lower ADMA blood levels were questionable,Citation101,119,120 studies using gene transfer to increase DDAH expression in humans are still hampered by serious technical problems and medical concerns. Thus, currently large prospective interventional studies are lacking that prove lowering of ADMA slows the rate of renal function decline in patients with CKD.
RENAL HYPOXIA RESPONSE: BAD AND GOOD COMPONENTS
Renal hypoxia leads to progression of CKD through an ongoing vicious circle mainly by two initiating factors hypoxia and ROS induced during hypoxia. These initiating events activate transcription factors, HIF, non-HIF, and a host of effectors classified as harmful or protective mediators with regard to the general outcome from the renal perspective. Their action terminates to subsequent physiopathologic processes that participate in the progression of CKD. Among the damaging effectors are the renin–angiotensin–aldosterone axis, endothelin, plasmin-activator inhibitor-I, adhesion molecules, and growth factors which are triggered by hypoxia and induce endothelial dysfunction, vasoconstriction, microvascular depletion, inflammation, fibrosis, and tubular and glomerular damage. Many hypoxia-triggered HIF-dependent protective effectors such as EPO and VEGF rise, which counteracts the effect of the harmful pathways, promoting angiogenesis, improving tissue oxygenation and cell survival, and facilitating homing of progenitor cells.Citation121 In fact, rats with chronic tubulointerstitial diseaseCitation122 or diabetesCitation123 have shown unpredicted resistance to acute hypoxic insults, possibly through hypoxia tolerance, induced by HIF-mediated pathways. However, diabetic kidneys subjected to extreme hypoxic conditions, such as warm ischemia reflowCitation124 or ex vivo perfusion with red cell-free perfusate,Citation123 are highly exposed to hypoxic damage, which suggests that HIF-mediated renoprotection is ineffective beyond a “window of opportunity” of moderate acute hypoxic stress.Citation125 Thus, the gross distinction between the harmful and protective effectors is not always easily done, but depends to a large extent on the intensity and persistency of their expression and on the concomitant action of other mediators. Some may have twofold actions: for example, NO synthase isoforms not only mediate vasodilation, but also lead to the formation of peroxynitrite.Citation126 Norman and FineCitation121 have recently provided details of the complex associations of the various effectors with kidney hypoxia. With respect to the complex structure of the kidney and its vasculature, evaluating the exact location of an upregulated system is highly important.
CONCLUSION
From compelling evidences and data available, inhibition of the activity of HIF hydroxylase enzymes offers a way of activating HIF-1 in normal tissues and of potentiating the HIF response in moderate hypoxia. Potential application of this may be to improve outcomes in ischemic conditions. It is appealing that a compound developed as an inhibitor of collagen prolyl-4-hydroxylase, which is also a potent inhibitor of HIF hydroxylases, was recently shown to improve outcome in a rat model of myocardial infarction.Citation93 However, a concern arises since such an approach would activate HIF-1 in all cells with widespread effects, but selectivity among the different enzymes will be achievable, which may allow a degree of targeting.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
REFERENCES
- Brezis M, Rosen S, Silva P, Epstein FH. Renal ischemia: A new perspective. Kidney Int. 1984;26:375–383.
- Lubbers DW, Baumgartl H. Heterogeneities and profiles of oxygen pressure in brain and kidney as examples of the pO2 distribution in the living tissue. Kidney Int. 1997;51:372–380.
- Zhang W, Edwards A. Oxygen transport across vasa recta in the renal medulla. Am J Physiol Heart Circ Physiol. 2002;283:H1042–H1055.
- Schurek HJ, Jost U, Baumgartl H, . Evidence for a preglomerular oxygen diffusion shunt in rat renal cortex. Am J Physiol. 1990;259:F910–F915.
- Brezis M, Rosen S. Hypoxia of the renal medulla—its implications for disease. N Engl J Med. 1995;332:647–655.
- Semenza GL. Hypoxia-inducible factor 1: Master regulator of O2 homeostasis. Curr Opin Genet Dev. 1998;8:588–594.
- Semenza GL. HIF-1, O(2), and the 3 PHDs: How animal cells signal hypoxia to the nucleus. Cell. 2001;107:1–3.
- Zhu H, Bunn HF. Signal transduction: How do cells sense oxygen? Science. 2001;292:449–451.
- Maxwell P. HIF-1: An oxygen response system with special relevance to the kidney. J Am Soc Nephrol. 2003;14:2712–2722.
- Carmeliet P, Dor Y, Herbert JM, . Role of HIF-1alpha in hypoxia-mediated apoptosis, cell proliferation and tumour angiogenesis. Nature. 1998;394:485–490.
- Greijer AE, Van Derwall E. The role of hypoxia inducible factor 1 (HIF-1) in hypoxia induced apoptosis. J Clin Pathol. 2004;57:1009–1014.
- Bruick RK, McKnight SL. A conserved family of prolyl-4-hydroxylases that modify HIF. Science. 2001;294:1337–1340.
- Epstein AC, Gleadle JM, McNeill LA, . C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell. 2001;107:43–54.
- Ivan M, Kondo K, Yang H, . HIF alpha targeted for VHL mediated destruction by proline hydroxylation: Implications for O2 sensing. Science. 2001;292:464–468.
- Jaakkola P, Mole DR, Tian YM, . Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292:468–472.
- Ehrismann D, Flashman E, Genn DN, . Studies on the activity of the hypoxia-inducible-factor hydroxylases using an oxygen consumption assay. Biochem J. 2007;401:227–234.
- Koivunen P, Hirsila M, Kivirikko KI, Myllyharju J. The length of peptide substrates has a marked effect on hydroxylation by the hypoxia-inducible factor prolyl 4-hydroxylases. J Biol Chem. 2006;281:28712–28720.
- Kaelin Jr, WG, Ratcliffe PJ. Oxygen sensing by metazoans: The central role of the HIF hydroxylase pathway. Mol Cell. 2008;30:393–402.
- Carbia-Nagashima A, Gerez J, Perez-Castro C, . RSUME, a small RWD-containing protein, enhances SUMO conjugation and stabilizes HIF-1alpha during hypoxia. Cell. 2007;131:309–323.
- Bae SH, Jeong JW, Park JA, . Sumoylation increases HIF-1alpha stability and its transcriptional activity. Biochem Biophys Res Commun. 2004;324:394–400.
- Berta MA, Mazure N, Hattab M, Pouyssegur J, Brahimi-Horn MC. SUMOylation of hypoxia-inducible factor-1alpha reduces its transcriptional activity. Biochem Biophys Res Commun. 2007;360:646–652.
- Cheng J, Kang X, Zhang S, Yeh ET. SUMO specific protease 1 is essential for stabilization of HIF1alpha during hypoxia. Cell. 2007;131:584–595.
- Mekhail K, Khacho M, Gunaratnam L, Lee S. Oxygen sensing by H+: Implications for HIF and hypoxic cell memory. Cell Cycle. 2004;3:1027–1029.
- Mekhail K, Gunaratnam L, Bonicalzi ME, Lee S. HIF activation by pH-dependent nucleolar sequestration of VHL. Nat Cell Biol. 2004;6:642–647.
- Ema M, Taya S, Yokotani N, Sogawa K, Matsuda Y, Fujii-Kuriyama Y. A novel bHLH-PAS factor with close sequence similarity to hypoxia-inducible factor 1alpha regulates the VEGF expression and is potentially involved in lung and vascular development. Proc Natl Acad Sci USA. 1997;94:4273–4278.
- Flamme I, Frohlich T, von Reutern M, Kappel A, Damert A, Risau W. HRF, a putative basic helix-loop-helix-PAS-domain transcription factor is closely related to hypoxia-inducible factor-1 alpha and developmentally expressed in blood vessels. Mech Dev. 1997;63:51–60.
- Hogenesch JB, Chan WK, Jackiw VH, . Characterization of a subset of the basic-helix-loop-helix-PAS superfamily that interacts with components of the dioxin signaling pathway. J Biol Chem. 1997;272:8581–8593.
- Tian H, McKnight SL, Russell DW. Endothelial PAS domain protein 1 (EPAS1), a transcription factor selectively expressed in endothelial cells. Genes Dev. 1997;11:72–82.
- O’Rourke JF, Tian YM, Ratcliffe PJ, Pugh CW. Oxygen-regulated and transactivating domains in endothelial PAS protein 1: Comparison with hypoxia-inducible factor-1alpha. J Biol Chem. 1999;274:2060–2071.
- Mahon PC, Hirota K, Semenza GL. FIH-1. A novel protein that interacts with HIF-1alpha and VHL to mediate repression of HIF-1 transcriptional activity. Genes Dev. 2001;15:2675–2686.
- Wiesener MS, Turley H, Allen WE, . Induction of endothelial PAS domain protein-1 by hypoxia: Characterization and comparison with hypoxia inducible factor-1alpha. Blood. 1998;92:2260–2268.
- Hewitson KS, McNeill LA, Riordan MV, . Hypoxia-inducible factor (HIF) asparagine hydroxylase is identical to factor inhibiting HIF (FIH) and is related to the cupin structural family. J Biol Chem. 2002;277:26351–26355.
- Lando D, Peet DJ, Gorman JJ, Whelan DA, Whitelaw ML, Bruick RK. FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev. 2002;16:1466–1471.
- Dayan F, Roux D, Brahimi-Horn MC, Pouyssegur J, Mazure NM. The oxygen sensor factor-inhibiting hypoxia-inducible factor-1 controls expression of distinct genes through the bifunctional transcriptional character of hypoxia-inducible factor-1alpha. Cancer Res. 2006;66:3688–3698.
- Koivunen P, Hirsila M, Gunzler V, Kivirikko KI, Myllyharju J. Catalytic properties of the asparaginyl hydroxylase (FIH) in the oxygen sensing pathway are distinct from those of its prolyl 4-hydroxylases. J Biol Chem. 2004;279:9899–9904.
- Appelhoff RJ, Tian YM, Raval RR, . Differential function of the prolyl hydroxylases PHD1, PHD2, and PHD3 in the regulation of hypoxia-inducible factor. J Biol Chem. 2004;279:38458–38465.
- Gardner HF. The use of cobaltous chloride in the anemia associated with chronic renal disease. J Lab Clin Med. 1953;41:56–64.
- Matsumoto M, Makino Y, Tanaka T, . Induction of renoprotective gene expression by cobalt ameliorates ischemic injury of the kidney in rats. J Am Soc Nephrol. 2003;14:1825–1832.
- Tanaka T, Matsumoto M, Inagi R, . Induction of renoprotective gene expression by cobalt ameliorates tubulointerstitial injury in the progressive Thy1 nephritis model. Kidney Int. 2005;68(6):2714–2725.
- Tanaka T, Kojima I, Ohse T, . Hypoxia-inducible factor (HIF) modulates tubular cell survival in cisplatin nephrotoxicity. Am J Physiol Renal Physiol. 2005;289:F1123–F1133.
- Tanaka T, Kojima I, Ohse T, . Cobalt promotes angiogenesis via hypoxia inducible factors and protects ischemic tubulointerstitium in the remnant kidney. Lab Invest. 2005;85:1292–1307.
- Warnecke C, Griethe W, Weidemann A, . Activation of the hypoxia-inducible factor-pathway and stimulation of angiogenesis by application of prolyl hydroxylase inhibitors. FASEB J. 2003;17:1186–1188.
- Ryan HE, Lo J, Johnson RS. HIF-1 alpha is required for solid tumor formation and embryonic vascularization. EMBO J. 1998;17:3005–3015.
- Iyer NV, Kotch LE, Agani F, . Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 alpha. Genes Dev. 1998;12:149–162.
- Doedens A, Johnson RS. Transgenic models to understand hypoxia-inducible factor function. Methods Enzymol. 2007;435:87–105.
- Peng J, Zhang L, Drysdale L, Fong GH. The transcription factor EPAS-1/hypoxia-inducible factor 2alpha plays an important role in vascular remodeling. Proc Natl Acad Sci USA. 2000;97:8386–8391.
- Maltepe E, Schmidt JV, Baunoch D, Bradfield CA, Simon MC. Abnormal angiogenesis and responses to glucose and oxygen deprivation in mice lacking the protein ARNT. Nature. 1997;386:403–407.
- Steenhard BM, Freeburg PB, Isom K, . Kidney development and gene expression in the HIF2alpha knockout mouse. Dev Dyn. 2007;236:1115–1125.
- Ferrara N, Carver-Moore K, Chen H, . Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature. 1996;380:439–442.
- Carmeliet P, Ferreira V, Breier G, . Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature. 1996;380:435–439.
- Kitamoto Y, Tokunaga H, Tomita K. Vascular endothelial growth factor is an essential molecule for mouse kidney development: Glomerulogenesis and nephrogenesis. J Clin Invest. 1997;99:2351–2357.
- Eremina V, Sood M, Haigh J, . Glomerular-specific alterations of VEGF-A expression lead to distinct congenital and acquired renal diseases. J Clin Invest. 2003;111:707–716.
- Eremina V, Quaggin SE. The role of VEGF-A in glomerular development and function. Curr Opin Nephrol Hypertens. 2004;13:9–15.
- Eremina V, Jefferson JA, Kowalewska J, . VEGF inhibition and renal thrombotic microangiopathy. N Engl J Med. 2008;358:1129–1136.
- Risdon RA, Sloper JC, De Wardener HE. Relationship between renal function and histological changes found in renal-biopsy specimens from patients with persistent glomerular nephritis. Lancet. 1968;2:363–366.
- Schainuck LI, Striker GE, Cutler RE, Benditt EP. Structural-functional correlations in renal disease: II. The correlations. Hum Pathol. 1970;1:631–641.
- Striker GE, Schainuck LI, Cutler RE, Benditt EP. Structural-functional correlations in renal disease: I. A method for assaying and classifying histopathologic changes in renal disease. Hum Pathol. 1970;1:615–630.
- Fine LG, Bandyopadhay D, Norman JT. Is there a common mechanism for the progression of different types of renal diseases other than proteinuria? Towards the unifying theme of chronic hypoxia. Kidney Int Suppl. 2000;75:S22–S26.
- Norman JT, Clark IM, Garcia PL. Hypoxia promotes fibrogenesis in human renal fibroblasts. Kidney Int. 2000;58:2351–2366.
- Manotham K, Tanaka T, Matsumoto M, . Transdifferentiation of cultured tubular cells induced by hypoxia. Kidney Int. 2004;65:871–880.
- Manotham K, Tanaka T, Matsumoto M, . Evidence of tubular hypoxia in the early phase in the remnant kidney model. J Am Soc Nephrol. 2004;15:1277–1288.
- Higgins DF, Kimura K, Bernhardt WM, . Hypoxia promotes fibrogenesis in vivo via HIF-1 stimulation of epithelial-to-mesenchymal transition. J Clin Invest. 2007;117:3810–3820.
- Rankin EB, Tomaszewski JE, Haase VH. Renal cyst development in mice with conditional inactivation of the von Hippel-Lindau tumor suppressor. Cancer Res. 2006;66:2576–2583.
- Raval RR, Lau KW, Tran MG, . Contrasting properties of hypoxia-inducible factor 1 (HIF-1) and HIF-2 in von Hippel-Lindau-associated renal cell carcinoma. Mol Cell Biol. 2005;25:5675–5686.
- Hu CJ, Iyer S, Sataur A, Covello KL, Chodosh LA, Simon MC. Differential regulation of the transcriptional activities of hypoxia inducible factor 1 alpha (HIF-1alpha) and HIF-2alpha in stem cells. Mol Cell Biol. 2006;26:3514–3526.
- Rosenberger C, Mandriota S, Jurgensen JS, . Expression of hypoxia-inducible factor-1alpha and -2alpha in hypoxic and ischemic rat kidneys. J Am Soc Nephrol. 2002;13:1721–1732.
- Tanaka T, Miyata T, Inagi R, . Hypoxia in renal disease with proteinuria and/or glomerular hypertension. Am J Pathol. 2004;165:1979–1992.
- Rosenberger C, Griethe W, Gruber G, . Cellular responses to hypoxia after renal segmental infarction. Kidney Int. 2003;64:874–886.
- Rosenberger C, Heyman SN, Rosen S, . Up-regulation of HIF in experimental acute renal failure: Evidence for a protective transcriptional response to hypoxia. Kidney Int. 2005;67:531–542.
- Mazzali M, Jefferson JA, Ni Z, . Microvascular and tubulointerstitial injury associated with chronic hypoxia-induced hypertension. Kidney Int. 2003;63:2088–2093.
- Johnson RJ, Herrera-Acosta J, Schreiner GF, Rodrigueziturbe B. Subtle acquired renal injury as a mechanism of saltsensitive hypertension. N Engl J Med. 2002;346:913–923.
- Oliver JA, Maarouf O, Cheema FH, . The renal papilla is a niche for adult kidney stem cells. J Clin Invest. 2004;114:795–804.
- Adelman DM, Gertsenstein M, Nagy A, . Placental cell fates are regulated in vivo by HIF-mediated hypoxia responses. Genes Dev. 2000;14:3191–3203.
- Ceradini DJ, Kulkarni AR, Callaghan MJ, . Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat Med. 2004;10:858–864.
- Pagel H, Jelkmann W, Weiss C. A comparison of the effects of renal artery constriction and anemia on the production of erythropoietin. Pflugers Arch. 1988;413:62–66.
- Hodgkiss RJ. Use of 2-nitroimidazoles as bioreductive markers for tumour hypoxia. Anticancer Drug Des. 1998;13:687–702.
- Matsumoto M, Tanaka T, Yamamoto T, . Hypoperfusion of peritubular capillaries induces chronic hypoxia before progression of tubulointerstitial injury in a progressive model of rat glomerulonephritis. J Am Soc Nephrol. 2004;15:1574–1581.
- Basile DP, Donohoe DL, Roethe K, Mattson DL. Chronic renal hypoxia after acute ischemic injury: Effects of L-arginine on hypoxia and secondary damage. Am J Physiol Renal Physiol. 2003;284:F338–F348.
- Priyadarshi A, Periyasamy S, Burke TJ, . Effects of reduction of renal mass on renal oxygen tension and erythropoietin production in the rat. Kidney Int. 2002;61:542–546.
- Prasad PV, Edelman RR, Epstein FH. Noninvasive evaluation of intrarenal oxygenation with BOLD MRI. Circulation. 1996;94:3271–3275.
- Bohle A, von Gise H, Mackensen-Haen S, Stark-Jakob B. The obliteration of the postglomerular capillaries and its influence upon the function of both glomeruli and tubuli. Functional interpretation of morphologic findings. Klin Wochenschr. 1981;59:1043–1051.
- Choi YJ, Chakraborty S, Nguyen V, . Peritubular capillary loss is associated with chronic tubulointerstitial injury in human kidney: Altered expression of vascular endothelial growth factor. Hum Pathol. 2000;31:1491–1497.
- Yuan H-T, Li X-Z, Pitera JE, Long DA, Woolf AS. Peritubular capillary loss after mouse acute nephrotoxicity correlates with down-regulation of vascular endothelial growth factor-A and hypoxia-inducible factor-1alpha. Am J Pathol. 2003;163:2289–2301.
- Kairaitis LK, Wang Y, Gassmann M, Tay YC, Harris DC. HIF-1alpha expression follows microvascular loss in advanced murine adriamycin nephrosis. Am J Physiol Renal Physiol. 2005;288:F198–F206.
- Ohashi R, Kitamura H, Yamanaka N. Peritubular capillary injury during the progression of experimental glomerulonephritis in rats. J Am Soc Nephrol. 2000;11:47–56.
- Norman JT, Orphanides C, Garcia P, Fine LG. Hypoxia induced changes in extracellular matrix metabolism in renal cells. Exp Nephrol. 1999;7:463–469.
- Tanaka T, Hanafusa N, Ingelfinger JR, Ohse T, Fujita T, Nangaku M. Hypoxia induces apoptosis in SV40-immortalized rat proximal tubular cells through the mitochondrial pathways, devoid of HIF-1-mediated upregulation of Bax. Biochem Biophys Res Commun. 2003;309:222–231.
- Futrakul N, Tohsukhowong P, Patumraj S, Siriviriyakuk P, Tipprukmas N, Futrakul P. Treatments of hemodynamic maladjustment and oxidative stress prevent renal disease progression in chronically severe glomerulonephritides. Ren Fail. 2003;25:839–844.
- Futrakul N, Vongthavarawat V, Sirisalipotch S, Chairatanarat T, Futrakul P, Suwanwalaikorn S. Tubular dysfunction and hemodynamic alteration in normoalbuminuric type 2 diabetes. Clin Hemorheol Microcirc. 2005;32:59–65.
- Lombardi D, Gordon KL, Polinsky P, Suga S, Schwartz SM, Johnson RJ. Salt-sensitive hypertension develops after short-term exposure to angiotensin II. Hypertension. 1999;33:1013–1019.
- Shao J, Nangaku M, Miyata T, . Imbalance of T-cell subsets in angiotensin II-infused hypertensive rats with kidney injury. Hypertension. 2003;42:31–38.
- Astor BC, Muntner P, Levin A, Eustace JA, Coresh J. Association of kidney function with anemia: The Third National Health and Nutrition Examination Survey (1988– 1994). Arch Intern Med. 2002;162:1401–1408.
- Iseki K, Ikemiya Y, Iseki C, Takishita S. Hematocrit and the risk of developing end-stage renal disease. Nephrol Dial Transplant. 2003;18:899–905.
- Mohanram A, Zhang Z, Shahinfar S, Keane WF, Brenner BM, Toto RD. Anemia and end-stage renal disease in patients with type 2 diabetes and nephropathy. Kidney Int. 2004;66:1131–1138.
- Adler S, Huang H. Impaired regulation of renal oxygen consumption in spontaneously hypertensive rats. J Am Soc Nephrol. 2002;13:1788–1794.
- Welch WJ, Baumgartl H, Lubbers D, Wilcox CS. Nephron pO2 and renal oxygen usage in the hypertensive rat kidney. Kidney Int. 2001;59:230–237.
- Palm F, Cederberg J, Hansell P, Liss P, Carlsson PO. Reactive oxygen species cause diabetes-induced decrease in renal oxygen tension. Diabetologia. 2003;46:1153–1160.
- Welch WJ, Blau J, Xie H, Chabrashvili T, Wilcox CS. Angiotensin-induced defects in renal oxygenation: Role of oxidative stress. Am J Physiol Heart Circ Physiol. 2005;288:H22–H28.
- Adler S, Huang H, Wolin MS, Kaminski PM. Oxidant stress leads to impaired regulation of renal cortical oxygen consumption by nitric oxide in the aging kidney. J Am Soc Nephrol. 2004;15:52–60.
- Ries M, Basseau F, Tyndal B, . Renal diffusion and BOLD MRI in experimental diabetic nephropathy. Blood oxygen level dependent. J Magn Reson Imaging. 2003;17:104–113.
- Kielstein JT, Fliser D. The past, presence and future of ADMA in nephrology. Nephrol Ther. 2007;3:47–54.
- Tanaka M, Sydow K, Gunawan F, . DDAH overexpression suppresses graft coronary artery disease. Circulation. 2005;112:1549–1556.
- Jacobi J, Sydow K, von Degenfeld G, . Overexpression of dimethylarginine dimethylaminohydrolase reduces tissue asymmetric dimethylarginine levels and enhances angiogenesis. Circulation. 2005;111:1431–1438.
- Leiper J, Nandi M, Torondel B, . Disruption of methylarginine metabolism impairs vascular homeostasis. Nat Med. 2007;13:198–203.
- Segarra G, Medina P, Ballester RM, . Effects of some guanidino compounds on human cerebral arteries. Stroke. 1999;30:2206–2210.
- Segarra G, Medina P, Vila JM, . Inhibition of nitric oxide activity by arginine analogs in human renal arteries. Am J Hypertens. 2001;14:1142–1148.
- Gardiner SM, Kemp PA, Bennett T, . Regional and cardiac hemodynamic effects of NG, NG,dimethyl-L-arginine and their reversibility by vasodilators in conscious rats. Br J Pharmacol. 1993;110:145714–145764.
- Kielstein JT, Impraim B, Simmel S, . Cardiovascular effects of systemic nitric oxide synthase inhibition with asymmetrical dimethylarginine in humans. Circulation. 2004;109:172–177.
- Achan V, Broadhead M, Malaki M, . Asymmetric dimethylarginine causes hypertension and cardiac dysfunction in humans and is actively metabolized by dimethylarginine dimethylaminohydrolase. Arterioscler Thromb Vasc Biol. 2003;23:1455–1459.
- Kielstein JT, Simmel S, Bode-Boger SM, . Subpressor dose asymmetric dimethylarginine (ADMA) modulates renal function in humans. Kidney Blood Press Res. 2004;27:143–147.
- Vallance P, Leone A, Calver A, . Accumulation of an endogenous inhibitor of nitric oxide synthesis in chronic renal failure. Lancet. 1992;339:572–575.
- Kielstein JT, Boger RH, Bode-Boger SM, . Marked increase of asymmetric dimethylarginine in patients with incipient primary chronic renal disease. J Am Soc Nephrol. 2002;13:170–176.
- Fliser D, Kronenberg F, Kielstein JT, . Asymmetric dimethylarginine and progression of chronic kidney disease: The mild to moderate kidney disease study. J Am Soc Nephrol. 2005;16:2456–2461.
- Ravani P, Tripepi G, Malberti F, Testa S, Mallamaci F, Zoccali C. Asymmetrical dimethylarginine predicts progression to dialysis and death in patients with chronic kidney disease: A competing risks modeling approach. J Am Soc Nephrol. 2005;16:2449–2455.
- Lajer M, Tarnow L, Jorsal A, . Plasma concentration of asymmetric dimethylarginine (ADMA) predicts cardiovascular morbidity and mortality in type 1 diabetic patients with diabetic nephropathy. Diabetes Care. 2008;31:747–752.
- Hanai K, Babazono T, Nyumura I, . Asymmetric dimethylarginine is closely associated with the development and progression of nephropathy in patients with type 2 diabetes. Nephrol Dial Transplant. 2009;24:1884–1888.
- Matsumoto Y, Ueda S, Yamagishi S, . Dimethylarginine dimethylaminohydrolase prevents progression of renal dysfunction by inhibiting loss of peritubular capillaries and tubulointerstitial fibrosis in a rat model of chronic kidney disease. J Am Soc Nephrol. 2007;18:1525–1533.
- Shibata R, Ueda S, Yamagishi S, . Involvement of asymmetric dimethylarginine (ADMA) in tubulointerstitial ischemia in the early phase of diabetic nephropathy. Nephrol Dial Transplant. 2009;24:1162–1169.
- Fliser D, Wagner KK, Loos A, Tsikas D, Haller H. Chronic angiotensin II subtype 1-receptor inhibition reduces (intra)renal vascular resistance in patients with type 2 diabetes mellitus. J Am Soc Nephrol. 2005;16:1135–1140.
- Aslam S, Santha T, Leone A, Wilcox C. Effects of amlodipine and valsartan on oxidative stress and plasma methylarginines in end-stage renal disease patients on hemodialysis. Kidney Int. 2006;70:2109–2115.
- Norman JT, Fine LG. Intrarenal oxygenation in chronic renal failure. Clin Exp Pharmacol Physiol. 2006;33:989–996.
- Goldfarb M, Rosenberger C, Abassi Z, . Acute-on-chronic renal failure in the rat: Functional compensation and hypoxia tolerance. Am J Nephrol. 2006;26:22–33.
- Rosenberger C, Goldfarb M, Khamaisi M, . Acute kidney injury in the diabetic rat: Studies in the isolated perfused and intact kidney. Am J Nephrol. 2008;28:831–839.
- Melin J, Hellberg O, Fellstrom B. Hyperglycaemia and renal ischemia-reperfusion injury. Nephrol Dial Transplant. 2003;18:460–462.
- Rosenberger C, Shina A, Rosen S, Goldfarb M, Eckardt K, Heyman SN. Hypoxia inducible factors and tubular cell survival in isolated perfused kidneys. Kidney Int. 2006;70:60–70.
- Doi K, Noiri E, Nakao A, Fujita T, Kobayashi S, Tokunaga K. Functional polymorphisms in the vascular endothelial growth factor gene are associated with development of end-stage renal disease in males. J Am Soc Nephrol. 2006;17:823–830.