Abstract
Renal impairment is a frequent accompaniment post-myocardial infarction (MI) heart failure. However, the mechanisms and predictors are yet poorly understood. The present study aimed to explore early markers for renal impairment and to test the hypothesis that angiotensin II type 1 receptor (AT1R) blocker exerted renoprotection by regulating local angiotensin II receptors post-MI heart failure. Sprague–Dawley rats underwent ligation of the left descending coronary artery and were treated with losartan (20 mg/kg/day) or vehicle for 3 or 9 weeks. Samples of urine, blood, and kidney were collected for assessment of renal function, histology, and protein changes. The current study revealed that blood cystatin C, rather than serum creatinine and blood urea nitrogen, as well as urine proteins, increased post-MI heart failure significantly. These changes were associated with increased immunohistochemical staining of AT1R and AT2R proteins, accompanied by increased renal fibrosis, tubular necrosis, and inflammatory cell infiltration. Treatment with losartan for MI rats significantly attenuated upregulated AT1R but not AT2R. Losartan also decreased blood cystatin C levels and attenuated renal fibrosis, tubular necrosis, and inflammatory cell infiltration. In conclusion, blood cystatin C may be a better marker for early renal impairment. AT1R blockers modulated local angiotensin II receptors, as well as inflammatory reaction and profibrotic effects, providing potential clinical application in the setting of cardiorenal syndrome post-MI.
INTRODUCTION
Post-myocardial infarction (MI) heart failure is a leading cause of mortality and morbidity in developed countries. Accelerated progressive decline in renal function is a frequent accompaniment of MI, and the presence of renal dysfunction will further increase approximately three times mortality more than those without renal impairment of MI patients during hospitalization.Citation1–3 Therefore, it is of paramount importance to explore early predictors for the onset of renal impairment and to provide effective strategies to prevent the progression of renal impairment post-MI heart failure.
However, the mechanisms underlying the renal impairment following MI with subsequent heart failure are yet not fully clear. Neurohormonal activation of the renin–angiotensin–system (RAS), including tissue-based RAS, in both MI patients and experimental MI animals, may be important modulators that have detrimental effects on post-MI renal function.Citation1,3,4 MI will significantly induce enhanced activation of the intrarenal RAS marked by increased renal levels of RAS components, such as renin mRNA, angiotensinogen mRNA, and angiotensin II (AngII) concentration.Citation4 Growing evidence has suggested that undue activation of the local RAS within the kidneys in an inappropriate fashion is an important contributor to the pathogenesis of renal injury, while intervention with RAS inhibitions will provide renoprotective effects after MI treatment.Citation5,6
Till now, data on renal pathology are mostly limited to functional hemodynamic changes in models of MI with subsequent heart failure. These are still devoid of studies exploring the role of intrarenal RAS in early renal impairment in MI subjects. Therefore, we hypothesized that MI-triggering activation of intrarenal AngII type 1 receptor (AT1R) contributed to renal impairment, which could be inhibited by AT1R blockers (ARBs). The present study aimed to determine which markers could be used to predict the onset of early renal impairment and the changes of local AngII receptors (AT1R and AT2R), as well as renal histopathology, in rat MI models with or without losartan treatment and to explore the potential mechanisms involved in the process of these changes.
MATERIALS AND METHODS
Animals and Experimental Myocardial Infarction
The animals used in these experiments were treated in accordance with the Guide for the Care and Use of Laboratory Animals (NIH Publication No. 85-23, revised 1996), and the study protocols were approved by the Animal Ethics Committee of Sun Yat-sen University.
Experimental MI in rats was induced by left coronary artery ligation by our method described previously with a minor modification.Citation7 Briefly, male Sprague–Dawley rats weighing 280–310 g were anesthetized with ketamine/xylazine (50/5 mg/kg). Adequate anesthesia was ensured by steady states of monitoring breathing rate, heart rate, and arterial blood pressure, as well as the absence of a withdrawal response to a hard pinch on the foot. After obtaining a steady anesthesia, the rats were intubated and then ventilated with a rodent respirator. The electrocardiogram leads were continuously monitored. A left thoracotomy was performed via the left fourth intercostal space and the heart was exposed. After the pericardium was opened, the left anterior descending coronary artery was ligated with a 6–0 silk suture. Successful ligation of coronary artery was confirmed by color change in the ischemic area and the elevation of the ST segment in electrocardiogram monitoring. The chest was then closed with a soft tube in the cavity in order to withdraw air or blood. After ventilation with the room air for 2–5 min, the animal was gradually weaned from the respirator once spontaneous respiration resumed and remained in a supervised setting until completely conscious. Sham-operated animals were performed the same surgical procedures except ligations.
Experimental Groups and Treatment
Forty-eight hours after surgery, survival MI animals were randomly allocated to treat with losartan (20 mg/kg/day) or vehicle. The male rats in this study were randomly divided into four groups: normal (n = 8 for 3 and 9 weeks, respectively), sham-operated (sham) (n = 8 for 3 and 9 weeks, respectively), MI (n = 8 for 3 and 9 weeks, respectively), and MI plus losartan (MI + los) (n = 10 for 3 and 9 weeks, respectively). Losartan treatment was initiated 2 days post-MI. The administration of drug was performed via gastric gavage daily for 3 or 9 weeks. Double distilled water was also given as vehicle by gastric gavage to rats without losartan treatment to avoid the possible physiological alterations associated with gavage-induced stress.
Biochemical Examinations in Experimental Animals
Trunk or tail vessel blood at different time points (2-day, 3-week, 6-week, and 9-week post-MI) was collected for creatinine and blood urea nitrogen (BUN) assessment. After these were obtained, the blood samples were centrifuged and then serum were stored at −80°C for further analysis. The serum creatinine detecting method employed picric acid reaction (Beijing Leadman Biochemistry Technology Co. Ltd., Beijing, China), and BUN was measured using a urease/glutamate dehydrogenase coupled enzymatic technique (Beijing Leadman Biochemistry Technology Co. Ltd., Beijing, China).
After a 24-h acclimatization period, the urine samples collected from metabolic cages at the same time points (2-day, 3-week, 6-week, and 9-week post-MI) were centrifuged and supernatants were stored at −20°C for urinary total protein analysis. Urinary concentration of total protein was determined by colorimetric assays using a protein assay kit (Beijing Leadman Biochemistry Technology Co. Ltd., Beijing, China).
Enzyme-Linked Immunosorbent Assay
The serum samples collected at each time point (2-day, 3-week, 6-week, and 9-week post-MI) were tested for cystatin C contents. Enzyme-linked immunosorbent assays were performed using commercial kits against cystatin C (MSCTC0, R&D Systems) according to the manufacturer’s instructions.
Histological and Immunohistochemical Examinations
After 3- or 9-week treatment period, rat kidney tissues were obtained for histological and immunohistochemical assessments. Immediately after anesthesia with overdose of sodium pentobarbital (100 mg/kg, i.p.), the left kidney tissue was cut and fixed in 4% formalin and embedded in paraffin for histological and immunohistochemical assays. Kidney sections (4 μm) were stained with hematoxylin and eosin (HE) for routine histological examination and with Masson’s trichrome (Mass Histology Service) reagents for renal fibrosis assessment. The blocks were processed for immunohistochemical staining in accordance with our previously described protocol.Citation8 The slides were deparaffinized in xylene, rehydrated through graded alcohol, immersed in 3% hydrogen peroxide for 10 min to block endogenous peroxidase activity, and antigen-retrieved by pressure cooking for 3 min in citrate buffer (pH = 6). For blocking nonspecific binding, the slides were preincubated with 10% normal goat serum at room temperature for 30 min. Subsequently, the slides were incubated respectively with anti-AT-1R (1:300; Abcam, Cambridge, MA) and anti-AT-2R (1:300; Abcam, Cambridge, MA) and stored overnight at 4°C. The slides were sequentially incubated with a secondary antibody (Envision, Dako, Denmark) for 1 h at room temperature and stained with DAB (3,3-diaminobenzidine). Finally, the sections were counterstained with Mayer’s hematoxylin, dehydrated, and mounted. A negative control was obtained by replacing the primary antibody with a normal murine or rabbit IgG. Histopathological and morphological changes in renal for each group were determined by a pathological expert in a blinded manner.
Other Parameters
Echocardiographic measurements were performed to determine post-MI heart failure 3 and 9 weeks after treatment, just before euthanasia, using a high-resolution echocardiographic imaging system equipped with a 16-MHz transducer (Vevo2100, Visualsonics, Canada). The rats were anesthetized with 3% isoflurane mixed with oxygen, and a two-dimensional short-axis view of the LV was obtained at the midpapillary level to record M-mode tracings. Systolic and diastolic anatomic parameters were obtained. After at least 15-min rest, systolic blood pressure (SBP), diastolic blood pressure (DBP), and heart rate during the 3-/9-week treatment period were measured five consecutive times in conscious rats by a tail-cuff plethysmograph (model BP-98A; Softron Co., Japan).
Statistical Analysis
All quantitative data are presented as means ± SD and were compared by a one-way ANOVA followed by LSD post-hoc test or by the Kruskal–Wallis test followed by the Mann–Whitney U-test, according to normality test results. Two-tailed p-values <0.05 were considered significant, and adjusted p-values were used between the subgroup comparison analyses. All statistical analyses were performed with the software package SPSS 16.0 for Windows.
RESULTS
General Characteristics of Animals
MI induced approximately 33.3% mortality within the first 48 h. Post-MI heart failure was determined in the animals that underwent a ligation of the left coronary artery by echocardiographic measurements (). Losartan significantly improved heart function post-MI treatment at both 3 and 9 weeks. At baseline, the animals had almost similar heart rate, SBP, and DBP among the four groups (). In comparison with normal animals, MI induced nonsignificant changes of heart rat, SBP, and DBP at each time point. ARB significantly reduced the levels of SBP and DBP, but had no significant effects on the changes of heart rate at each time point.
Figure 1. Echocardiographic measurements determined ejection fraction in normal, sham, myocardial infarction (MI), and MI+ losartan (los) rats. (A) Ejection fraction at 3 week (p < 0.001). (B) Ejection fraction at 9 week (p < 0.001). Data are mean ± SD. Notes: ###p < 0.008 versus normal; ***p < 0.008 versus sham; †††p < 0.008 versus MI.
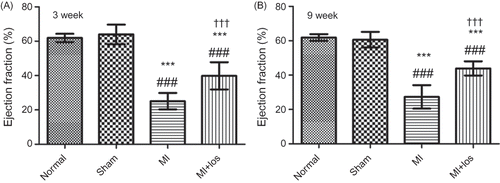
Figure 2. Tail-cuff plethysmograph determined heart rate, systolic blood pressure (SBP) and diastolic blood pressure (DBP) in normal, sham, myocardial infarction (MI), and MI+ losartan (los) rats. (A1) Baseline heart rate (p = 0.180). (A2) Heart rate at 3 week (p = 0.324). (A3) Heart rate at 6 week (p = 0.102). (A4) Heart rate at 9 week (p = 0.943). (B1) Baseline SBP (p = 0.687). (B2) SBP at 3 week (p < 0.001). (B3) SBP at 6 week (p < 0.001). (B4) SBP at 9 week (p < 0.001). (C1) Baseline DBP (p = 0.164). (C2) DBP at 3 week (p < 0.001). (C3) DBP at 6 week (p < 0.001). (C4) DBP at 9 week (p < 0.001). Data are mean ± SD. Notes: ###p < 0.008 versus normal; ***p < 0.008 versus sham; †††p < 0.008 versus MI.
Changes of Renal Function
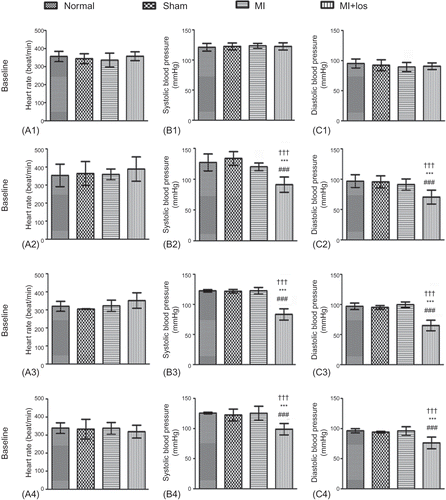
The operation of MI seemed to induce increase of serum creatinine 2 days post-MI (A), but there was no significant difference of serum creatinine among the four groups during the treatment period (3, 6, and 9 weeks) (A). BUN levels were also revealed to increase 2 days and 3 weeks post-MI, but these increased trends normalized at 6 and 9 weeks (B). Losartan treatment did not demonstrate significant effects on the changes of serum creatinine and BUN at each time point post-MI.
Figure 3. Changes of serum creatinine and blood urea nitrogen (BUN) in normal, sham, myocardial infarction (MI), and MI+ losartan (los) rats. (A1) Serum creatinine 2 days post-MI (p = 0.002). (A2) Serum creatinine at 3 week (p = 0.249). (A3) Serum creatinine at 6 week (p = 0.073). (A4) Serum creatinine at 9 week (p = 0.848). (B1) BUN 2 days post-MI (p < 0.001). (B2) BUN at 3 week (p = 0.004). (B3) BUN at 6 week (p = 0.127). (B4) BUN at 9 week (p = 0.409). Data are mean ± SD. Notes: ###p < 0.008 versus normal; ***p < 0.008 versus sham; †††p < 0.008 versus MI.
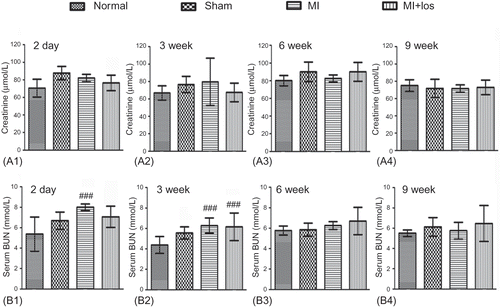
Figure 4. Changes of serum cystatin C and 24 h urine protein in normal, sham, myocardial infarction (MI), and MI+ losartan (los) rats. (A1) Serum cystatin C 2 days post-MI (p < 0.001). (A2) Serum cystatin C at 3 week (p < 0.001). (A3) Serum cystatin C at 6 week (p < 0.001). (A4) Serum cystatin C at 9 week (p < 0.001). (B1) Urine protein 2 days post-MI (p = 0.622). (B2) Urine protein at 3 week (p = 0.110). (B3) Urine protein at 6 week (p = 0.443). (B4) Urine protein at 9 week (p = 0.069). Data are mean ± SD. Notes: ###p < 0.008 versus normal; ***p < 0.008 versus sham; †††p < 0.008 versus MI.
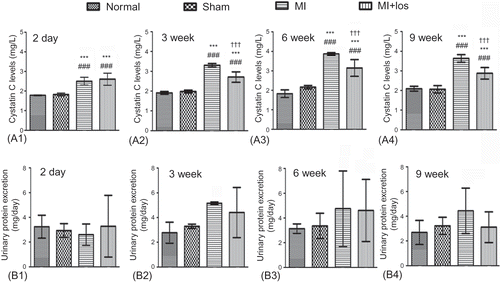
MI begeted enhanced levels of serum cystatin C early 2-day post-MI, and significant increases of cystatin C maintained or even further increased during the following treatment periods (A). Losartan treatment significantly decreased serum concentrations of cystatin C after MI, but could not restore cystatin C to levels as those in normal or sham animals. Despite not within a pathological range and with a great variability, MI tended to result in increased urine proteins, and losartan tended to decrease urine protein excretion (B). However, there was no significant difference among the four groups at each time point.
Renal Histological Changes
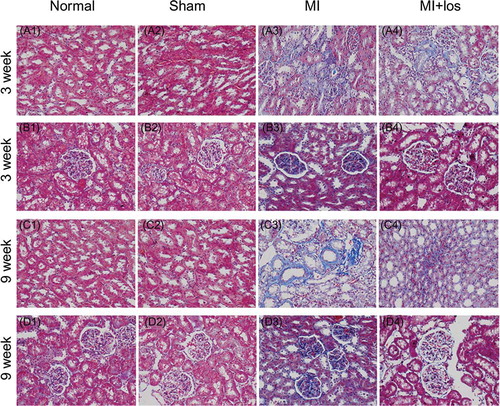
In normal or sham animals, HE stains revealed few inflammatory cells in renal cortical tissue, including renal glomerulus and tubules, at both 3 and 9 weeks (). Much more inflammatory cell infiltrations were demonstrated within renal tubular interstitium and surrounding renal glomerulus in MI animals at both time points, especially at 9 weeks. Treatment with losartan significantly ameliorated MI-induced inflammatory cell infiltration, especially within renal tubular interstitium at 9 weeks. MI surgery-induced tubular necroses were also demonstrated by HE staining at 3 and 9 weeks, which could be ameliorated by losartan administration (E–F). Masson’s trichrome staining demonstrated significant renal fibrosis within tubular tissue and glomerulus in MI animals when compared with both normal and sham animals. However, losartan could exert antifibrotic effects to a large extent post-MI treatment ().
Figure 5. Representative inflammatory cell infiltration and tubular necrosis by hematoxylin and eosin (HE) staining (original magnification, ×400) in normal, sham, myocardial infarction (MI), and MI + losartan (los) rats. Inflammatory cells increased significantly in renal cortical tissue, mainly within the tubular interstitium (A3) and surrounding the renal glomerulus (B3) 3 week post-MI heart failure. Further more inflammatory cells were demonstrated 9 week post-MI heart failure (C3, D3). The infiltration of inflammatory cells could be significantly attenuated by the treatment of losartan both at 3 (A4, B4) and 9 week (C4, D4). HE staining suggesting tubular necrosis (arrows) were also revealed in animals with heart failure 3 (E3) and 9 week (F3) post-MI, while losartan treatment significantly prevented tubular necrosis at both time points (E4, F4).
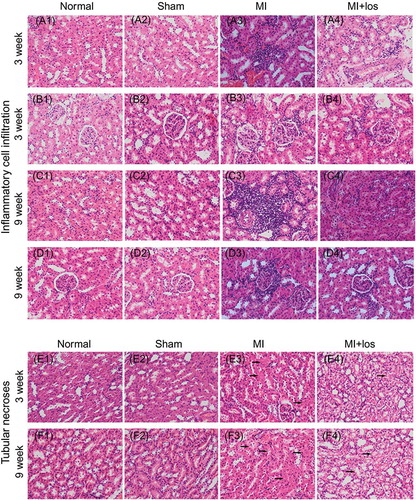
Figure 6. Masson’s trichrome staining (original magnification, ×400) suggesting renal fibrosis (blue staining) in normal, sham, myocardial infarction (MI), and MI + losartan (los) rats. MI induced significantly increase renal fibrosis, including within the tubular interstitium (A3, C3) and glomerulus (B3, D3) at both treatment periods. However, increased renal fibrosis seemed to be attenuated by losartan (B4, C4, and D4).
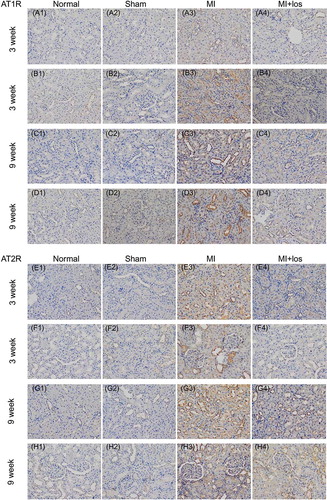
Local Changes of Angiotensin II Receptors
Local changes of AT1R and AT2R were detected by immunohistochemical staining (). The proteins of AT1R and AT2R in renal cortical tissue were found to be significantly higher in MI animals than those in normal animals, while sham animals showed few stains of AT1R and AT2R protein similar to normal animals. Furthermore, the upregulation of AT1R protein mainly focused on renal cortical tissue surrounding the renal glomerulus, including the glomerulus themselves, at 3 (B) and 9 weeks (D). However, despite the upregulation of AT2R protein surrounding the renal glomerulus, markedly staining suggesting AT2R protein were revealed within the renal cortical tissue remote from the glomerulus (E–H3). Treatment with losartan significantly prevented the augmentation of AT1R protein at each time point, but the expression of AT2R was still high after the treatment of losartan, especially in the long-term treatment period.
Figure 7. Immunohistochemical staining determined expressions of angiotensin II type 1/2 receptor (AT1R/AT2R) proteins (brown staining) in normal, sham, myocardial infarction (MI), and MI + losartan (los) rats. At both time points, the upregulated expression of AT1R was revealed mainly to locate surrounding the renal glomerulus (B3, D3), which decreased significantly post losartan treatment (B4, D4). However, the upregulated expression of AT2R protein mainly located within the tubular interstitium and surrounding the glomerulus, especially in the renal cortical tissue remote from the glomerulus (E3, G3), and was still high after the treatment of losartan (H4).
DISCUSSION
The present study demonstrates that blood cystatin C, rather than blood creatinine and BUN, is a preferential marker for the early onset of renal impairment post-MI heart failure. MI-inducing activation of local AT1R, inflammatory reaction, and renal fibrosis may account for the early renal impairment, all of which could be significantly attenuated by early losartan treatment post-MI. The beneficial effects of losartan may also be mediated by upregulated AT2R.
Concomitant and significant renal impairment is common in subjects with heart failure. Current evidence suggests that renal damage conventionally defined by increased levels of blood creatinine markedly deteriorates after a first MI.Citation5,9,10 Cystatin C is a 13-kDa nonglycosylated protein and a member of a family of competitive inhibitors of lysosomal cysteine protease produced by all nucleated cells of the body and released into the bloodstream at a constant rate. Cystatin C filtrates freely in the glomerulus, is reabsorbed completely in the proximal tubule, and is affected by age, gender, diet, and other factors to a less extent as creatinine, which makes it an interesting predictor than serum creatinine level and creatinine-based estimations of glomerular filtration rate for the development of early renal impairment in heart failure.Citation11,12 Increased levels of cystatin C occur frequently and have an independent association with higher mortality in both chronic and acute heart failure.Citation11,12 Consistently our findings demonstrate increased serum cystatin C but not creatinine or BUN in animals with post-MI heart failure, and increased serum cystatin C is demonstrated early 2 days after a surgery of coronary artery ligation. Proteinuria may outweigh blood creatinine to sensitively predict the early renal impairment. The present study reveals that post-MI heart failure had an incremental tendency of 24-h urine protein. However, consistent with other findings,Citation13 no significant urine protein were observed in the present study. Therefore, our findings further suggest that blood cystatin C, rather than blood creatinine or urine protein, should be used as an early marker for renal impairment following MI with subsequent heart failure.
Increased blood cystatin C levels were revealed to be associated with upregulated AT1R within the kidney in the present study. A growing body of evidence has suggested that activation of the intrarenal RAS in a deleterious fashion is an important contributor to the pathogenesis of renal injury. It is of paramount importance that all components of RAS are present in the kidney. Homma et al. have unraveled that renal activation of AT1R seems to be independent of systemic RAS activation and aldosterone signaling.Citation13 Local enhanced AngII-mediated adverse effects may be independent of hemodynamic changesCitation14 and maintained even when the systemic vascular AT1Rs are effectively blocked.Citation15 The renal activation of AT1R has also been revealed to be an important cause of renal injury in the chronic MI rat model.Citation13 Activation of AT1R following heart failure could contribute to oxidative stress, apoptosis, and fibrosis.Citation16 The current study reveals enhanced activation of tissue-based AT1R in rats following MI. Furthermore, in good agreement with other observations,Citation2,5,6 the common renal damage identified by increased serum cystatin C, renal fibrosis, and tubular necrosis could be significantly prevented by RAS intervention, such as losartan, in the present study. The effect of an ARB for the prevention of increased cystatin C levels has also been demonstrated in patients with hypertension.Citation17–19 Increased inflammatory cell infiltration occurring post-MI heart failure has been significantly attenuated by losartan in the present study, which is consistent with other authors’ findings.Citation13,20 Collectively, these findings further support the concept that MI-induced activation of local AT1R may contribute to the pathogenesis of progressive renal impairment in cardiorenal syndrome and could be prevented by ARBs.
ARB may also elicit renoprotective effects via mechanisms independent of AT1R. ARBs preferentially block AT1R and thus much of the beneficial effects of ARB may be mediated by unopposed AngII on AT2R. It is of note that AT2R will exert its beneficial effects by counteracting AT1R when activated. Direct AT2R stimulation reduces early renal inflammatory responses independent of blood pressure reduction.Citation21 Therefore, the beneficial effects on renal injury achieved with ARBs are contributed to not only by blockade of the AT1R but also by increasing AngII effects transduced through AT2R.Citation22 Our findings demonstrated upregulated expressions of AT2R protein following MI, and these upregulated changes were not restored significantly by losartan administration. However, further studies are needed to answer the question whether AngII stimulating the unopposed AT2R contributes to the renoprotective effect after AT1R blockade by ARBs.
In summary, MI could induce activation of local AT1R in renal cortical tissue coupled with renal impairment identified by increased blood cystatin C, renal fibrosis, and tubular necrosis. Therapeutic choice of losartan could attenuate renal impairment by inhibiting AT1R-mediated cascade reaction post-MI. Despite exerting anti-inflammatory and antifibrotic effects to a large degree, losartan restores cystatin C incompletely. Reduced blood pressure may discount the renoprotection of losartan, and the reduced renal damage in the present study may also be a consequence of improved cardiac function by losartan. Therefore, further exploration of other underlying mechanisms may contribute to the discovery of renoprotective therapies.
ACKNOWLEDGMENT
This work was supported by the National Natural Science Foundation of China (no.: 30971262/81270212), Academic Scholarship of Ministry of National Education for Doctoral Candidates (no.: 81000-3191001), and Doctoral Student Innovation Personnel Training Projects of Sun Yat-sen University.
Declaration of Interest The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
REFERENCES
- Lekawanvijit S, Kompa AR, Zhang Y, Wang BH, Kelly DJ, Krum H. Myocardial infarction impairs renal function, induces renal interstitial fibrosis and increases renal kim-1 expression: implications for cardiorenal syndrome. Am J Physiol Heart Circ Physiol. 2012;302(9):H1884–H1893.
- Mashima Y, Konta T, Ichikawa K, Rapid decline in renal function after acute myocardial infarction. Clin Nephrol. 2012;79:15–20.
- Liang KV, Williams AW, Greene EL, Redfield MM. Acute decompensated heart failure and the cardiorenal syndrome. Crit Care Med. 2008;36(1 Suppl):S75–S88.
- Schunkert H, Tang SS, Litwin SE, Regulation of intrarenal and circulating renin-angiotensin systems in severe heart failure in the rat. Cardiovasc Res. 1993;27(5):731–735.
- Hillege HL, van Gilst WH, van Veldhuisen DJ, Accelerated decline and prognostic impact of renal function after myocardial infarction and the benefits of ace inhibition: the cats randomized trial. Eur Heart J. 2003;24(5):412–420.
- Windt WA, Eijkelkamp WB, Henning RH, Renal damage after myocardial infarction is prevented by renin-angiotensin-aldosterone-system intervention. J Am Soc Nephrol. 2006;17(11):3059–3066.
- Zheng S, Zhou C, Weng Y, Improvements of cardiac electrophysiologic stability and ventricular fibrillation threshold in rats with myocardial infarction treated with cardiac stem cells. Crit Care Med. 2011;39(5):1082–1088.
- Cai MY, Hou JH, Rao HL, High expression of h3k27me3 in human hepatocellular carcinomas correlates closely with vascular invasion and predicts worse prognosis in patients. Mol Med. 2011;17(1–2):12–20.
- va Dokkum RP, Eijkelkamp WB, Kluppel AC, Myocardial infarction enhances progressive renal damage in an experimental model for cardio-renal interaction. J Am Soc Nephrol. 2004;15(12):3103–3110.
- Windt WA, Henning RH, Kluppel AC, Xu Y, de Zeeuw D, van Dokkum RP. Myocardial infarction does not further impair renal damage in 5/6 nephrectomized rats. Nephrol Dial Transplant. 2008;23(10):3103–3110.
- Lassus J, Harjola VP. Cystatin c: a step forward in assessing kidney function and cardiovascular risk. Heart Fail Rev. 2012;17(2):271–282.
- Lassus JP, Nieminen MS, Peuhkurinen K, Markers of renal function and acute kidney injury in acute heart failure: definitions and impact on outcomes of the cardiorenal syndrome. Eur Heart J. 2010;31(22):2791–2798.
- Homma T, Sonoda H, Manabe K, Activation of renal angiotensin type 1 receptor contributes to the pathogenesis of progressive renal injury in a rat model for chronic cardiorenal syndrome. Am J Physiol Renal Physiol. 2012;302(6):F750–F761.
- Kobori H, Nangaku M, Navar LG, Nishiyama A. The intrarenal renin-angiotensin system: from physiology to the pathobiology of hypertension and kidney disease. Pharmacol Rev. 2007;59(3):251–287.
- Wang CT, Zou LX, Navar LG. Renal responses to at1 blockade in angiotensin ii-induced hypertensive rats. J Am Soc Nephrol. 1997;8(4):535–542.
- Bock JS, Gottlieb SS. Cardiorenal syndrome: new perspectives. Circulation. 2010;121(23):2592–2600.
- Koc Y, Mazi E, Sakaci T, Effect of olmesartan on serum cystatin c levels in the patients with essential hypertension. Eur Rev Med Pharmacol Sci. 2011;15(12):1389–1394.
- Watanabe S, Okura T, Kurata M, Valsartan reduces serum cystatin c and the renal vascular resistance in patients with essential hypertension. Clin Exp Hypertens. 2006;28(5):451–461.
- Albayrak S, Ordu S, Ozhan H, Effect of olmesartan medoxomil on cystatin c level, left ventricular hypertrophy and diastolic function. Blood Press. 2009;18(4):187–191.
- Izuhara Y, Nangaku M, Inagi R, Renoprotective properties of angiotensin receptor blockers beyond blood pressure lowering. J Am Soc Nephrol. 2005;16(12):3631–3641.
- Matavelli LC, Huang J, Siragy HM. Angiotensin at (2) receptor stimulation inhibits early renal inflammation in renovascular hypertension. Hypertension. 2011;57(2):308–313.
- Naito T, Ma LJ, Yang H, Angiotensin type 2 receptor actions contribute to angiotensin type 1 receptor blocker effects on kidney fibrosis. Am J Physiol Renal Physiol. 2010;298(3):F683–F691.