Abstract
Background: Cardiac stem cell (CSC) dysfunction exists in various kinds of cardiovascular diseases, and may be responsible for the insufficient regeneration of cardiac myocytes and coronary vessels. However, whether chronic renal failure (CRF) affected CSC is unknown. Method: CRF was induced in adult male mice by 5/6 nephrectomy. The mice were killed at 12 weeks after operation. C-kit+ CSC numbers was evaluated by flow cytometer. Apoptosis and DNA damage of C-kit+ CSC in the control and CRF mice was analyzed by immunohistochemistry. In the in vitro study, normal medium, and medium with uremic rat serum were used for the CSC culture. Results: CSC counts attenuated significantly in the chronic renal failure model, whereas apoptosis cells and 8-OHdG-positive cells significantly increased. CSC derived form 5/6 nephrectomy mice showed an impaired anti-oxidant potential. In the cultured cells, CSCs subjected to uremic rat serum showed a higher frequency of TUNEL stain-positive and 8-OHdG-positive cells. The uremia rat serum reduced the expression of hepatocyte growth factor (HGF) and vascular endothelial growth factor (VEGF) in CSC. Conclusions: The current study elucidated that CSC number and function disorders existed in mice with chronic renal insufficiency. Apoptosis, oxidative stress and reduced angiogenic factors secretion caused by uremic toxins in serum are contributors to CSC dysfunction.
Introduction
Patients with chronic renal insufficiency suffer from higher morbidity than the general population.Citation1 Heart structural abnormalities in chronic kidney diseases, such as left ventricular hypertrophy, myocardial interstitium fibrosis, and microvessel lesions, may be attributed to the main factors affecting cardiac perfusion, especially the difference between cardiac myocytes and capillaries distribution.Citation2 As previously reported, capillary density decreases in the heart of mouse with chronic renal failure, followed by obvious cardiac myocyte hypertrophy and DNA peroxidation injury.Citation3,Citation4 The underlying mechanisms, however, remain unclear.
Previous studies have found that human and rodent heart contains clusters of CSCs that are intimately connected by gap junctions and adherens junctions to myocytes and fibroblasts; myocytes and fibroblasts represent the supporting cells within the cardiac niches. The distribution of niche is asymmetrical in heart. Most of niche located in atrial and apical myocardium. In the niches, CSCs and committed cells were negative for hematopoietic markers. These findings support the notion of a resident CSC compartment.Citation5 Recent observations indicate a defective CSC compartment during heart failure, heart senescence, and diabetic cardiopathy.Citation6 Therefore, our study assumed that CSC deficiency has a relationship with devastating heart function, and that uremia causes CSC impairment, resulting in decompensated angio adaption.
Materials and methods
Experimental chronic renal failure
C57BL/6 J mice weighing 23 g ± 2 g at 8 weeks were taken from the Shanghai Institute of Zoology, Chinese Academy of Sciences. The mice were randomly divided into three groups: normal (n = 12), sham-operated (n = 18), and 5/6 nephrectomy (n = 18). Before the surgical procedures, the anesthetic pentobarbital at 80 mg/kg body weight was administered intraperitoneally. 5/6 nephrectomy was performed by dissecting the left kidney to expose and ablate the upper and lower poles; the right intact kidney was then removed after 1 week. The sham-operated group was subjected to anesthesia, skin incision, and kidney dissection, but no kidney ablation. The mice were killed at 12 weeks after operation.
Physiological and biochemical parameters
At the end of the study, echocardiograms were measured using an echocardiographic system (Aloka Tokyo, Japan), equipped with a 7.5MHz imaging transducer. The mice were killed, and blood was obtained from the retro-orbital venous plexus in microtubes (West China Medical University Affiliated Instrument Factory). Blood samples were centrifuged at 12 × 103 g for 10 min at 4 °C. Blood urea nitrogen, and creatinine were detected using an automatic biochemistry analyzer (Hitachi 7600–210, Tokyo, Japan).
Histopathological assessment
Heart tissues were collected from 6 mice every group and were embedded in 10% buffered formalin for morphological analysis. Serial sections (4 µm) were cut and stained with hematoxylin and eosin for general assessment. FLK-1 antibody (Santa Cruz, CA, USA) staining was conducted to evaluate myocardial capillary density.
CSC isolation and purification
CSCs were isolated from the murine heart as previously described.Citation7 Briefly, 6 mice per preparation were anesthetized using pentobarbital and the heart was cannulated through the aortic arch and perfused at 37 °C in oxygenated basic buffer (J-MEM, 0.7 g/L HEPES, 1.25 g/L taurine, 20 U/L insulin, Pen/Strep/Glutamine, Amphotericin, Gentamicin, pH 7.3) on a Radnotti apparatus. The heart was cut into small piece then digested for 10 min at 37 °C in 320 units/mL collagenase II in oxygenated basic buffer. Afterward, the heart was minced in basic buffer containing 0.5% bovine serum albumin and the cardiomyocytes were pelleted and discarded. Remaining cells in the supernatant were passed through a 25 -m filter. The single cell suspension obtained stained with anti-CD117-FITC, a C-kit + stem cell marker, antibody (Miltenyi Biotech, Bergisch Gladbach, Germany) and magnetic activated cell sorting(MACS) (BD FACS Aria, USA) was performed to separated and quantified C-kit positive cells. P1 cells were used for apoptosis and 8-OHdG immunohistochemistry (IHC) experiment, and 0.3% H2O2 stimulated experiment.
In situ terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL)
TUNEL assay was performed in cultured C-kit positive cells using a TUNEL kit (Roche, Mannheim, Germany) principally according to the manufacturer’s instructions. Positive cells were counted on 10 non-overlapping fields in cultured cells grown on cover glass (×400 magnification).
8-OHdG detection
The cultured C-kit positive cells were first incubated with primary antibody anti-8-OHdG (Millipore, USA) at a dilution ratio of 1:1000, and then incubated with ChemMate EnVision reagent, (Gene Tech, Shanghai, China), a peroxidase-conjugated polymer carrying antibodies to rabbit and mouse immunoglobulins, following the manufacturer’s instructions. Immunostaining was evaluated on 10 non-overlapping fields in the cultured cells (×400 magnification).
Cell culture
The obtained healthy C-kit positive cells were grown in 35-mm dishes coated with 200 μg/mL fibronectin (Sigma) and cultured in CSC media (DMEM/F12, 10% embryonic stem cell–grade fetal bovine serum (FBS), PSG, insulin-transferrin-selenium, 1000 U/mL LIF, 40 ng/mL EGF, 20 ng/mL bFGF) as normal control, and CSC media +10% uremic serum obtained from 5/6 nephrectomy Male Lewis rat as experimental group. After reaching a confluence of 80%, the cells were rinsed three times with cold Dulbecco's phosphate buffered saline and collected for subsequent experiments.
Western blot analysis
Cultured cells were homogenized with RIPA lysis buffer (Beyotime, Nanjing, China). After incubation on ice for 10 min, the homogenates were centrifuged at 12 × 103 g for 10 min at 4 °C. Proteins were prepared and separated in 8%, 10%, and 20% polyacrylamide gel electrophoresis, and then transferred into polyvinylidene fluoride membranes. Primary antibodies against insulin-like growth factor (IGF), hepatocyte growth factor (HGF), vascular endothelial growth factor (VEGF), and angiopoietin at a dilution ratio of 1:750 were employed. IGF and HGF antibodies were purchased from Santa Cruz, VEGF and angiopoietin antibody were bought from Abcam (Cambridge, England), and the DAB kit (Biobid, Kibbutz Beit Hemek, Israel) was employed to detect the protein signal.
Statistic analysis
Values were expressed as mean ± standard error of the mean. Analysis of data between two groups and among several groups was conducted using unpaired two-tailed t-test and ANOVA, respectively. Differences with p values < 0.05 were considered statistically significant. All statistical analyses were completed using SPSS 16.0 software.
Results
As reported in , 12 weeks after operation, 5/6 nephrectomy mice showed elevated serum creatinine and blood urea nitrogen, indicating the establishment of CRF. 5/6 nephrectomy mice showed worsened left ventricular end-diastolic diameter (mm) and left ventricular fractional shortening (%). FLK-1 immunostain, a marker used to evaluate density of blood vessels, reported in , shown that 5/6 nephrectomy caused a decrease of capillary density in the heart compared with the levels of control and sham groups. Since CSC incompetence may be one of the contributors; thus, CSC number, apoptosis, and oxidative stress in the mice subjected to 5/6 nephrectomy were detected.
Figure 1. Density of blood vessels decreased in mice subjected to 5/6 nephrectomy. Representative sections of the heart for density of blood vessels by FLK-1 immunohistochemistry (×400), assessed at 12 weeks after 5/6 nephrectomy in normal mice (n = 6), sham-operated mice (n = 6) and 5/6 nephrectomy mice (n = 6) *p < 0.05 versus normal group, †p < 0.05 versus sham-operated mice.
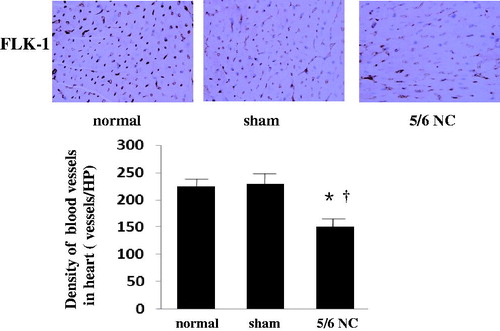
Table 1. Left ventricular function and renal function assessed in mice 12 weeks after 5/6 nephrectomy.
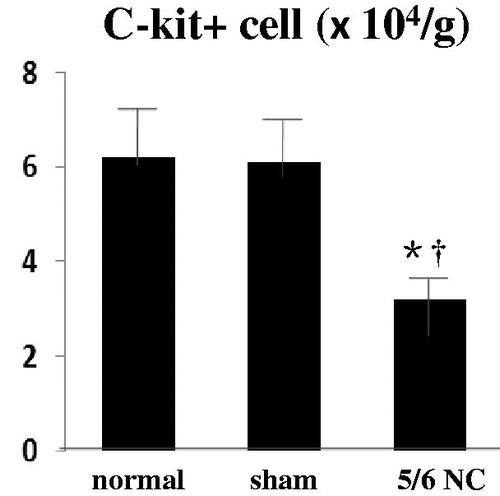
Three overlapping subgroups were detected in the reservoir of the resident CSCs, including C-kit+, sca-1+, and multiple drug resistant cells. The current study focused on the characteristics of the C-kit+ cells, because these cells process the potential of long-term expansion. No standard criteria exist for cardiac stem cell count evaluation among different individuals; therefore, the number of cells per gram heart tissue was used as reference. As shown in , In the normal and sham-operated group, 6 × 104/g C-kit+ cells were found, whereas only 3 × 104/g C-kit+ cells in the mice subjected to 5/6 nephrectomy. The gap among the different groups was statistically significant.
Figure 2. Number impairment in C-kit+ cells stemmed from 5/6 nephrectomy mice. Flow cytometry counts of C-kit positive cells in normal (n = 6), sham-operated (n = 6), and 5/6 nephrectomy mice (n = 6). *p < 0.05 versus normal group, †p < 0.05 versus sham-operated mice.
Because increased oxidative stress was found in the end-stage renal disease, free radicals produced by enhanced oxidative stress could lead to cell senescence and apoptosis, which may impair several CSCs. We detected apoptosis and DNA damage ratio in primary cultured C-kit+ cells. In the in vitro cell culture, C-kit+ CSCs derived from CRF mice presented a higher frequency of apoptosis ( and ) and a higher reactive oxygen species (ROS)-mediated DNA damage expressed by 8-OHdG stain positive ratio (). The oxidative stress tolerance of the CSCs derived from 5/6 nephrectomy decreased when exposed to 0.3% H2O2 ( and ).
Figure 3. Function impairment in C-kit+ cells stemmed from 5/6 nephrectomy mice. (a) Cultured C-kit+ cells obtained from the mice subjected to 5/6 nephrectomy (n = 6) and sham-operated (n = 6) mice were Immunohistochemical analysis revealing the TUNEL positive cell and 8-OhdG positive cell; TUNEL, 8-OHdG, green; C-kit, red; nuclei stained with Hoechst, blue; C-kit and TUNEL, C-kit and 8-OHdG overlap, yellow. (b) Apoptosis percentage and 8-OHdG positive ratio. (c) Effects of 5/6 nephrectomy on 0.3% H2O2-induced oxidative stress. in cultured C-kit+ cells. *significantly different from the sham group. (d) Effects of 5/6 nephrectomy on 0.3% H2O2-induced apoptosis in cultured C-kit+ cells. *significantly different from the sham group. (e) Effects of uremic rat serum on cultured C-kit+ cells apoptosis and oxidative stress. *statistically and significantly different from the conditional medium group.
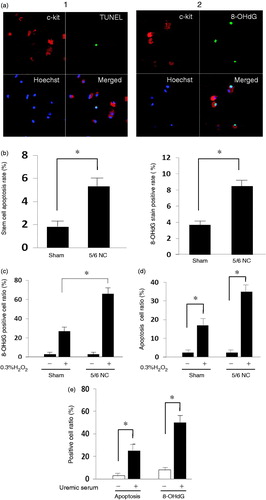
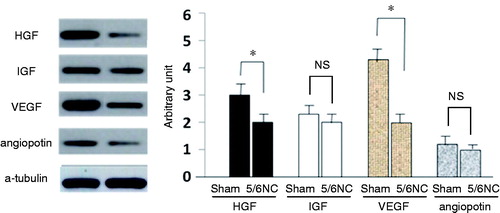
As to assess effects of chronic renal failure on the tolerance of healthy C-kit+ CSCs, we added uremic serum to medium as a substitute for fetal bovine serum. Since serum obtained from mice were not enough for cell culture; therefore, uremic rat was created through 5/6 nephrectomy to obtain serum for cell culture. Uremic serum treatment increased the apoptosis rate and 8-OHdG positive percentages from 1.87% and 4.75% in the baseline to 20.9% and 52.1%, respectively, in the cultured C-kit+ cells isolated from the normal mice (). Significant difference was found between groups. Western blot analysis showed that uremic serum treatment significantly decreased the expression of pro-angiogenic factors HGF and VEGF ().
Figure 4. Secretion of proangiogenic factors reduced in 5/6 nephrectomy mice CSC. Immunoblot analysis of Cultured cells expression of proangiogenic factors of HGF, IGF, VEGF, and angiopoietin. *p < 0.05 versus sham-operated mice (n = 6), NS stands for no significance.
Discussion
Findings of the current study demonstrated that end-stage renal diseases decreased CSC counts, which resulted in CSC function impairment characterized by reduced oxidative stress tolerance and decreased proangiogenic factors.
Previous studies have reported that uremia decreases endothelial progenitor cell number in peripheral blood and causes angiogenic effect deficiency.Citation8,Citation9 Endothelial progenitor cell has an important role in the maintenance of angiogenesis and integrity of vessels. Compared to the normal control, the rat uremia model showed decreased angiogenic potency and reduced VEGF expression in ischemic areas.Citation10 These results demonstrate that angiogenic function impairment and VEGF production reduction in ischemic regions are related to cardiovascular diseases in uremia. Aside from the reduced expression of erythropoietin, inflammation and oxidative stress attributable to uremia are considered related to these endothelial progenitor cell changes.Citation11
CSC process the potential possibility of differentiating into endothelial cells, vascular smooth muscle cells, and cardiomyocytes.Citation6 However, with the development of research, cardiac function improvement through stem cell therapy cannot be readily attributed to stem cell differentiation and proliferation, but to the paracrine effects of transplanted cells.Citation12 The current study focused on VEGF, HGF, IGF-1, and angiopoietin, which are not only strong promoters of angiogenesis, but are also involved in some paracrine signal pathways that may bring about positive effects on injured myocardium.Citation13
Local administration of VEGF and HGF could promote the circulation of CSC migration into impaired parts and repair myocardial infarction and ischemia/reperfusion.Citation14,Citation15 CSCs with elevated expression of IGF-1 exert pro-survival effects on cardiomyocytes via IGF-1 signal pathway.Citation16 HGF processes the potent anti-apoptotic capacity, and can also induce CSC migration to injured areas.Citation17,Citation18
Excessive ROS injures DNA and telomeres, leading to telomere shortening, cellular senescence, and cell death,Citation19 which is regulated by the reduction in telomere-related proteins.Citation20 8-OHdG is an established marker of ROS-mediated DNA damage. 8-OHdG positive cells were observed to prevail in the 5/6-nephrectomy model and in CSCs treated with uremic rat serum. Studies on hematopoietic stem cells (HSCs) showed that an elevated ROS level damages the repopulating potency and self-renewal capacity of HSCs.Citation21 Enhanced oxygen toxicity in diabetes alter the cardiac progenitor cell (CPC) function, and this effect is implemented via ROS-mediated telomere shortening and CPC apoptosis.Citation22 ROS also inhibits cellular adhesion of the engrafted mesenchymal stem cells implanted into ischemic myocardium.Citation23
The current study demonstrated that uremia decreased CSC number and reduced cytokine-stimulated angiogenesis. To identify the relationship between these changes and uremia toxins, an in vitro study was further conducted. CSC apoptosis and oxidative damage increased after being subjected to uremia rat serum; in addition, the synthesis of mediators involving angiogenesis and arteriogenesis reduced. CSCs isolated from uremia mice were also shown to exert less tolerance when exposed to oxidative stress. These data strengthen the hypothesis that CSC paracrine effect deficiency plays a significant role in the development and progression of uremic cardiomyopathy.
Cardiovascular disease is a major cause of mortality in chronic kidney disease patients. Recent study found that serum indoxyl sulfate (IS) may have a significant role in the vascular disease and higher mortality, through induce oxidative stress and inhibits NO production, observed in CKD patients.Citation24,Citation25 Different toxins exist under the chronic renal insufficiency condition. Hence, further studies are needed to identify the exact toxins and mechanisms that contribute to CSC injury.
However, some limitations were present in the present study. CSC numbers and functions were only evaluated at the end-stage renal disease. Whether CSC number and function dysfunction occur early in the progress of renal disease is unknown. Metabolic products and toxins remove dysfunction appears early in the procession of chronic kidney diseases. Constricted by experimental conditions, hemodialysis could not be applied on the mice subjected to 5/6-nephrectomy. Therefore, identifying the effects of uremic toxin removal by blood purification on CSCs, as well as identifying whether CSC injury is a reversible factor, is difficult.
Our current study elucidated that CSC number and function disorders existed in mice with chronic renal insufficiency. Apoptosis, oxidative stress and reduced angiogenic factors secretion caused by uremic toxins in serum are contributors to CSC dysfunction.
Declaration of interest
This work was supported by Research Grants of Chinese National Science Fund (81070583) and Research Grants of the Science and Technology Department of Zhejiang province, China (2008R10024).
References
- Tonelli M, Wiebe N, Culleton B, et al. Chronic kidney disease and mortality risk: a systematic review. J Am Soc Nephrol. 2006;17:2034–2047
- Guerin AP, Adda H, London GM, Marchais SJ. Cardiovascular disease in renal failure. Minerva Urol Nefrol. 2004;56:279–288
- Li Y, Takemura G, Okada H, et al. Molecular signaling mediated by angiotensin II type 1A receptor blockade leading to attenuation of renal dysfunction-associated heart failure. J Card Fail. 2007;13:155–162
- Li Y, Wu J, He Q, et al. Angiotensin (1--7) prevent heart dysfunction and left ventricular remodeling caused by renal dysfunction in 5/6 nephrectomy mice. Hypertens Res. 2009;32:369–374
- Urbanek K, Cesselli D, Rota M, et al. Stem cell niches in the adult mouse heart. Proc Natl Acad Sci USA. 2006;103:9226–9231
- Kajstura J, Urbanek K, Rota M, et al. Cardiac stem cells and myocardial disease. J Mol Cell Cardiol. 2008;45:505–513
- Beltrami AP, Barlucchi L, Torella D, et al. Adult cardiac stem cells are multipotent and support myocardial regeneration. Cell. 2003;114:763–777
- de Groot K, Bahlmann FH, Sowa J, et al. Uremia causes endothelial progenitor cell deficiency. Kidney Int. 2004;66:641–646
- Choi JH, Kim KL, Huh W, et al. Decreased number and impaired angiogenic function of endothelial progenitor cells in patients with chronic renal failure. Arterioscler Thromb Vasc Biol. 2004;24:1246–1252
- Jacobi J, Porst M, Cordasic N, et al. Subtotal nephrectomy impairs ischemia-induced angiogenesis and hind limb re-perfusion in rats. Kidney Int. 2006;69:2013–2021
- Yao EH, Yu Y, Fukuda N. Oxidative stress on progenitor and stem cells in cardiovascular diseases. Curr Pharm Biotechnol. 2006;7:101–108
- Fazel S, Cimini M, Chen L, et al. Cardioprotective C-kit+ cells are from the Bone marrow and regulate the myocardial balance of angiogenic cytokines. J Clin Invest. 2006;116:1865–1877
- Li Y, Takemura G, Kosai K, et al. Postinfarction treatment with an adenoviral vector expressing hepatocyte growth factor relieves chronic left ventricular remodeling and dysfunction in mice. Circulation. 2003;107:2499–2506
- Rota M, Padin-Iruegas ME, Misao Y, et al. Local activation or implantation of cardiac progenitor cells rescues scarred infarcted myocardium improving cardiac function. Circ Res. 2008;103:107–116
- Guzman MJ, Crisostomo PR, Wang M, et al. Vascular endothelial growth factor improves myocardial functional recovery following ischemia/reperfusion injury. J Surg Res. 2008;150:286–292
- Kawaguchi N, Smith AJ, Waring CD, et al. C-kitpos gata-4 high rat cardiac stem cells foster adult cardiomyocyte survival through igf-1 paracrine signalling. PLoS One. 2010;5:e14297
- Urbanek K, Rota M, Cascapera S, et al. Cardiac stem cells possess growth factor-receptor systems that after activation regenerate the infarcted myocardium, improving ventricular function and long-term survival. Circ Res. 2005;97:663–673
- Morishita R, Aoki M, Hashiya N, et al. Therapeutic angiogenesis using hepatocyte growth factor (hgf). Curr Gene Ther. 2004;4:199–206
- Kawanishi S, Oikawa S. Mechanism of telomere shortening by oxidative stress. Ann N Y Acad Sci. 2004;1019:278–284
- Opresko PL, Fan J, Danzy S, et al. Oxidative damage in telomeric DNA disrupts recognition by trf1 and trf2. Nucleic Acids Res. 2005;33:1230–1239
- Ito K, Hirao A, Arai F, et al. Reactive oxygen species act through p38 mapk to limit the lifespan of hematopoietic stem cells. Nat Med. 2006;12:446–451
- Rota M, LeCapitaine N, Hosoda T, et al. Diabetes promotes cardiac stem cell aging and heart failure, which are prevented by deletion of the p66shc gene. Circ Res. 2006;99:42–52
- Song H, Cha MJ, Song BW, et al. Reactive oxygen species inhibit adhesion of mesenchymal stem cells implanted into ischemic myocardium via interference of focal adhesion complex. Stem Cells. 2010;28:555–563
- Barreto FC, Barreto DV, Liabeuf S, et al. Serum indoxyl sulfate is associated with vascular disease and mortality in chronic kidney disease patients. Clin J Am Soc Nephrol. 2009;4:1551–1558
- Tumur Z, Niwa T. Indoxyl sulfate inhibits nitric oxide production and cell viability by inducing oxidative stress in vascular endothelial cells. Am J Nephrol. 2009;29:551–557