Abstract
Primary hyperoxaluria is a rare autosomal recessive disorder. Type 1 PH is the most common form and develops due to a defect in a liver specific enzyme the alanine aminotransferase enzyme. As a result of the enzyme deficiency, there is an overproduction of oxalate and excessive urinary excretion. Recurrent urolithiasis and nephrocalcinosis are the most important findings of the disorder and often at the beginning end-stage renal disease develops. This report presents a case backed up by literature of a patient with end stage renal failure and erythropoietin-resistant anaemia whose bone marrow biopsy showed crystal deposition which received delayed diagnosis of oxalosis.
Introduction
Primary hyperoxalurias (PH) are rare autosomal recessive diseases of the oxalate metabolism.Citation1 Primary hyperoxaluria type 1 (PH1) occurs due to the functional defect of the liver enzyme alanine aminotransferase (AGT).Citation2–4 Primary hyperoxaluria type 2 (PH2) is rare and develops secondary to failure of the cytosolic enzyme D-glycerate dehydrogenase.Citation3 Both primary hyperoxaluria types result in excessive production of oxalate and are clinically characterized by hyperoxaluria.Citation5 Oxalate is an end product and it is not metabolized.Citation1,Citation6 The kidneys play the most important role in excreting oxalate from the systemic circulation and due to the hyperoxaluria recurrent kidney stone formation, nephrocalcinosis and progressive renal failure develops.Citation3,Citation5,Citation7 The kidney dysfunction and decreased urinary excretion results in a rise in plasma oxalate and systemic deposition of over produced oxalate. Deposition is most common in the bones but can be found in all organs and tissues except the liver.Citation5 Bone marrow involvement is characterized by the calcium oxalate intraosseous tophi and the bone marrow eliminating granuloma structure.Citation5 Retinal deposition of oxalate can be easily recognized and can be the first symptom of systemic oxalosis.Citation5,Citation8 Other fundamental oxalate deposition areas are medias of arteries, peripheric nervous system (neuropathy), myocardia (AV block), thyroid and skin (livedo reticularis).Citation5
This report presents the case of a patient with chronic renal failure and oxalosis diagnosed with massive bone marrow involvement who underwent a bone marrow biopsy due to the erythropoietin (EPO) resistant anaemia. This case is presented with relevant literature.
Case report
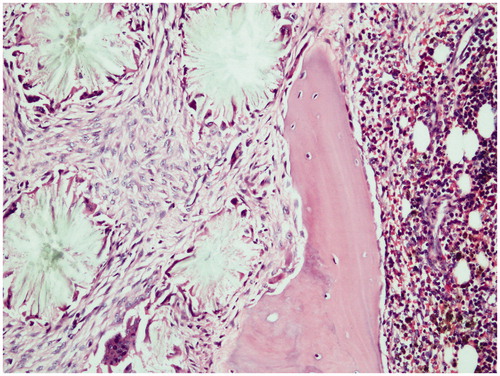
The case is a 34-year-old male patient. At his first consultation at the end of 2007, he complained of fatigue. As a result of the investigations carried out during the consultation, he was diagnosed with end-stage renal disease (ESRD) and started hemodialysis in early 2008. The patient does not have a known history of systemic diseases, hypertension or gall stones. Approximately 1 year after the start of the dialysis the patient became anuric. On determination of the erythropoietin treatment resistant anemia during the hemodialysis treatment the patient was referred to a nephrology clinic. At the next consultation, it was observed that he was pale. The patient informed the doctors that he had been experiencing increasing bone and muscle pain in the last 2 months. The patient’s laboratory tests did not reveal any vitamin deficiency. Following a consultation with a hematologist, it was decided to carry out a bone marrow biopsy. The bone marrow aspirate was dry tap due to fibrosis. A small number of giant cells and their crystal structures were present in imprint materials. Histopathologic examination of the bone marrow biopsy revealed a granulomatous reaction that filled a large section of the intertrabecular area (). The granuloma structures were seen to be light yellow, sequential in radial pattern and included double light refracting crystals under a polarized microscope (). Epithelioid histiocytes were present around these crystals (). As a result of these findings the cause of the primary renal disease was thought to be primary hyperoxaluria and further investigations were carried out. The direct urinary system graph indicated increased density in the areas of both kidney canals. Ultrasonographic examination indicated increased diffuse echogenicity in line with cortical nephrocalcinosis in both kidneys and echogenicity of the multiple stones that filled the calyx structures of the renal collecting systems were identified. Biochemical examination of the patient’s urine could not be carried out due to the patient being anuric for 3 years. The diagnosis of PH was confirmed with a mutational analysis of the AGXT gene. Val 321 Asp homozygous mutation was detected.
Figure 1. Foreign body granulomatous reaction filling almost all the intertrabecular area (H&E, ×100).
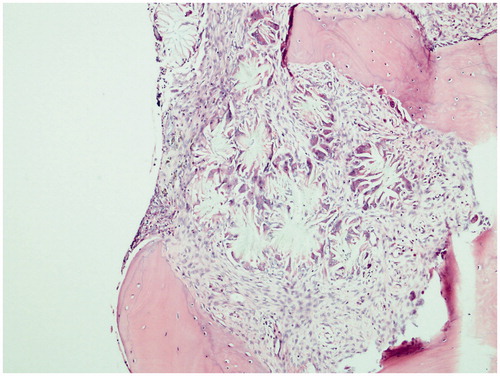
Figure 2. Double light refracting crystals under a polarized microscope (H&E, ×100).
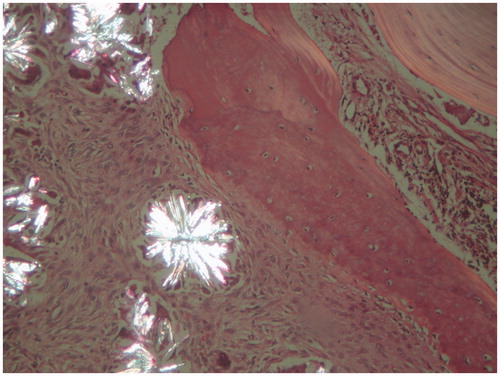
Figure 3. Numerous crystalline deposits are seen in the center of granulomas (H&E, ×200).
Discussion
Primary hyperoxaluria is an extremely rare autosomal recessive disease. The incidence is 1/120,000/year. It is usually identified in childhood or adolescence with urolithiasis. However, it can also be diagnosed following the development of ESRD and there are cases that were diagnosed in the 6th decade.Citation2 The case presented in this report is in the 4th decade, 4 years after the development of ESRD, bone marrow biopsy was carried out due to the EPO resistant anaemia. Following the clinical research and the calcium oxalate crystals identified in the bone marrow biopsy, he was diagnosed with oxalosis.
Literature defines PH as a heterogeneous group in terms of age at onset and progression of renal failure. The infantile form is characterized by the development of systemic oxalosis on average in 6 months and rapid progression to ESRD. The juvenile form is characterized by symptoms associated with renal stones and often leads to renal failure. Adult forms similar to the case presented have been identified. In this form, the onset of symptoms is delayed and the patients have developed ESRD and have been diagnosed with symptoms of systemic oxalosis.Citation9 Our case was diagnosed with symptoms of systemic oxalosis 4 years after the development of ESRD.
Hyperoxaluric syndromes can be primary as in the overproduction of oxalate or they can be secondary as in increased intestinal absorption or overconsumption by diet.Citation5 PH are rare autosomal recessive diseases. There are three subtypes depending on the hereditary enzyme deficiencies, type 1 (PH1), type 2 (PH2) and type 3 (PH3).Citation8 PH1 is characterized by the lack of the AGT enzyme which normally would detoxify the glyoxylate found in hepatic peroxisomes. Due to the lack of this enzyme peroxisomal glyoxylate turns into oxalate and due to the poor solubility of calcium oxalate, crystallization occurs primarily in the kidneys and other organs.Citation10 The deficient enzyme in PH2 is glyoxylate reductase/hydroxy pyruvate reductase (GRHPR) which results in the excessive production of oxalate and glycerate.Citation2
PH3 is phenotypically similar to PH1 and PH2 however there is no enzyme deficiency of AGT and GRHPR. There have only been a few cases of PH3 in literature and no specific enzyme deficiency has been identified.Citation1,Citation11,Citation12 PH1 is the most serious cause of primary hyperoxaluria. Recurrent urolithiasis, progressive nephrocalcinosis are the main manifestations of PH. Consequently, liver damage, reduction in glomerular filtration rate, ESRD and systemic oxalosis develops.Citation8
Oxalosis is the final stage of the hyperoxaluric syndrome. Different diagnostic and therapeutic approaches are available for this condition in both developed and developing countries.Citation6 The diagnostic methods tend to be more difficult for technical and financial purposes.Citation6 Serum and urine oxalate examinations cannot be carried out in routine biochemical laboratories; furthermore the AGT measurement of the liver biopsy which is the gold standard for the diagnosis can only be done by a few laboratories.Citation7 Due to the additional development of ESRD, the application of biochemical tests becomes more difficult.Citation7 In the presented case, due to the 4-year ESRD history and due to the case being anuric, the urinary oxalate measurements could not be carried out. Furthermore, the hepatic enzyme activities could not be analyzed and the calcium oxalate crystal depositions have led to the possible diagnosis of PH. Through clinical, radiological/sonographical findings combined with mutational analysis of the AGXT gene a final diagnosis of PH has been made.
In PH systemic involvement is most common in the bones.Citation5,Citation9 However, the bone lesions are less specific and can mimic clinical renal osteodystrophy.Citation9 However, the dispersion of the skeletal changes in osteodystrophy are different to the dispersion of hyperoxaluria. The crystal depositions are more prominent in hypervascular areas such as the metaphysis segments of the tubular bones.Citation9 In the given case, the crystal depositions are localized in the hypervascular areas. In hand radiographics pathologic fractures were observed and radiographics indicate dense metaphyseal bands that match the metaphyseal areas and diaphyseal dense areas belonging to the oxalate deposits. The clinical manifestations of oxalate osteopathy are pain, spontaneous fractures and EPO resistant anaemia. All of the bone related symptoms were found in the case pertaining to this report.
The presence of calcium oxalate crystals in the bone marrow is a common finding of systemic oxalosis. Some researchers stress the importance of bone marrow biopsy for diagnosing systemic oxalosis or when PH is suspected.Citation3,Citation12 Literature also presents similar cases of PH with massive bone marrow involvement.Citation13–15 The development of pancytopenia as a result of intensive crystal deposition in the bone marrow is most rare.Citation3 The rarity of pancytopenia in PH cases is thought to be related to the; genetic, enzymatic and phenotypic heterogeneity, the excessive systematic weight of the calcium oxalate crystals or the clinically late occurrence of pancytopenia.Citation3 In the presented case, a 2-year history of EPO resistant anaemia has been defined which had developed after 1 year of hemodialysis treatment and pancytopenia appears to be a late finding.
In PH the primary medical treatment of fluid support includes daily pyridoxine treatment and crystallization inhibitors.Citation9 Apart from the medical treatment the ideal course of action would be to plan organ transplantation prior to the development of ESRD and systemic oxalosis.Citation8 Isolated kidney or combined kidney–liver transplantations are implemented for the treatment of primary hyperoxaluria however; transplantation strategies need to be developed in order to protect the kidney from oxalate damage.Citation16 The transplantation options given to PH1 cases include isolated kidney, isolated liver and combined kidney and liver transplantations.Citation4 Isolated kidney transplantation would result in significant decrease in oxalate levels however, due to the continued excessive production of oxalate; ESRD will quickly develop in the transplanted kidney.Citation17 Isolated liver transplantation is only implemented prior to the development of any kidney damage.Citation4 The other transplantation option for PH1 cases is the combined kidney–liver transplantation and is the most common cause of kidney–liver transplantations in literature.Citation17 The transplantation procedure especially for infants and cases of ESRD are first liver then kidney transplantations.Citation17 Currently the most frequently applied procedure is simultaneous liver–kidney transplantation however, practices are changed depending on patient preferences.Citation17
In summary, primary hyperoxaluria is a rare metabolic disease that leads to end stage renal disease. Early diagnosis and aggressive conservative treatment could prevent the development of ESRD thus the possibility of primary hyperoxaluria should always be borne in mind in cases with nephrocalcinosis and oxalate disease.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Mookadam F, Smith T, Jiamsripong P, et al. Cardiac abnormalities in primary hyperoxaluria. Circ J. 2010;74:2403–2409. Epub 2010 Sep 29
- Karadag S, Gursu M, Aydin Z, et al. Primary hyperoxaluria in an adult presenting with end-stage renal failure together with hypercalcemia and hypothyroidism. Hemodial Int. 2011;15:573–576. Epub 2011 Jul 29
- Sud K, Swaminathan S, Varma N, et al. Reversal of pancytopenia following kidney transplantation in a patient of primary hyperoxaluria with bone marrow involvement. Nephrology (Carlton). 2004;9:422–425
- Celik G, Sen S, Sipahi S, et al. Regressive course of oxalate deposition in primary hyperoxaluria after kidney transplantation. Ren Fail. 2010;32:1131–1136
- Leumann E, Hoppe B. The primary hyperoxalurias. J Am Soc Nephrol. 2001;12:1986–1993
- Doganavsargil B, Akil I, Sen S, Mir S, Basdemir G. Autopsy findings of a case with oxalosis. Pediatr Dev Pathol. 2009;12:229–232
- Spasovski G, Beck BB, Blau N, Hoppe B, Tasic V. Late diagnosis of primary hyperoxaluria after failed kidney transplantation. Int Urol Nephrol. 2010;42:825–829. Epub 2009 Dec 18
- Harambat J, Fargue S, Bacchetta J, Acquaviva C, Cochat P. Primary hyperoxaluria. Int J Nephrol. 2011;2011:864580. 11pp. Epub 2011 Jun 16
- Shang MH, Jun H, Fan Y, et al. Recurrence of primary hyperoxaluria after kidney transplantation: the report of two cases. Chin Med J (Engl). 2009;122:2794–2797
- Malakoutian T, Asgari M, Houshmand M, et al. Recurrence of primary hyperoxaluria after kidney transplantation. Iran J Kidney Dis. 2011;5:429–433
- Belostotsky R, Seboun E, Idelson GH, et al. Mutations in DHDPSL are responsible for primary hyperoxaluria type III. Am J Hum Genet. 2010;87:392–399
- Cochat P. Primary hyperoxaluria type 1. Kidney Int. 1999;55:2533–2547
- Tapia G, Navarro JT, Navarro M. Leukoerythroblastic anemia due to oxalosis with extensive bone marrow involvement. Am J Hematol. 2008;83:515–516
- Walter MJ, Dang CV. Pancytopenia secondary to oxalosis in a 23-year-old woman. Blood. 1998;91:4394
- Mete O, Dogan O. Pancytopenia due to massive bone marrow involvement in a patient with primary hyperoxaluria. Int J Surg Pathol. 2010;18:213. Epub 2009 Dec 24
- Bergstralh EJ, Monico CG, Lieske JC, et al. Transplantation outcomes in primary hyperoxaluria. Am J Transplant. 2010;10:2493–2501. Epub 2010 Sep 17
- Cochat P, Fargue S, Harambat J. Primary hyperoxaluria type 1: strategy for organ transplantation. Curr Opin Organ Transplant. 2010;15:590–593