Abstract
Gitelman’s syndrome (GS) is a rare disease with autosomal recessive trait, characterized by hypokalemia, hypomagnesemia, metabolic alkalosis, hypocalciuria and hyperkinemic hyperaldosteronism. While muscle weakness, tetany, stomachache, nausea and fever are very common, it could sometimes be completely asymptomatic as is the case in our patient. It is generally benign, but some severe complications like growth retardation and, though rare, paralysis and cardiac arrest could also be seen. A 57-year-old male patient sent to our hospital for further examination because of hypokalemia was diagnosed with GS as a result of clinical and laboratory assessments. Potassium and magnesium replacement was started. We are presenting our case seeing that GS is not a syndrome to be overlooked as it bears a risk of severe complications, although it might be asymptomatic until advanced ages.
Introduction
Known as hypokalemia--hypomagnesemia, Gitelman’s syndrome (GS) is a salt-losing renal tubulopathy with autosomal recessive trait.Citation1 It occurs as a result of a mutation in the SLC12A3 gene, which codes thiazide-sensitive Na/Cl cotransporter on the apical membrane of the distal convoluted tubule in kidneys. Na/Cl cotransporter is inactivated as a result of the mutation.Citation2,Citation3 Decrease in NaCl reabsorption in the distal tubule causes hypervolemia. Hypervolemia activates the renin–angiotensin–aldosterone system. Aldosterone increases the sodium reabsorption through the epithelial sodium channel in the cortical collecting tubule. Secretion of potassium and hydrogen ions increases and as a result hypokalemia and metabolic alkalosis develop. Its prevalence is 1/40,000, and it is generally diagnosed at an adult age.Citation4 The most common symptom is tetany seen especially in periods of fever or with diarrhea and vomiting, which cause loss of magnesium. Paresthesia in the face is also very common. Muscle weakness, fatigue, stomachache, nausea and, though rare, sudden cardiac arrests could be seen.Citation4–6 In some patients chondrocalcinosis due to hypomagnesaemia might develop. Chondrocalcinosis shows itself with swelling, local heat and tenderness in the affected joint.Citation7,Citation8 GS is diagnosed with clinical and biochemical findings (hypokalemia, metabolic alkalosis, hypomagnesemia and hypocalciuria).Citation4
Case
A 56-year-old patient was sent to our clinic for further examination as hypopotassemia was found in his blood tests done by coincidence. It was learned that in his history he had swelling and pain in both knees which recuperated in a few days. Varicosis was diagnosed in both legs by the doctor he visited that time, and he was recommended to wear surgical stockings. The patient did not have any complaints or diseases known other than that. Nor did he have a history of usage of diuretics or laxatives, vomiting or diarrhea. Blood pressure was measured as 100/60 mmHg and pulse as 72/min. No pathological findings could be observed in the physical examination. The patient’s biochemical data are summarized in . Urine analysis was normal and there was no glucosuria or abnormal proteinuria. His electrocardiogram (ECG) showed sinus rhythm and QT interval was normal.
Table 1. Biochemical data of patient.
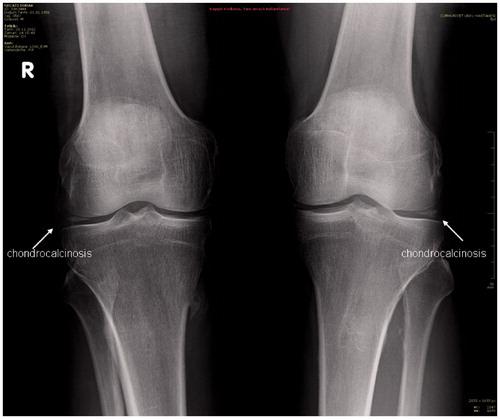
Because the patient stated that there had been swelling and pain in his knees two years ago, both knees were X-rayed and chondrocalcinosis was diagnosed in both of his knee joints ().
Figure 1. Chondrocalcinosis in X-ray of the patient’s knees.
The patient was diagnosed with GS as his blood pressure was normal and he had hypokalemia, hypomagnesemia, hypochloremia, metabolic alkalosis, increased amount of sodium, potassium and magnesium in the urea and hypocalciuria. The patient was recommended a sodium- and potassium-rich diet. A 100 mEq a day potassium chloride infusion and 2 × 1830 mg a day magnesium citrate sachet treatment was started. Five days later, potassium level rose to 4.1 mEq/L and magnesium level up to 1.89 mg/dL. He was given 4 g/day potassium citrate and 2 × 1830 mg magnesium citrate orally and discharged to come for control.
Discussion
Vomiting, diarrhea, anorexia and diuretic and laxative usage history should first be questioned in the distinctive diagnosis of a hypokalemic patient who is not hypertensive. After these causes are excluded, renal tubular acidosis, Bartter syndrome (BS) or GS should be taken into consideration.Citation9,Citation10 Our patient did not have a history of vomiting, diarrhea, anorexia or drugs usage. Because metabolic alkalosis was found in his tests, a distinctive diagnosis between BS and GS was made. The common clinical findings of BS and GS are hypokalemic metabolic alkalosis, normal or low blood pressure, loss of salt and increased levels of plasma renin and aldosterone.Citation10 And the difference between them is that, while there is hypomagnesemia and hypocalciuria in GS, these usually do not exist in BS. However, some cases with mutations in the Na/Cl cotransporter do not show hypomagnesemia or hypocalciuria.Citation11 Meanwhile, some cases of BS type 3 have been reported to show hypocalciuria or hypomagnesemia.Citation12 Therefore, it is difficult to distinguish BS and GS only by biochemical parameters; the distinctive diagnosis could be done with renal clearance tests. Giving the patients furosemide and thiazide in two phases, changes in fractional excretions of sodium, chloride and potassium are measured. If no increase is observed in the fractional excretion of sodium, chloride and potassium after furosemide is given, the problem is thought to be in the ascending limb of Henle. If there is not any increase in the fractional excretions of sodium, chloride and potassium after thiazide is given, then the problem is thought to be in the thiazide-sensitive Na/Cl cotransporter.Citation13 Bettinelli and his friends stated the criteria for the diagnosis of GS as hypomagnesemia, hypokalemia and hypocalciuria.Citation14 BS typically emerges before the age of 6 with severe symptoms like dehydration, nephrocalcinosis and growth retardation, whereas GS usually appears at an adult age and with neuromuscular symptoms.Citation4 The main disorder in BS is the one in the sodium reabsorption in the thick ascending limb of the loop of Henle. There is mutation in the Na--K 2 Cl cotransporter in some types of the disease and in basolateral chloride channels and rat outer medullas channels (ROMK) in some others. Calcium and magnesium absorption in the thick ascending limb of Henle is also blocked; but as some of the magnesium is reabsorbed from the distal convoluted tubule loss of magnesium reduces.Citation15–17 Typically, BS is seen as similar to furosemide effect while GS resembles thiazide effect. Due to hypovolemia, in BS and GS, blood pressure is lower than the general population. In BS, renal production of vasodilator prostaglandins (prostaglandin E2) is increased, which contributes to hypotension. In GS, amount of urinary prostaglandin E2 is low.Citation18 As our patient is of an advanced age and has hypomagnesaemia and hypocalciuria, he was diagnosed with GS. A genetic analysis could not be done because he did not accept that.
GS occurs as a result of the mutation in the SLC12A3 gene, which codes the thiazide-sensitive Na/Cl cotransporter in the distal convoluted tubule and is localized in the shorter limb of the 16th chromosome.Citation19 In adult patients, the presence of mutations of the SLC12A3 gene is present in 80% of cases.Citation20,Citation21 Sodium reabsorption disorder in the distal tubule causes loss of sodium and hypovolemia. Renin–angiotensin–aldosterone system is activated, thus increasing the secretion of the potassium and hydrogen ions and as a result hypokalemia and metabolic alkalosis develop. Passive calcium reabsorption in the proximal tubule and decrease in the epithelial magnesium channels localized in the distal collective tubule are observed in these patients. Thiazides are known to inhibit the Na/Cl cotransporter system. As there is a phenotypic similarity between the GS and chronic thiazide treatments, it is very possible that similar mechanisms can be effective in the pathogenesis of hypocalciuria and hypomagnesemia seen in GS.Citation22
The clinical evidence of GS is muscle cramps due to hypokalemia and hypomagnesemia, fatigue, tetany and paralysis. However, serious complications like rhabdomyolysis and sudden cardiac arrest have also been reported.Citation4,Citation23,Citation24 The fact that such serious complications could develop, although the patients are generally asymptomatic, indicates the importance of early diagnosis in GS. Diagnosis of GS is determined with clinical and biochemical findings like hypokalemia, metabolic alkalosis, hypomagnesemia and hypocalciuria in our patient. Increase in the levels of serum renin and aldosterone is not as clear as in BS and these levels are only slightly higher than normal. Renin and aldosterone level was normal in our patient. ECGs of half of the patients with GS have shown that QT interval is prolonged. Blood pressure is usually measured as normal or low. In the ECG of our case, QT interval was not prolonged and blood pressure was normal. GS symptoms do not correlate with the degree of the laboratory abnormalities. In some patients, the only evidence could be the chondrocalcinosis. Chondrocalcinosis occurs when calcium pyrophosphate dehydrate crystals are stored in the synovium or synovial fluid as a result of magnesium deficiency and can be healed by magnesium replacement.Citation4 Although our patient was asymptomatic too, he described an arthritis attack in both knees two years ago. Chondrocalcinosis was detected in the X-rays of his knees. GS is generally benign. Chronic hypokalemia may cause renal tubular vacuolization and tubulointerstitial nephropathy. Progression to renal failure is very rare in GS. Only one patient so far has been reported to have developed an end-stage renal failure.Citation25
Treatment of GS is a lifelong potassium and magnesium replacement and potassium-sparing diuretics usage. Spironolactone is one of the most frequently used potassium-sparing diuretics, but as it also binds to androgen and progesterone receptors, it has some side effects such as gynecomastia, erectile dysfunction, menstrual cycle disorder and hirsutism. As these patients need a lifelong treatment, drugs with fewer side effects should be chosen. Eplerenone, a drug causing selective aldosterone blockade, has a less affinity to androgen and progesterone receptors than spironolactone.Citation26 There are publications stating that eplerenone, which is used more commonly in cardiovascular diseases and heart failure, is also beneficial in GS.Citation27–29 Angiotensin-converting enzyme inhibitors and angiotensin receptor blockers could also be used in hypokalemia control. As, contrary to BS, prostaglandin levels are normal in GS, nonsteroid inflammatory drugs are not effective in GS treatment.Citation18
In conclusion, GS is a hereditary disease characterized by mild symptoms like fatigue and muscle cramps and diagnosed at an adult age. But, it also bears the risk of serious complications like paralysis and sudden cardiac arrest. Therefore, the early diagnosis of these patients is very important. Sometimes patients, as in our case, might come with a chondrocalcinosis-related arthritis table. In this case, if the cause of the arthritis cannot be explained, a biochemical analysis should be done and GS should be taken into consideration in the differential diagnosis if hypokalemia and hypomagnesemia detected.
Declaration of interest
Authors declare no conflict of interest. We have not financial relationship with the organization that sponsored the research.
References
- Gitelman HJ, Graham JB, Welt LG. A new familial disorder characterized by hypokalemia and hypomagnesemia. Trans Assoc Am Physicians. 1966;79:221–235
- Shaher AJ. Inherited primary renal tubular hypokalemic alkalosis: a review of Gitelman and Bartter syndromes. Am J Med Sci. 2001;322:316–332
- Lemmink HH, Knoers NV, Károlyi L, et al. Novel mutations in the thiazide sensitive NaCl cotransporter gene in patients with Gitelman syndrome with predominant localization to the C terminal domain. Kidney Int. 1998;54:720–730
- Knoers NVAM, Levtchenko EN. Gitelman syndrome. Orphanet J Rare Dis. 2008;3:221–226
- Karolyi L, Ziegler A, Pollak M. Gitelman’s syndrome is genetically distinct from other forms of Bartter’s syndrome. Pediatr Nephrol. 1996;10:551–554
- Lee YT, Wang IF, Lin TH, et al. Gitelman syndrome: report of three cases and literature review. Kaohsiung J Med Sci. 2006;22:357–362
- Riveira-Munoz E, Chang Q, Godefroid N, et al. Belgian network for study of gitelman syndrome. Transcriptional and functional analyses of SLC12A3 mutations: new clues for the pathogenesis of Gitelman syndrome. J Am Soc Nephrol. 2007;1:1271–1283
- Cruz DN, Shaer AJ, Bia MJ, Lifton RP, Simon DB. Gitelman's syndrome revisited: an evaluation of symptoms and health-related quality of life. Kidney Int. 2001;59:710–717
- Eren MA, Tabur S, Sezgin B, Sabuncu T. A rare cause of hypokalemia: Gitelman syndrome. Eur J Gen Med. 2011;8(2):154–156
- Nakamura A, Shimizu C, Nagai S, et al. Problems in diagnosing atypical Gitelman's syndrome presenting with normomagnesaemia. Clin Endocrinol. 2010;72(2):272–276
- Lin SH, Cheng NL, Hsu YJ, Halperin ML. Intrafamilial phenotype variability in patients with Gitelman syndrome having the same mutations in their thiazide-sensitive sodium/chloride cotransporter. Am J Kidney Dis. 2004;43:304–312
- Jeck N, Konrad M, Peters M, Weber S, Bonzel KE, Seyberth HW. Mutations in the chloride channel gene, CLCNKB, leading to a mixed Bartter--Gitelman phenotype. Pediatr Res. 2000;48:754–758
- Yagi H, Yahata K, Usui T, Hasegawa C, Seta K, Sugawara A. Inheritance of an autosomal recessive disorder, Gitelman's syndrome, across two generations in one family. Intern Med. 2011;50(11):1211–1214
- Bettinelli A, Bianchetti MG, Girardin E, et al. Use of calcium excretion values to distinguish two forms of prımary renal tubular hypokalemic alkalosis: Bartter and Gitelman syndromes. J Pediatr. 1992;120:38–43
- Simon DB, Lifton RP. The molecular basis of inherited hypokalemic alkalosis Bartter’s and Gitelman’s syndrome. Am J Physiol. 1996;271:F961–F966
- Akçakaya M, Oymak O, Ünal A, Kavuncuoğlu F, Tokgöz B, Utaş C. Familial hypokalemic alkalosis: Gitelman’s Syndrome. Turkish Nephrol Dial Transplant J. 2009;18(2):97–99
- Peters M, Jeck N, Reinalter S, et al. Clinical presentation of genetically defined patients with hypokalemic salt losing tubulopathies. Am J Med. 2002;112:183–190
- Emmett M. Bartter and Gitelman Syndromes; 2013. Available at: http://www.uptodate.com
- Zelikovic I, Szargal R, Hawash A, et al. A novel mutation in the chloride channel gene CLCNKB as a cause of Gitelman and Bartter syndrome. Kidney Int. 2003;63:24–32
- Nakhoul F, Nakhoul N, Dorman E, Berger L, Skorecki K, Magen D. Gitelman’s syndrome: a pathophysiological and clinical update. Endocrine. 2012;41:53–57
- Vargas-Poussou R, Dahan K, Kahila D, et al. Spectrum of mutations in Gitelman Syndrome. J Am Soc Nephrol. 2011;22:693–703
- Nijenhuis T, Vallon V, Kemp AWCM, Loffing J, Hoenderop JG, Bindels RJ. Enhanced passive Ca2+ reabsorption and reduced Mg2+ channel abundance explains thiazide-induced hypocalciuria and hypomagnesemia. J Clin Invest. 2005;115:1651–1658
- Ng HY, Lin SH, Hsu CY, Tsai YZ, Chen HC, Lee CT. Hypokalemic paralysis due to Gitelman syndrome: a family study. Neurology. 2006;67:1080–1082
- Scognamiglio R, Negut C, Calò LA. Aborted sudden cardiac death in two patients with Bartter's/Gitelman's syndromes. Clin Nephrol. 2007;67:193–197
- Bonfante L, Davis PA, Spinello M, et al. Chronic renal failure, end-stage renal disease, and peritoneal dialysis in Gitelman's syndrome. Am J Kidney Dis. 2001;38:165–168
- Ito Y, Yoshida M, Nakayama M, et al. Eplerenone improved hypokalemia in a patient with Gitelman's syndrome. Intern Med. 2012;51(1):83–86
- Pitt B, Remme W, Zannad F, et al. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med. 2003;348:1309–1321
- Zannad F, McMurray JJ, Krum H, et al. Eplerenone in patients with systolic heart failure and mild symptoms. N Engl J Med. 2011;364:11–21
- Morton A. Eplerenone in the treatment of Gitelman's syndrome. Intern Med J. 2008;38(5):377