
Abstract
Introduction: The tricho-rhino-phalangeal syndrome type III (TRPS III) is a rare autosomal dominantly inherited condition. The main clinical features are sparse and slow-growing hair and nails, a pear-shaped nose with a bulbous tip, elongated and flat philtrum, thin upper lip, cone-shaped epiphyses of the phalanges, and short stature. All patients have a point mutation in the TRPS1 gene. Case report: In this paper, we present a 13-year-old female with the typical clinical features of TRPS III, extreme growth retardation, severe deformities of both proximal radii resulting in limited extension of the elbows, and chronic renal failure (CRF) in addition. Molecular diagnostics revealed a missense mutation in exon 6 of TRPS1 that she inherited from her father who is also affected with TRPS III, but does not have CRF. In the index patient, the CRF was found to be due to bilateral renal hypodysplasia (RHD). Conclusion: Beside the renal dysplasia, the girl had severe deformities of the proximal radii – findings which have not been reported so far in TRPS III.
Introduction
The tricho-rhino-phalangeal syndromes (TRPS type I, OMIM#190350, type II, OMIM#150230, and type III, OMIM#190351) are rare autosomal dominantly inherited conditions characterized by a triad of hair, craniofacial, and skeletal abnormalities. The common features are sparse and slow-growing hair and nails, a pear-shaped nose with a bulbous tip, elongated and flat philtrum, thin upper lip, and bone deformities like Perthes-like hip disease and, in particular, cone-shaped epiphyses of the phalanges. TRPS I and III mainly differ in the severity of the microsomia, whereas TRPS II is a contiguous gene syndrome that combines the features of TRPS with multiple cartilaginous exostoses. TRPS I is caused by a mutation in or a deletion of the TRPS1 gene, which is located on the chromosome 8q24.1 and encodes the 1281 amino acids zinc-finger transcription factor protein TRPS1.Citation1 Patients with TRPS III almost always have missense mutations in the GATA-type zinc-finger domain (ZnF) of TRPS1.Citation2,Citation3 In patients with TRPS II, at least the interval from TRPS1 through EXT1, which causes the exostoses, is deleted. Patients with TRPS II and deletions extending to outside this interval are often developmentally retarded. Patients with any type of TRPS may also have endocrine, cardiac, or renal manifestation. In this paper, we present a 13-year-old girl, who in addition to the characteristic features of TRPS III presents with chronic renal failure and malformed proximal radii, findings that have not been reported in TRPS before and that were not present in her father, who also has TRPS III. Our report augments the clinical spectrum of TRPS and demonstrates the clinical variability of this condition.
Case report
A very communicative and friendly 13-year-old girl was referred to the Department of Nephrology from an orthopedic hospital for diagnostic evaluation of chronic renal failure. She had extreme short stature [height 109 cm (−5.7 SD)] and weight of 16.2 kg (−3.6 SD)]. She had previously undergone surgery for congenital dysplasia of the right hip (). At that time, her growth retardation had not attracted the attention of any attending physician. The parents also have never cared about her stature, because both parents and many relatives on the paternal side were of a short stature. The index patient had neither a history of urinary tract infections nor episodes of unmotivated fever.
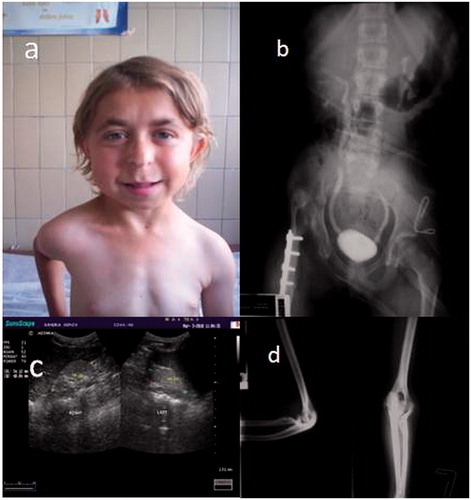
Figure 1. Clinical and radiological features of the index patient. (a) Facial appearance of the patient, (b) voiding urethrocystography reveals bilateral vesicoureteral reflux. Congenital dysplasia of the right hip (after surgery) (c) kidney ultrasound scan reveals bilateral hypodysplasia and (d) X-ray shows deformity of the proximal radius.
The laboratory analyses revealed mild normochromic anemia and normal routine urinalysis. Her serum biochemistry was as follows: urea 16.4 mmol/L (normal < 6.8 mmol/L), creatinine 264 µmol/L (normal< 88 µmol/L), clearance according to Schwartz formula 15 mL/min/1.73 m2 (normal >90 mL/min/1.73 m2), Na 142 mmol/L, K 4.1 mmol/L, Ca 2.21 mmol/L, Mg 1.0 mmol/L, P 1.54 mmol/L, total protein 64 g/L, albumin 36 g/L, alkaline phosphatase 157 U/L, PTH 123 pg/mL (normal 10–65). Liver enzymes were within normal ranges. There was mild acidosis: pH 7.27 (normal 7.35–7.47), HCO3 16.0 mmol/L (normal 22–26 mmol/L), BE-9.6 mmol/L (normal <|1.5| mmol/L). She was polyuric (diuresis = 1750 mL) and her proteinuria was 1.3 g/d (normal <150 mg/d). T4 (7.3 mcg/mL) and TSH (0.914 microU/mL) were within referent limits. Measurement of growth hormone during the night and after stimulation with clonidine revealed normal levels. Detailed ophthalmologic and audiometric examination revealed normal results. On echocardiography, a non-significant mitral valve prolapse was evidenced. She had normal blood pressure values on repeated measurements.
Kidney ultrasound demonstrated small and hyperechogenic kidneys without clear corticomedullary differentiation in favor of bilateral hypodysplasia (). The bladder wall had normal thickness. Voiding urethrocystography revealed bilateral moderate vesicoureteral reflux grade III (). Since there was no clear history of febrile urinary tract infections, the etiology of chronic renal failure in this young girl was bilateral RHD. The severe growth retardation was not in concert with the degree of renal insufficiency and the absence of severe acid base and electrolyte abnormalities. Since the patient had family history for short stature and facial dysmorphy, she and her first degree relatives underwent careful dysmorphologic evaluation. The index patient had sparse and slowly growing hair. Her eyebrows were medially thick and laterally thin. She had a pear-shaped nose with a bulbous tip, a long and flat philtrum, thin upper lip, and protruding ears (). Her digits were short. The nails were also small, slowly growing and she rarely need to cut them. X-ray of her hands showed shortened metacarpals and cone-shaped epiphyses of medial phalanges. The maturation corresponded to the age of 7 years. She had limited extensibility in both elbows, and X-ray of the elbows revealed marked deformity of both proximal radii ().
Her 36-year-old mother was also of a short stature (140 cm), but did not have dysmorphic stigmata. The both male siblings were of normal stature for the age and did not have facial dysmorphism. The father, who was 40 years old, was of short stature (147 cm). Besides short stature he had impressively short fingers and small nails on both hands and feet. There was prominence of the second toe on the right foot; at a lesser extent on the left side. He also had prominent bulbous nose, long philtrum, thin upper lip, very thick eyebrows medially, and sparse laterally. He has normal kidney ultrasound and serum creatinine (66 μmol/L). Paternal mother and several relatives on her side had short stature and digits, suggesting autosomal dominant inheritance of the syndrome in this family. No consanguinity was reported by the parents. Ultrasound examination of the urinary tract did not demonstrate any abnormality in the parents and both siblings.
Mutational analysis of the TRPS1 gene in the index and first degree relatives was done after obtaining informed consent. Individual exons of the TRPS1 gene were amplified by polymerase chain reaction (PCR) and sequenced essentially as described elsewhere,Citation1,Citation2 with the exception that the following primers: TRPS_H_05 (5′-ctcctgggttgatttggtct-3′) and TRPS_H_06 (5′-agccagggaatgggacttat-3′) were used to amplify and sequence exon 6 that contains the mutation. Sequencing of the entire coding region of the TRPS1 gene revealed a heterozygous missense mutation in the exon 6 c.2756C > T [p.A919V] in the index patient and her father. The mother and both healthy siblings carried only the wild-type sequence.
Discussion
The most common affection of inner organs in the TRPS is that of the urogenital tract, cardiac, and endocrine system. Several heart defects have been described, e.g., ventricular septal defect, truncus arteriosus with severe pulmonary hypertension, mitral valve prolapse. We diagnosed mitral valve prolapse in our patient, but it was hemodynamically not significant.
Besides cardiac defects, different renal and urogenital abnormalities are described in TRPS, among them vesicoureteral reflux is reported most often (). Lu et al.Citation4 reported a male neonate with tricho-rhino-phalangeal syndrome type II and interstitial deletion of chromosome 8 with karyotype 46, XY, del (8) (q24.11–>q24.13). The baby had hypotrichosis of the scalp hair, bulbous nose, and redundant skin. In addition, he had aplasia of the epiglottis and congenital nephrotic syndrome (non-Finish type) and died at the age of 11 d. Yáñez et al.Citation5 reported a 21-year-old female with TRPS (most likely type 3) who was found to have bilateral ureterovesical junction obstruction at the age of 7. She had recurrent urinary tract infections, nephrolithiasis, and progressed to chronic renal insufficiency. Wilson et al.Citation6 reported a patient with interstitial deletion of chromosome 8q and bilateral vesicoureteral reflux. Rossi et al.Citation7 reported a familial case with TRPS and missense mutation in the nuclear localization signal (R952C) of TRPS1. Only one of the three affected family members was reported to have a history of vesicoureteral reflux. Ramos et al.Citation8 described a female patient with persistent cloaca and clinical features of TRPS type II as a result of 8q interstitial deletion. She was found to have vesicoureteral reflux as a component of Prune Belly sequence which is extremely rare in females. Other urogenital malformations which are described in TRPS are hydroureter, megacystic–megaureter, polycystic ovary, vaginal agenesis, arcuate uterus, and hemosalpinx. Although vesicoureteral reflux (VUR) is the most common urinary tract malformation in TRPS, it appears to the authors that a considerable amount of patients remains unreported.
Table 1. Spectrum of renal/urogenital abnormalities in patients with tricho-rhino-phalangeal syndrome.
All clinical features of our family fit well with TRPS type III. Only VUR/renal hypodysplasia/chronic renal failure is exceptional finding in the index case. We did not find any urinary tract abnormality in the first degree relatives at ultrasound screening. It is well known that VUR and renal dysplasia have genetic basis and increased prevalence among family relatives, particularly VUR.Citation9–11 Although systematic screening has not been performed in TRPS, it seems that VUR phenotype is not fully penetrant. This is in agreement with the findings in our patient's family and this is not unusual in syndromal RHD, in general. Weber et al.Citation11 observed significant variability in the renal phenotype in two families affected by a dominant EYA1 mutation and deletion. The index patients presented with complete phenotype of branchio-oto-renal syndrome, while their parents had deafness, ear tags, and cervical fistulas without a renal phenotype. This is in favor that additional genetic factors modify the variable expressiveness of the renal phenotype in patients with well-defined genetic syndromes.
There is evidence that Trps1 is expressed in the lungs, gut, kidneys, and other tissues during embryonic development. In the urogenital system, Trps1 is expressed in the stroma of the kidney, and the mesenchyme of the mesonephric duct. Gai et al.Citation12 recently investigated the role of Trps1 in nephrogenesis in animal models. They generated Trps1 deficient mice and found that newborns had fewer tubules and glomeruli, expanded renal interstitium, and numerous uninduced metanephric mesenchymal cells, which resulted in fewer nephrons. Trps1 acts downstream of bone morphogenic protein 7 (Bmp7), which is essential for normal renal development. In Trps1-deficient kidneys, Bmp7-induced mesenchymal to epithelial transition is impaired; there are low levels of Pax2 and Wt1, which are important for differentiation of cap mesenchyme to renal vesicles. The authors concluded that Trps1 is required during early renal development for normal formation of nephrons.
Concerning the growth and renal function, our patient was medically neglected patient. The fact that her father and paternal relatives also had short stature did not justify delay in medical examination. In contrast, minor dysmorphic stigmata in the index case and her father did not attract medical attention and probably many mild cases of TRPS go unrecognized. In the literature, there are reports of endocrine disturbance which may affect the growth;Citation13,Citation14 in our case, growth hormone levels basely and after stimulation were in the normal ranges. Chronic renal insufficiency significantly worsens growth parameters, but we do not have data about our patient’s renal function in the past. It is known that some TRPS patients may spontaneously improve their growth in the puberty.Citation15
Mutational analysis in the index patient and her father revealed a single base substitution in the exon 6 of the TRPS1 gene. This mutation has already been reported as disease causing mutation in two reportsCitation3,Citation16 and was absent in the exome variant server (http://evs.gs.washington.edu/EVS) which include >6500 individuals. This base substitution causes an amino acids exchange in the zinc coordinating region of the GATA-type zinc finger domain, and must be regarded as a causative mutation.Citation2 Mutant proteins as a result of missense mutations are able to enter the nucleus. These mutants are thought to have a decreased affinity to DNA due to alteration in GATA zinc fingers ZnF, and manifest a dominant negative effect as a component of a multimeric protein complex, in contrast to haploinsufficiency supposed for the remaining mutations. Therefore, patients with missense mutation have a more severe clinical phenotype regarding the skeletal changes and stature as was the case in our index patient and her father.
In conclusion, we present a young female with TRPS who had two new features: bone deformities of both proximal radii resulting in marked limitation of extension of the elbows and bilateral RHD leading to chronic renal failure. The prevalence of renal abnormalities in TRPS is not known. Therefore, non-invasive ultrasound screening should be offered to all patients with TRPS and vice versa – one should look for typical hair, facial, and skeletal features in patients with congenital anomalies of the kidney and urinary tract.
Declaration of interest
The authors report no conflict of interests. This study was supported in part by the Bundesministerium für Bildung und Forschung (BMBF Grant 01GM0878).
Acknowledgments
We are grateful to M. Heitmann for expert technical assistance.
References
- Momeni P, Glöckner G, Schmidt O, et al. Mutations in a new gene, encoding a zinc-finger protein, cause tricho-rhino-phalangeal syndrome type I. Nat Genet. 2000;24:71–74
- Lüdecke H-J, Schaper J, Meinecke P, et al. Genotypic and phenotypic spectrum in tricho-rhino-phalangeal syndrome types I and III. Am J Hum Genet. 2001;68:81–91
- Hilton MJ, Sawyer JM, Gutiérrez L, Hogart A, Kung TC, Wells DE. Analysis of novel and recurrent mutations responsible for the tricho-rhino-phalangeal syndromes. J Hum Genet. 2002;47:103–106
- Lu FL, Hou JW, Tsai WS, Teng RJ, Yau KI, Wang TR. Tricho-rhino-phalangeal syndrome type II associated with epiglottic aplasia and congenital nephrotic syndrome. J Formos Med Assoc. 1997;96:217–221
- Yáñez S, Hernández-Vicente I, Armijo M. Tricho-rhino-phalangeal syndrome. Int J Dermatol. 1992;31:706–709
- Wilson WG, Wyandt HE, Shah H. Interstitial deletion of 8q. Am J Dis Child. 1983;137:444–448
- Rossi A, Devirgiliis V, Panasiti V, et al. Missense mutation in exon 7 of TRPS1 gene in an Italian family with a mild form of tricho-rhino-phalangeal syndrome type I. Br J Dermatol. 2007;157:1021–1024
- Ramos FJ, McDonald-McGinn DM, Emanuel BS, Zackai EH. Tricho-rhino-phalangeal syndrome type II (Langer–Giedion) with persistent cloaca and prune belly sequence in a girl with 8q interstitial deletion. Am J Med Genet. 1992;44:790–794
- Sanna-Cherchi S, Caridi G, Weng PL, et al. Localization of a gene for nonsyndromic renal hypodysplasia to chromosome 1p32–33. Am J Hum Genet 2007;80:539–549
- Sanna-Cherchi S, Caridi G, Weng PL, et al. Genetic approaches to human renal agenesis/hypoplasia and dysplasia. Pediatr Nephrol. 2007;22:1675–1684
- Weber S, Moriniere V, Knüppel T, et al. Prevalence of mutations in renal developmental genes in children with renal hypodysplasia: results of the ESCAPE study. J Am Soc Nephrol. 2006;17:2864–2870
- Gai Z, Zhou G, Itoh S, et al. Trps1 functions downstream of Bmp7 in kidney development. J Am Soc Nephrol. 2009;20:2403–2411
- Riedl S, Giedion A, Schweitzer K, et al. Pronounced short stature in a girl with tricho-rhino-phalangeal syndrome II (TRPS II, Langer–Giedion syndrome) and growth hormone deficiency. Am J Med Genet A. 2004;131:200–203
- Stagi S, Bindi G, Galluzzi F, Lapi E, Salti R, Chiarelli F. Partial growth hormone deficiency and changed bone quality and mass in type I tricho-rhino-phalangeal syndrome. Am J Med Genet A. 2008;146A:1598–1604
- Naselli A, Vignolo M, Di Battista E, et al. Tricho-rhino-phalangeal syndrome type I in monozygotic twins discordant for hip pathology. Report on the morphological evolution of cone-shaped epiphyses and the unusual pattern of skeletal maturation. Pediatr Radiol. 1998;28:851–855
- Kantaputra P, Miletich I, Lüdecke HJ, et al. Tricho-rhino-phalangeal syndrome with supernumerary teeth. J Dent Res. 2008;87:1027–1031
- Howell CJ, Wynne-Davies R. The tricho-rhino-phalangeal syndrome. A report of 14 cases in 7 kindreds. J Bone Joint Surg Br. 1986;68:311–314
- Lemke T, Pirsig W. The tricho-rhino-phalangeal syndrome. Laryngol Rhinol Otol (Stuttg) 1978;57:1112–1115
- Braga D, Manganoni AM, Gavazzoni R, Pasolini G, De Panfilis G. A case of trichorhinophalangeal syndrome, type I. Cutis. 1994;53:92–94
- Graybeal LS, Baum VC, Durieux ME. Anaesthetic management of a patient with tricho-rhino-phalangeal syndrome. Eur J Anaesthesiol. 2005;22:400–402