
Abstract
Adriamycin (ADR) is commonly used for many solid tumor treatments. Its clinical utility is, however, largely limited by the adverse reactions, are known to be nephrotoxic. The mechanism by which it induces kidney damage is still not completely understood, but its nephrotoxicity might relate to increase reactive oxidant status (ROS), mitochondrial dysfunction. Until now, neurohormonal activation of it is unclear. ADR might activate the renin angiotensin system. Angiotensin-II also induced ROS and mitochondrial dysfunction. The aim of this study was to investigate whether angiotensin-II production inhibition has the protective effect on attenuation of mitochondrial function in rats with acute ADR-nephrotoxicity or not. Rats were divided into five groups as a control, ADR, co-treated ADR with captopril (CAP), co-treated ADR with Aliskren, co-treated ADR with both CAP and Aliskren groups. Creatinine kinase (CK) levels were measured at the end of treatment period. The kidneys were homogenized and biochemical measurements were made in mitochondria, cytosol. Mitochondria membrane potential (MMP) and ATP levels were determined. ADR increased CK levels and oxidative stress in mitochondria too (p < 0.05). ADR significantly decreased MMP and ATP level in kidney mitochondria (p < 0.05). Co-administration with ADR and Aliskren and CAP improved the dissipation of MMP (p < 0.05). The decrease in ATP level was restored by treatment with inhibitors of ACE and renin. We concluded that inhibitors of angiotensin-II are effective against acute ADR induced nephrotoxicity via the restoration of MMP and ATP production and prevention of mitochondrial damage in vivo.
Introduction
Adverse drug reaction (ADR) has commonly been utilized for a variety of cancers, including breast cancer and childhood leukemias. Unfortunately this broadly successful antineoplastic agent has also been shown to be toxic in many tissues including kidney, heart, liver, or testis.Citation1 The anthracycline anti-tumor drug ADR causes severe nephrotoxicity in a variety of experimental animals.Citation2,Citation3 ADR’s nephrotoxicity represents a model very close to human progressive chronic renal disease. The results of the renal function test revealed that ADR administration produced intrinsic renal failure, which was evident from the elevated levels of serum urea and creatinine.Citation4 It was, however, shown that its nephrotoxicity could initially start by accumulation of the ADR in renal proximal tubular cells. The tubular cells undergo necrosis accompanied by the corresponding of kidney dysfunctions.
The molecular mechanism by which Adriamycin causes renal damage is not still completely understood. ADR-induced toxicity might appear to be multifactorial. A critical component may be the generation of free radicals and the presence of redox-related damage, which occur through both enzymatic and nonenzymatic pathways. ADR-generated free radicals induce lipid peroxidation, which in turn, causes diverse oxidative damage on critical cellular components and membrane lipids in the plasma membranes and mitochondria.Citation5–9 In addition, mitochondrial damage, increased Ca2+ current along with inhibition of sarcoplasmic reticulum function and decreased activity of Na, K-ATPase, have all been implicated in ADR-induced toxicity.Citation10
The main target of ADR-induced toxicity in cardiomyocytes is mitochondria which accumulate ADR over time.Citation11 The exact mechanisms of ADR inhibition of mitochondrial electron transport and oxidative phosphorylation have not been clearly clarified.Citation12 Most evidence on this concept indicates to mediate oxygen-free radicals interference with mitochondrial bioenergetics and calcium regulation as being definitive in the pathogenic process.Citation13 When mitochondrial permeability transition (MPT) is promoted by oxidative stress and by increases in the cytosolic Ca+2 concentration, the release of cytochrome c is associated with MPT and results in a loss of the H+ gradient and mitochondria membrane potential (MMP) across the mitochondrial inner membrane, an increase in the matrix volume, and disruption of the outer membrane.Citation14 Additional indications of oxidative injury to mitochondria include membrane lipid peroxidation,Citation15 inhibition of respiration and oxidative phosphorylation, decreased mitochondrial ATPase activity, and a net decrease in the redox state of respiratory carriers.Citation16
Recently, ADR treatment has been shown to activate the renin angiotensin system. Angiotensin II (Ang-II) is the most important active factor in the renin–angiotensin system and has close relationship with glomerulosclerosis.Citation17 Previous studies have also reported that Ang-II plays a key role in the process of ADR-induced nephrotoxicity.Citation18–20 However, studies have suggested that angiotensin-converting enzyme inhibitorCitation20 and angiotensin receptor blockerCitation18 exert a protective role against ADR-induced nephrotoxicity.Citation21,Citation22
Ang-II is able to increase oxidative stress by stimulating the generation of both NO and NAD(P)H oxidase-derived superoxide, thereby enhancing reactive nitrogen species.Citation23 It was additionally observed in studies that Ang-II stimulates mitochondrial ROS production in vascular smooth muscle cells, in endothelial cells, and in rat aorta in vivo. A link between Ang-II related ROS/RNS production and mitochondrial function was suggested by a report showing that antioxidants inhibit the regulatory effects of Ang-II on the AP-1 signaling pathway. Since AP-1, whose activity responds to oxidation/reduction, regulates cytochrome c expression, it was suggested that Ang-II may facilitate changes in mitochondrial cytochrome c content.Citation24 Additionally, in mice, acute and chronic Ang-II infusions showed that in the depression of mitochondrial energy metabolism, Ang-II lowers MMP as a result of stimulation of mitochondrial ROS production.Citation24
As a result, both ADR and Ang-II have an induction of ROS via redox cycling on the mitochondrial electron transport chain. This increase in ROS leads to several damaging events in the mitochondrion. Attenuation of mitochondrial function leads to decreased ATP-production. Thus, the hypothesis of this study was to investigate whether Ang-II production inhibition by using ACE and/or renin inhibitors have the protective effect on diminish mitochondrial function in rats with acute ADR-nephrotoxicity.
Materials and methods
Animals
All experimental protocols were approved by the Medical Faculty Ethics Committee on Animal Research at Erciyes University. Thirty-five male Sprague-Dawley rats were housed individually in polypropylene cages under hygienic and standard environmental conditions (24 ± 1 °C, humidity 60–70%, 12 h light/dark cycle). The animals were allowed a standard diet and water ad libitum. Animals were randomly assigned into five different treatment groups: Control (n = 7), ADR (n = 7), CAP (Captopril, n = 7), AL (n = 7), and CAP plus AL (Aliskren, n = 7). ADR (Adriamycin HCl, Adriblastina vial 10 mg, Pharmacia, Milan, Italy) was treated in four equal injections (each intraperitoneal injection containing 4 mg/kg in saline, for a total dose of 16 mg/kg) during 8 days, and the control group received the same volume of physiological saline. Captopril was intragastrically treated (10 mg/kg, every day for 8 days). Aliskren was intragastrically treated (50 mg/kg, every day for 8 days).
After last treatment of drugs, blood samples were taken to measure creatinine kinase (CK) as an index of kidney damage and separated plasma. Kidney tissues were collected then kept at −80 °C until use, and later separated cytosol and mitochondria by centrifugation and used for biochemical assays.
Preparation of mitochondria and cytosol
Mitochondria from left rat kidneys were isolated as described. The kidneys were homogenized in an ice-cold buffer A (250 mM sucrose, 2 mM EGTA, 5 mM Tris HCl) with a homogenizer and centrifuged at 2000g for 8 min at 4 °C. The supernatant (cytosol) was further centrifuged at 12,000 g for 10 min at 4 °C in a new tube. The pellet of mitochondria was suspended in an ice-cold buffer B (140 mM potassium, 20 mM Tris HCl). The mitochondria and cytosols were kept at −80 °C until use.
Measurement of MMP in mitochondria from kidney
MMP was assessed by using the fluorescent mitochondrial-specific cationic dye 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethyl benzimidazole carbocyanine iodide (JC-1). JC-1 undergoes potential-dependent accumulation in the mitochondria. Mitochondria with a normal MMP aggregated JC-1 (red fluorescence) and in depolarized mitochondria, JC-1 formed monomers (green fluorescence). Mitochondria were added to each well, pre incubated with 10 μM JC-1 for 20 min at 37 °C in the dark; then the mitochondria were analyzed by a fluorescent plate reader (Biotek, Synergy HT, Gaziantep, Turkey). In healthy cells, JC-1 forms J-aggregates which displayed strong fluorescent intensity with excitation and emission at 560 and 595 nm, respectively. In apoptotic or unhealthy cells, JC-1 existed as monomers which showed strong fluorescence intensity with excitation and emission at 485 and 535 nm, respectively. The ratio of fluorescent intensity of J-aggregates to fluorescent intensity of monomers was used as an indicator of cell health.
Determination of ATP content in mitochondria from kidney
To measure ATP content in rat kidney with ADR-nephrotoxicity, a bioluminescent kit was used. The kit is based upon the bioluminescent of ATP that is present in all metabolically active cells. The bioluminescent method uses an enzyme, luciferase, which catalyzes the formation of light from ATP. Briefly, a 10 μL aliquot of a mitochondria sample was mixed with 100 μL cell lysis reagent and incubated at room temperature for 10 min to extract ATP from cells. Following the addition of 100 μL ATP Monitoring Reagent, luminescence was measured using a luminometer (Biotek, Synergy HT). A standard curve was generated from known concentrations of ATP and used to calculate the concentration of ATP in each sample. Luminescence increased linearly with the negative log of the ATP concentration in the samples over the range of measured concentrations. Mitochondrial ATP content from each probe was assessed in duplicate.
Biochemical studies
Creatine kinase assay
The creatine kinase (CK) level was estimated by kinetic determination using the commercial kits of Bechman by Bechman Coulter LX-2000 (Winooski, VT).
Measurement of total antioxidant status
Total antioxidant status of the plasma, myocytes cytosol, and mitochondria were measured using Rel Assay Kit (Paris, France). Total antioxidant status (TAS) method, which is based on the bleaching of the characteristic color of a more stable 2,2′-azino-bis (3-ethylbenz-thiazoline-6-sulfonic acid) (ABTS) radical cation by antioxidants. The results were expressed in mmol Trolox equiv./L.
Measurement of total oxidant status
Total oxidant status (TOS) of the plasma, myocytes cytosol and mitochondria were measured using Rel Assay Kit. TOS method is based on the oxidation of ferrous ion to ferric ion in the presence of various oxidative species in an acidic medium and the measurement of the ferric ion by xylenol orange. The results were expressed in μmol H2O2/L.
Statistical data analysis
Statistical analyses were conducted using Excel and SPSS version 16.0 (Chicago, IL). All results were expressed as mean ± SEM. Comparison among different groups were made using multiple analyses of variance (ANOVA), followed by a post hoc protected Tukey test. Nonparametric test including the Kruskal–Wallis one-way analysis of variance and Mann–Whitney U test were applied to find the statistical significance and a Bonferroni-adjusted alpha was used for the test. In all cases, p < 0.05 was considered to be significant.
Result
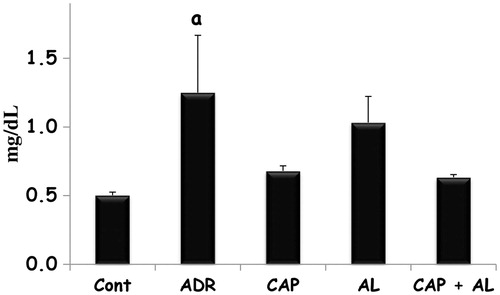
The effect of angiotensin inhibition on Adriamycin changed creatinine kinase levels in rats with nephrotoxicity
ADR is known to have nephrotoxicity. Therefore, CK level was first investigated as an indicator of kidney tissue damage. ADR caused to increase CK level (a: p < 0.05 vs. Cont). Renin and/or ACE inhibition slightly decreased CK level compared to ADR but not significant ().
Figure 1. The effect of renin–angiotensin II inhibitors on plasma creatinine kinase levels in rats with nephrotoxicity induced by ADR. TOS: Total oxidant status, Cont: Control group, ADR: Adriamycin group, CAP: Captopril group, AL: Aliskren group, CAP + AL: Captopril plus Aliskren group. a: p < 0.05 versus Cont. All data were expressed as mean ± SEM.
The effect of angiotensin inhibition on Adriamycin changed total oxidant status (TOS), and total antioxidant (TAS) status in rats with nephrotoxicity
The proposed principal mechanisms of ADR cardiotoxicity is well documented to increase oxidative stress, as evident from increased levels of reactive oxygen species, decreased levels of antioxidants. So, the level of oxidative stress was estimated by total oxidant status to total antioxidant status ratio. TAS and TOS were assed type of mitochondria, and cytosol from kidney tissue. ADR increased mitochondrial and cytosolic TOS (p < 0.05; ). Attenuation of Ang-II production by ACE and renin co-inhibitions had strong effect on mitochondrial TOS (p < 0.05; ), and cytosolic TOS as well (p < 0.05; ). Only renin inhibition decreased cytosolic TOS (p < 0.05; ). CAP had better healing effect on the ADR’s nephrotoxicity, resulting from decreasing cytosolic TOS () and increasing mitochondrial TAS (). But, CAP decreased cytosolic TAS (p < 0.05; ).
Table 1. The effect of renin–angiotensin II inhibitors on mitochondrial and cytosolic TOS in rats with nephrotoxicity induced by ADR.
Table 2. The effect of renin–angiotensin II inhibitors on mitochondrial and cytosolic TAS in rats with nephrotoxicity induced by ADR.
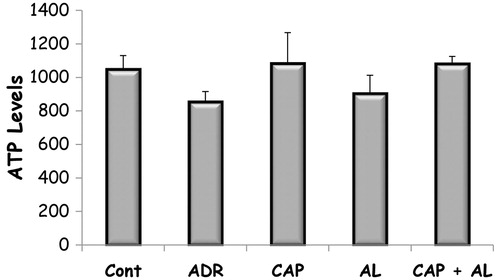
The effect of angiotensin inhibition on Adriamycin changed MMP and ATP level in rats with nephrotoxicity
The mitochondria are the most extensively and progressively injured subcellular organelles of ADR-induced toxicity.Citation25 Prevention of mitochondria function impairment will prevent kidney dysfunction. So, next step was determination of mitochondrial function impairment by using MMP and ATP production. MMP was measured using the fluorescent mitochondrial-specific cationic dye (JC-1). JC-1 could aggregate in normal mitochondria and present red fluorescence. The ratio of red and green fluorescence was used to demonstrate ADR’s toxicity in mitochondria and effect of angiotensin II inhibition on ADR’s toxicity. As expected, ADR was the dissipation of MMP (p < 0.05; ). Depolarized MMP by ADR did not accompany to decrease ATP production (). However, inhibition of Ang-II production by CAP and/or AL completely restored the dissipation of MMP (p < 0.05; ). Only ACE inhibition has slightly increased ATP level in kidney mitochondria, but not significant (). These data demonstrated that target of ADR was mitochondria in the kidney.
Figure 2. The effect of renin–angiotensin II inhibitors on mitochondrial membrane potential in rats with nephrotoxicity induced by ADR. MMP: Mitochondrial membrane potential, Cont: Control group, ADR: Adriamycin group, CAP: Captopril group, AL: Aliskren group, CAP + AL: Captopril plus Aliskren group. a: p < 0.05 versus CONT, b: p < 0.001 versus ADR, c: p < 0.01versus CAP + AL. All data were expressed as mean ± SEM.
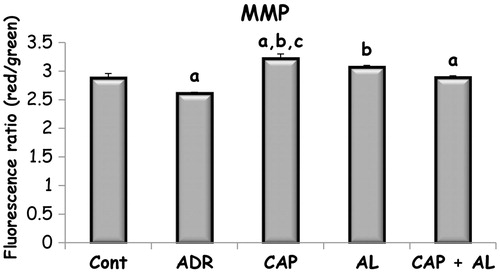
Figure 3. The effect of renin–angiotensin II inhibitors on ATP level in rats with nephrotoxicity induced by ADR. Cont: Control group, ADR: Adriamycin group, CAP: Captopril group, AL: Aliskren group, CAP + AL: Captopril plus Aliskren group. All data were expressed as mean ± SEM.
Discussion
ADR treatment has been reported to results in glomerular morphological changes that are similar to minimal change disease in human and suggested that toxic oxygen metabolites were responsible for Adriamycin toxicity in kidney tissue.Citation26,Citation27 It was also suggested that ADR-mediated ROS markedly increase the peroxidation of membrane polyunsaturated phospholipids and this lipid peroxidation level in vivo may be involved in the nephrotoxicity of ADR.Citation28
At the end of the treatment period of drug, we observed that the ADR treatment significantly reduced the MMP and slightly ATP level. In this study, the tissue oxidative stress in the cytosol and mitochondria were markedly increased by ADR, as the TOS was significantly increased and the TAS was dramatically decreased. Under physiological condition, intracellular ROS acts as a second messenger to a variety of growth factors.Citation7 However, when the ROS is excessively produced, for instance, in the setting of ADR treatment, it becomes deleterious and causes damage on cellular components and membrane lipids in the plasma membranes and mitochondria. The lipid peroxidation occurs in cellular components and membrane lipids when the ROS levels are elevated, leading to the release of reactive aldehydes, such as MDA.Citation7
Neurohormonal activation of ADR treatment is not clearly understood. However, ADR treatment has been shown to activate the renin angiotensin system (RAS).Citation19,Citation29–31 Furthermore, the effectiveness of blockade of the renin–angiotensin system in slowing the progression of proteinuria of kidney diseases is well recognized. Therefore, we investigated the interaction between the RAS and the ADR nephrotoxicity in this study.
Oxidative damage is the final result of an unbalance between free-radical production and the entity of antioxidant defenses, and the onset of the damage may be accounted to free-radical overproduction, antioxidant deficiency, or both. In the present study, ADR caused oxidative damage by increasing free radical products (TOS) and by slightly decreasing antioxidant status (TAS). Additional indications of oxidative injury of ADR in the kidney were increased apoptosis (MMP), and decreased energy production (ATP) in mitochondria of kidney tissue as it has been reported in other studies. The mechanism involved in the ADR-induced oxidative damage to the tissues appears to involve the generation of ROS which initiate free-radical-mediated chain reactions, resulting in the oxidation of unsaturated fatty acids of the membrane into lipid peroxides. This may explain the higher levels of MDA, an index of lipid peroxidation, in the kidneys of rats in the ADR.Citation32 It was found in a study that MDA as an index of oxidative damage was a significant increase and glutathione concentrations, an antioxidant, had a significant decrease in the renal cells after treatment with the ADR. These results are consistent with previous studies reported by other investigators that ADR induced nephrotoxicity in rats. According to Ibrahim et al.,Citation18 the renal protective effects of CAP were accompanied with attenuation of ADR-induced alteration of some antioxidants levels in kidney tissue. A single dose of captopril given 1 h before ADR was found to be protecting the kidney from damage induced by ADR, resulted from a decrease of serum creatinine, urea and by an increase in total proteins and albumin. The possibility that CAP protects the renal tissues against the ADR-induced toxicity by scavenging the superoxide anion radicals is uncertain.Citation29 It was shown in vitro study that ACE inhibition has reactive radical removal effect due to structure of CAP having SH group which is detoxified ROS.Citation31 Several agents have been investigated in an attempt to reduce Adriamycin toxicity. One of the most used agent was vitamin E (Vit-E).Citation33 It was postulated that Vit-E is a free radical scavenger and it can minimize cellular peroxidation after an injection of ADR.Citation34 Mimnaugh et al.Citation28 observed that beneficial effect on Adriamycin-enhanced microsomal and mitochondrial membrane lipid peroxidation could be potentially ameliorated by Vit-E in rat kidney tissue.
ROS production plays an important role in the pathogenesis of nephrotocity model.Citation35 Elevated Ang-II level induces the activation of NADPH oxidase via the AT1 receptor, and this represents an important source of ROS, which eventually lead to increase of superoxide generation and other ROS. In our current study, and consistent with those reports, animals with ADR-induced nephrotoxicity showed overproduction of TOS; this effect was diminished by ACE and/or renin inhibitors, suggesting an antioxidant effect of those inhibitors, probably due to the inhibition of an Ang-II NAD(P)H oxidase inducer effect with further decreased production of TOS.Citation36 Another possibility of this inhibitor’s effect on decreased TOS might be that the nephrotic state as such reduced the bradykinin-induced endothelium-dependent increase in coronary flow and enhanced the aortic constrictor response to Ang-II associated with a reduced basal NO release, as apparent from the comparison of Adriamycin nephrotic rats.Citation37
It has been observed that there is a large accumulation of ADR in kidney than in any other organ. The ADR is accumulated in the kidney, and its concentration in the kidney nuclei and mitochondria is much higher than that observed in the heart.Citation2,Citation3 Mitochondria may have been suggested to be a primary target of ADR-induced toxicity at the subcellular level.Citation6,Citation9,Citation38 The ADR also has the tremendous ability to inhibit oxidative phosphorylation, which may account for the impairment of energy metabolism of the cell inhibition of electron transport complexes, and disruption of calcium homeostasis.Citation38 Mitochondrial degeneration and dysfunction has been reported on ADR-induced toxicity.Citation2,Citation3,Citation6,Citation9 The impaired mitochondrial function may contribute to the pathogenesis of ADR nephrotoxicity, so that we now provided evidence for an additional mechanism of ACE inhibitor to ameliorate ADR nephrotoxicity via the restoration of mitochondrial function and attenuation of ROS in the present study. ADR-treated rats with both ACE and renin inhibitors were observed to have higher levels of MMP, slightly depletion of mitochondrial ATP levels, and less oxidative stress as compared to animals’ treatment of ADR alone. ACE was mostly able to attenuate the harmful effects of ADR to near control levels in each of these respects, and prevented deterioration in kidney as it has been reported in other studies. In a study, they found that ADR caused to decrease in state 3 respiration, which resulted in decreasing in the mitochondrial respiratory efficiency. The important observation here was that enalapril which is one of ACE inhibitors also prevented the decrease in state 4 and, more importantly, in state 3 oxygen consumption, thereby maintaining mitochondrial bioenergetics at normal levels.Citation38 We, however, found that cellular ATP levels were slightly lowered following ADR treatment, and that the inhibition of Ang-II production by ACE and/or renin inhibitors was slightly able to maintain cellular ATP levels at near control values. Furthermore, the lowered potential of these mitochondria to produce ROS may contribute to the attenuation of dysfunction in rats with ADR-induced nephrotoxicity.
Angiotensin II is also able to be produced via alternative pathways which are independent of ACE activity. Chymase which is a tissue-specific enzyme originally discovered in mast cells is the most important alternative pathway. Its activity has been well known to convert angiotensin I to angiotensin II. Although the importance of these alternative pathways is still controversial, some researchers have suggested that chymase is the major route of angiotensin II synthesis in tissues and may be more important in the pathophysiologic condition. Thus, ACE inhibitor treatment may only partially inhibit angiotensin II production.Citation39 Therefore, other potent drugs inhibiting to the RAS was used that direct renin inhibitor was called aliskiren in our current study. Since the rate-limiting step in angiotensin II production reported the renin, it has been argued that direct renin inhibitors are more efficient RAS inhibitors than ACE inhibitors or angiotensin receptor blockers.Citation40 We found that the treatment of AL (AL and CAP + AL groups) caused some important recovery in kidney mitochondria function including MMP, lightly ATP production due to decreasing mitochondrial and cytosolic TOS. So far, there only a few studies investigating about the effect of aliskiren on ADR-induced nephrotoxicity have been published.Citation19,Citation30,Citation41 They reported that plasma renin activity increased in rats with ADR-induced nephrotoxicity. They also found that ADR-induced nephrotoxicity was manifested by elevation in the plasma levels of BUN and creatinine. However, pretreatment with AL caused a significant reduction in creatinine, BUN and MDA levels, and a significant rise in some antioxidant such as GSH, CAT, and SOD levels which reflects the possible antioxidant effect of AL in rats with ADR-induced nephrotoxicity.Citation19,Citation30,Citation41 We could not find rigorous effect on CK, TAS, and TOS in AL groups (AL and CAP + AL). These discrepancies may be explained by some reasons. One of these reasons was that they found this beneficial AL effect against ADR-induced nephrotoxicity when AL was treated with 100 mg/kg in a day for 42 days and in a day for 14 days, but not treatment with 50 mg/kg in a day for 42 days. AL was treated as a daily dose of 50 mg/kg in our current study. Another reason for these discrepancies was different treatment periods in these two studies. Lastly, it should be recognized that renin is a species-specific enzyme that human renin inhibitors are able to inhibit weakly in the rodent enzymes.Citation42 There has also been limited pre-clinical evaluation of Aliskren in animal models.Citation43 So, AL might fail to completely inhibit Ang-II production.
Conclusion
ADR treatment might activate the renin angiotensin system. Our new data demonstrate that Adriamycin seems to be the Ang-II that plays a key role in the process of ADR-induced nephrotoxicity. Angiotensin-converting enzyme and renin inhibitors exert a protective role towards ADR-induced nephrotoxicity. The mechanism underlying the beneficial effects of Ang-II inhibition in the ADR nephrotoxicity is not only due to decreasing oxidative stress, but also protecting mitochondrial membrane potential and improving kidney mitochondria energy production. It is well-known that mitochondria play an important role in the pathogenesis of ADR-induced toxicity. Prevention of mitochondria dysfunction will prevent kidney dysfunction. Our results suggest that Captopril and Aliskren have protective effects on ADR-induced cardiotoxicity and may be an available agent to protect the kidney in ADR chemotherapy.
Declaration of interest
This study was supported financially by the Research Foundation of Erciyes University.
The authors have no conflicts of interest to disclose.
Acknowledgments
The authors wish to thank Assoc. Prof. Dr. Mukerrem Betul Yerer Aycan for helping to use a fluorescent plate reader (Biotek, Synergy HT).
References
- Berthiaume JM, Wallace KB. Adriamycin-induced oxidative mitochondrial cardiotoxicity. Cell Biol Toxicol. 2007;23(1):15–25
- Malarkodi KP, Balachandar AV, Varalakshmi P. Protective effect of lipoic acid on Adriamycin induced lipid peroxidation in rat kidney. Mol Cell Biochem. 2003;247(1–2):9–13
- Malarkodi KP, Balachandar AV, Varlakshmi P. The influence of lipoic acid on Adriamycin-induced hyperlipidemic nephrotoxicity in rats. Mol Cell Biochem. 2003;247(1–2):139–145
- Ajith TA, Aswathy MS, Hema U. Protective effect of Zingiber officinale roscoe against anticancer drug doxorubicin-induced acute nephrotoxicity. Food Chem Toxicol. 2008;46(9):3178–3181
- Dursun N, Taskin E, Ozturk F. Protection against Adriamycin-induced cardiomyopathy by carnosine in rats: role of endogenous antioxidants. Biol Trace Elem Res. 2011;143(1):412–424
- Dursun N, Taskin E, Yerer Aycan MB, Sahin L. Selenium-mediated cardioprotection against Adriamycin-induced mitochondrial damage. Drug Chem Toxicol. 2011;34(2):199–207
- Hou XW, Jiang Y, Wang LF, et al. Protective role of granulocyte colony-stimulating factor against Adriamycin induced cardiac, renal and hepatic toxicities. Toxicol Lett. 2009;187(1):40–44
- Ozdogan K, Taskin E, Dursun N. Protective effect of carnosine on Adriamycin-induced oxidative heart damage in rats. AKD Anatolian J Cardiol. 2011;11(1):3–10
- Taskin E, Dursun N. The protection of selenium on Adriamycin-induced mitochondrial damage in rat. Biol Trace Elem Res. 2012;147(1–3):165–171
- Harake D, Franco VI, Henkel JM, Miller TL, Lipshultz SE. Cardiotoxicity in childhood cancer survivors: strategies for prevention and management. Future Cardiol. 2012;8(4):647–670
- Konorev EA, Kennedy MC, Kalyanaraman B. Cell-permeable superoxide dismutase and glutathione peroxidase mimetics afford superior protection against doxorubicin-induced cardiotoxicity: the role of reactive oxygen and nitrogen intermediates. Arch Biochem Biophys. 1999;368(2):421–428
- Chandran K, Aggarwal D, Migrino RQ, et al. Doxorubicin inactivates myocardial cytochrome c oxidase in rats: cardioprotection by Mito-Q. Biophys J. 2009;96(4):1388–1398
- Wallace KB, Starkov AA. Mitochondrial targets of drug toxicity. Annu Rev Pharmacol Toxicol. 2000;40:353–388
- Morin D, Barthelemy S, Zini R, Labidalle S, Tillement JP. Curcumin induces the mitochondrial permeability transition pore mediated by membrane protein thiol oxidation. FEBS Lett. 2001;495(1–2):131–136
- Mimnaugh EG, Trush MA, Bhatnagar M, Gram TE. Enhancement of reactive oxygen-dependent mitochondrial membrane lipid peroxidation by the anticancer drug Adriamycin. Biochem Pharmacol. 1985;34(6):847–856
- Ji LL, Mitchell EW. Effects of Adriamycin on heart mitochondrial function in rested and exercised rats. Biochem Pharmacol. 1994;47(5):877–885
- Ji Z, Huang C, Liang C, Chen B, Chen S, Sun W. Protective effects of blocking renin-angiotensin system on the progression of renal injury in glomerulosclerosis. Cell Mol Immunol. 2005;2(2):150–154
- Ibrahim MA, Ashour OM, Ibrahim YF, El-Bitar HI, Gomaa W, Abdel-Rahim SR. Angiotensin-converting enzyme inhibition and angiotensin AT(1)-receptor antagonism equally improve doxorubicin-induced cardiotoxicity and nephrotoxicity. Pharmacol Res. 2009;60(5):373–381
- Rashikh A, Pillai KK, Ahmad SJ, Akhtar M, Najmi AK. Aliskiren alleviates doxorubicin-induced nephrotoxicity by inhibiting oxidative stress and podocyte injury. J Renin Angiotensin Aldosterone Syst. 2013;14(1):14–22
- Han P, Sun H, Xu Y, et al. Lisinopril protects against the adriamycin nephropathy and reverses the renalase reduction: potential role of renalase in adriamycin nephropathy. Kidney Blood Press Res. 2013;37(4–5):295–304
- Arozal W, Watanabe K, Veeraveedu PT, et al. Effect of telmisartan in limiting the cardiotoxic effect of daunorubicin in rats. J Pharm Pharmacol. 2010;62(12):1776–1783
- Nakamae H, Tsumura K, Terada Y, et al. Notable effects of angiotensin II receptor blocker, valsartan, on acute cardiotoxic changes after standard chemotherapy with cyclophosphamide, doxorubicin, vincristine, and prednisolone. Cancer. 2005;104(11):2492–2498
- Tacar O, Sriamornsak P, Dass CR. Doxorubicin: an update on anticancer molecular action, toxicity and novel drug delivery systems. J Pharm Pharmacol. 2013;65(2):157–170
- de Cavanagh EM, Inserra F, Ferder M, Ferder L. From mitochondria to disease: role of the renin-angiotensin system. Am J Nephrol. 2007;27(6):545–553
- Octavia Y, Tocchetti CG, Gabrielson KL, Janssens S, Crijns HJ, Moens AL. Doxorubicin-induced cardiomyopathy: from molecular mechanisms to therapeutic strategies. J Mol Cell Cardiol. 2012;52(6):1213–1225
- Okasora T, Takikawa T, Utsunomiya Y, et al. Suppressive effect of superoxide dismutase on Adriamycin nephropathy. Nephron. 1992;60(2):199–203
- Bertani T, Cutillo F, Zoja C, Broggini M, Remuzzi G. Tubulo-interstitial lesions mediate renal damage in Adriamycin glomerulopathy. Kidney Int. 1986;30(4):488–496
- Mimnaugh EG, Trush MA, Gram TE. A possible role for membrane lipid peroxidation in anthracycline nephrotoxicity. Biochem Pharmacol. 1986;35(23):4327–4335
- Mansour MA, El-Kashef HA, Al-Shabanah OA. Effect of captopril on doxorubicin-induced nephrotoxicity in normal rats. Pharmacol Res. 1999;39(3):233–237
- Rashikh A, Abul Kalam N, Akhtar M, Mahmood D, Pillai KK, Ahmad SJ. Protective effects of aliskiren in doxorubicin-induced acute cardiomyopathy in rats. Human Exp Toxicol. 2011;30(2):102–109
- Boucek RJ Jr, Steele A, Miracle A, Atkinson J. Effects of angiotensin-converting enzyme inhibitor on delayed-onset doxorubicin-induced cardiotoxicity. Cardiovasc Toxicol. 2003;3(4):319–329
- Giri SN, Al-Bayati MA, Du X, Schelegle E, Mohr FC, Margolin SB. Amelioration of doxorubicin-induced cardiac and renal toxicity by pirfenidone in rats. Cancer Chemother Pharmacol. 2004;53(2):141–150
- Yamanaka N, Kato T, Nishida K, Fujikawa T, Fukushima M, Ota K. Elevation of serum lipid peroxide level associated with doxorubicin toxicity and its amelioration by [dl]-alpha-tocopheryl acetate or coenzyme Q10 in mouse (doxorubicin, toxicity, lipid peroxide, tocopherol, coenzyme Q10). Cancer Chemother Pharmacol. 1979;3(4):223–227
- Wang YM, Madanat FF, Kimball JC, et al. Effect of vitamin E against Adriamycin-induced toxicity in rabbits. Cancer Res. 1980;40(4):1022–1027
- Pandir D, Kara O. Cisplatin-induced kidney damage and the protective effect of bilberry (Vaccinium myrtillus L.): an experimental study. Turkish J Med Sci. 2013;43(6):951–956
- Munoz M, Rincon J, Pedreanez A, Viera N, Hernandez-Fonseca JP, Mosquera J. Proinflammatory role of angiotensin II in a rat nephrosis model induced by Adriamycin. J Renin Angiotensin Aldosterone Syst. 2011;12(4):404–412
- Ulu N, Buikema H, van Gilst WH, Navis G. Vascular dysfunction in Adriamycin nephrosis: different effects of Adriamycin exposure and nephrosis. Nephrol Dial Transplant. 2008;23(6):1854–1860
- Hiona A, Lee AS, Nagendran J, et al. Pretreatment with angiotensin-converting enzyme inhibitor improves doxorubicin-induced cardiomyopathy via preservation of mitochondrial function. J Thorac Cardiovasc Surg. 2011;142(2):396–403 e393
- Dagenais NJ, Jamali F. Protective effects of angiotensin II interruption: evidence for antiinflammatory actions. Pharmacotherapy. 2005;25(9):1213–1229
- Westermann D, Riad A, Lettau O, et al. Renin inhibition improves cardiac function and remodeling after myocardial infarction independent of blood pressure. Hypertension. 2008;52(6):1068–1075
- Rashikh A, Pillai KK, Najmi AK. Protective effect of a direct renin inhibitor in acute murine model of cardiotoxcity and nephrotoxicity. Fundam Clin Pharmacol. 2013. [Epub ahead of print]. doi: 10.1111/fcp.12054
- St-Jacques R, Toulmond S, Auger A, et al. Characterization of a stable, hypertensive rat model suitable for the consecutive evaluation of human renin inhibitors. JRAAS. 2011;12(3):133–145
- Muller DN, Derer W, Dechend R. Aliskiren – mode of action and preclinical data. J Mol Med (Berl). 2008;86(6):659–662