
Abstract
The objective of this study is to identify ATP6V1B1, ATP6V0A4 and SLC4A1 genes mutations and assess audiologic characteristics in six Chinese children with primary distal renal tubular acidosis from four unrelated families between the ages of 2 and 13 years. Both ATP6V0A4 and ATP6V1B1 genes were preferentially screened in all index cases by direct sequence analysis. If inconclusive then SLC4A1 gene should be analyzed for mutation. Their clinical features, hearing status and inner ear imaging structure were also investigated. Six loss-of-function mutations were identified in six patients. Two novel mutations were identified in either of ATP6V0A4 and ATP6V1B1 genes, respectively. Two probands from different kindreds with mutations in ATP6V1B1 presented early onset profound sensorineural hearing loss (SNHL) and enlarged vestibular aqueduct (EVA). Two from different families carrying ATP6V0A4 mutations manifested early onset moderate mixed HL and moderate SNHL, respectively, the former comorbid with EVA, while the latter not; however, both their elder sisters showed normal hearing and inner ear. These findings expand the spectrum of mutations in the ATP6V0A4 and ATP6V1B1 genes associated with primary dRTA. Our study confirms the association of EVA and mutations in either of these two genes. More studies are necessary to clarify the relationship between dRTA, SNHL, EVA, and gene mutations.
Introduction
Distal renal tubular acidosis (dRTA) results from the impaired secretion of hydrogen ions from the distal nephron, causing metabolic acidosis, often with hypokalemia, nephrocalcinosis, and/or nephrolithiasis.Citation1–3
Both autosomal dominant (AD) and autosomal recessive (AR) inheritance patterns have been reported in primary dRTA. Mutations in the ATP6V1B1 and ATP6V0A4 genes, encoding subunits B1 and a4 of apical H+-ATPase, cause recessive forms of dRTA.Citation1,Citation4–6 ATP6V1B1 mutations have been associated with early sensorineural hearing loss (SNHL, OMIM 267300), whereas ATP6V0A4 mutations are classically associated with either late-onset SNHL or normal hearing (OMIM 602722). Enlarged vestibular aqueduct (EVA) was described in patients with recessive dRTA and SNHL, and recently, this abnormality has been associated with mutations in the ATP6V1B1 gene.Citation7 However, there is more and more evidence that these two recessive forms of dRTA are genetically more heterogeneous in the auditory phenotype than previously assumed. A few cases harbored ATP6V1B1 mutations without SNHL have been described.Citation8,Citation9 Recent genetic analyses have revealed that some individuals with mutations in the ATP6V0A4 gene also have early-onset severe SNHL.Citation9 And most recently, Andreucci et al. described a child carrying ATP6V0A4 mutations with early profound SNHL and unexpected EVA.Citation10 Furthermore, this auditory phenotype of severe hearing impairment and enlarged endolymphatic compartments was verified by an Atp6v0a4 knockout mouse model.Citation11 Mutations in the SLC4A1 gene encoding the human AE1 have been associated with either AD or AR dRTA.Citation12,Citation13 Additionally, the presence of SNHL in a patient who showed no mutation in ATP6V1B1 and ATP6V0A4 genes do not exclude a priori the possibility of a form of dRTA related to a mutation in SLC4A1 gene, as the development of SNHL may be the consequence of many different causes, such as other genetic factors, ototoxic agents, neonatal distress and electrolyte disequilibrium. Therefore, lack of definite phenotype-genotype correlation asks for the need of genetic analysis in dRTA patients.
Herein, we performed mutation analysis in six Chinese patients from four unrelated kindreds with dRTA. On the other hand, their audiologic phenotype and genotype correlations were also investigated.
Materials and methods
Patients
We studied six children (three females and three males) with dRTA belonging to four unrelated families; their clinical features and representative biochemical data are shown in . Diagnosis was based on the presence of the following necessary criteria: Metabolic acidosis with a normal anion gap and inappropriate alkaline urine (pH > 6.0) in a context of acidosis (CO2CP < 18 mmol/L); normal fractional excretion rate of bicarbonate (FEHCO3, normal value < 5%), the FEHCO3 is calculated after adequate alkalization; and/or nephrocalcinosis. Additional optional criteria were hypercalciuria, hypokalemia, polyuria, and failure to thrive. Audiological data and inner ear radiologic finding of all patients were collected. Hearing was assessed by pure-tone audiometry (PTA) and/or automated auditory brainstem response (AABR) test. The severity of hearing loss was ranked as mild (20–40 dB), moderate (41–55 dB), moderately severe (56–70 dB), severe (71–90 dB) and profound (91 dB or greater). In addition, medical records were reviewed to document the results of hearing screening tests that children may have received. Screening devices, test parameters, and test results are listed in . The parents of the patients in families II and III were consanguineous. Patients III-1 and III-2, and patients IV-1 and IV-2 were siblings. The study protocol was approved by the Ethics Committee on Human Studies at the affiliated hospital of medical college Qingdao University. Informed consent was obtained from each patient and his or her parents.
Table 1. Clinical characteristic at diagnosis of six children with distal renal tubular acidosis.
Table 2. Hearing status and inner ear radiologic characteristics of these children with distal renal tubular acidosis.
Mutation analysis
Both ATP6V0A4 and ATP6V1B1 genes were preferentially screened in all index cases, if inconclusive (no mutation or only one was identified in either gene) then SLC4A1 gene should be analyzed for mutation. Genomic DNA was extracted from peripheral blood of these six patients, their family members, and 100 normal healthy controls by the GenElute blood genomic DNA kit (Sigma, NA2010, St. Louis, MO). Twenty pairs of oligonucleotide primers were generated to amplify all coding exons (from exon 3 to 22) and flanking intronic regions of the ATP6V0A4 gene (Supplemental ). The coding region and adjacent intronic segments of the ATP6V1B1 and SLC4A1 genes were amplified using of previously described primer pairs, respectively.Citation2,Citation14 PCRs were performed in 50 μL of solution containing 0.2 mM dNTP, 0.03 U/μL Taq polymerase (Takara EX Taq Hot start version, DRR006B, Osaka, Japan), 2.0 mM MgCl2, 2.5 μL 10 × PCR Mg2+-free buffer (Takara), approximately 50 ng genomic DNA and 1 mM of each primer. PCR was performed with an initial denaturation step at 95 °C for 5 min, subsequently followed by 33 cycles with denaturation at 95 °C for 45 s, annealing at 52–66 °C for 45 s and elongation at 72 °C for 45 s. PCR samples were subjected to bidirectional sequencing. The sequence reactions were run on an ABI Prism 3730 DNA Analyzer (Applied Biosystems, Carlsbad, CA).
Results
Clinical findings
A total of six children from four unrelated kindreds were enrolled in this study. summarizes the clinical and biochemical manifestations leading to the diagnosis of dRTA in each patient. As shown in , all children showed nephrocalcinosis confirmed by abdominal ultrasonography, however, only two of six presented hypercalciuria. In addition, two of six children had hypercalcemia (patient I-1 and IV-1). Of both with hypercalcemia, hyperparathyroidism and vitamin D intoxication were excluded because of normal values of intact parathyroid hormone, 1,25(OH)-vitamin D and 25-OH-vitamin D. The calcium level of patient IV-1 normalized in the first 72 h with correction of metabolic acidosis and dehydration, whereas this similar phenomenon did not occur in patient I-1 who kept a mild elevated calcium level for nearly 1 year.
Alkali therapy and potassium supplement (sodium citrate and potassium citrate) was effective for metabolic acidosis, hypokalemia and delayed growth in all patients, with this therapy improving their growth to almost normal range.
Mutation analysis
Three variants including two novel mutations in the ATP6V1B1 gene were identified in two of the four kindreds (, ). Patient I-1 was a compound heterozygote with a recurrent duplication of c.1155dupC (p.Ile386Hisfs*56)Citation1,Citation3,Citation5,Citation9 and a novel deletion of c.1356delT. This novel deletion led to a frameshift that resulted in premature termination of the protein at codon 486 (p.Phe452Leufs*35). Patient II-1 showed homozygosity for a novel splicing variant of c.786-1G > C (loss of splice acceptor site of intron 8).
Figure 1. Novel mutations identified in ATP6V1B1 and ATP6V0A4 of patients with distal renal tubular acidosis. The mutant nucleotides are marked with asterisks.
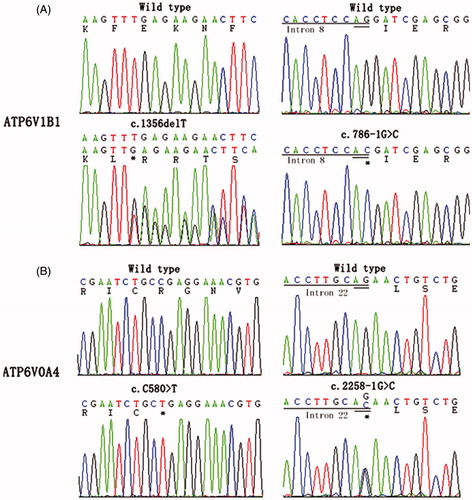
Table 3. Detected mutations in ATP6V1B1 and ATP6V0A4 of the six children with distal renal tubular acidosis.
We also identified three distinct mutations including two novel aberrations in ATP6V0A4 gene in the other two families (, ). A novel homozygous nonsense variant c.C580 > T (p.R194*) was revealed in both the sister III-1 and the brother III-2. While a novel splicing variant c.2258-1 G > C (loss of splice acceptor site of intron 21) and a previously reported mutation c.2137delG (p.Glu713fs*53) were identified in the sister IV-1 and the brother IV-2,Citation9 both parents were heterozygous for the two mutations. Direct sequencing analysis failed to find above-mentioned mutations in 100 unrelated healthy subjects.
We did not perform the SLC4A1 gene analysis since two definite variants were found in each patient.
Audiological phenotype and genotype correlation
presented the hearing status of all patients. Following the mutation testing, both patient I-1 and II-1 who harbored ATP6V1B1 gene mutations underwent a full confirming test of hearing status at the age of 9 months and 7 months, respectively. No ABR and no emissions could be elicited in both ears of them, and auditory multiple-frequency steady state evoked response (ASSR) test showed that both patients had very little residual hearing restricted to the low frequencies in both ears. And the normal Type “A” tympanogram suggested their normal middle ears function. Based on the above findings, both patient I-1 and II-1 was diagnosed with profound SNHL. The cerebral magnetic resonance imaging (MRI) scan performed at the age of 1 year showed dilation of the endolymphatic sac in patient I-1, while the high-resolution computed tomography (CT) of temporal bone carried out at the same age demonstrated EVA in patient II-1. Subsequently, both patients underwent cochlear implantation which helped to bring the achievement of almost normal spoken language development during the follow-up period. Patient III-1 has had no audio graphic evidence of HL to date; her brother who possesses the same mutation in the ATP6V0A4 gene presented with early-onset bilateral moderate HL however, which was confirmed by ABR at age 3 years. High-resolution CT and MRI of his temporal bones showed bilateral EVA with no other inner and middle ear malformations (). Since then, he had begun to use bilateral hearing aids. During the follow-up period, from the age of 5 to the age of 8 years, he experienced four times of attacks of rotatory vertigo, and his hearing decreased gradually with fluctuating exacerbation which was associated with common cold infections. PTA demonstrated that he suffered from bilateral mixed HL characterized by air-bone gaps at lower frequencies, and his right pure-tone averages (PTAs, PTAs is calculated from the audiometric thresholds at the frequencies of 500, 1000, and 2000 Hz) deteriorated from 56.3 dB to 78.3 dB, while his left PTAs worsened from 63.3 dB to 80 dB (). Patients IV-1 and IV-2 are also siblings and harbor the same ATP6V0A4 mutations. However, only the little brother (patient IV-2) manifested with early-onset bilateral moderate SNHL which was verified with PTA at age 5 years (), while his 13-year-old sister (IV-1) has had no audiologic evidence of HL up to now. No abnormal middle and inner ear imaging findings was found by the cerebral MRI scan performed at the age of 7 years in sister and at the age of 5 years in brother, respectively. And the bilateral PTAs of patient IV-2 maintained stable throughout the follow-up period of 6 years.
Figure 2. High-resolution computed tomography (CT) and magnetic resonance imaging (MRI) of the temporal bones in patient III-2. Bilateral enlargement of the vestibular aqueduct and endolymphatic sac (arrows) is evident.
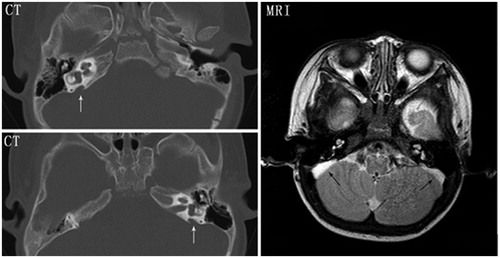
Figure 3. Audiograms of patients III-2 and IV-2. (A) Audiogram of patient III-2 at age of 5 years. (B) Audiogram of patient III-2 at age of 8 years. (C) Audiogram of patient IV-2 at age 5 years. Bilateral air–bone gaps are recognized, especially at lower frequencies in patient III-2. Circle or × indicate unmasked examination of air conduction for the right or left ear, triangle or square indicate masked examination of air conduction for right or left ear, < or > indicate unmasked bone conduction for right or left ear, [ or ] indicate masked bone conduction for right or left ear.
![Figure 3. Audiograms of patients III-2 and IV-2. (A) Audiogram of patient III-2 at age of 5 years. (B) Audiogram of patient III-2 at age of 8 years. (C) Audiogram of patient IV-2 at age 5 years. Bilateral air–bone gaps are recognized, especially at lower frequencies in patient III-2. Circle or × indicate unmasked examination of air conduction for the right or left ear, triangle or square indicate masked examination of air conduction for right or left ear, < or > indicate unmasked bone conduction for right or left ear, [ or ] indicate masked bone conduction for right or left ear.](/cms/asset/a956e013-ecf2-4d56-9567-52e4f799c828/irnf_a_930332_f0003_b.jpg)
Among the six children, all of them received newborn hearing screening test, five passed hearing screening in both ears, and one passed the screening in only one ear (patient II-1). Among the patients with HL, 50% of patients (1/2) with ATP6V1B1 mutations and 100% of patients (2/2) with ATP6V0A4 variants passed the screening test in both ears at birth.
Discussion
We have identified three ATP6V1B1 mutations (one duplication, one deletion, and one splicing mutation) and three ATP6V0A4 mutations (one deletion, one nonsense, and one splicing mutation) in patients with primary dRTA. All of the six above mentioned mutations may result in translational frame shifting, truncated proteins, or unstable mRNA, causing the abnormal function of H+-ATPase and dRTA. These findings expand the spectrum of mutations in the ATP6V0A4 and ATP6V1B1 genes associated with primary dRTA, especially in Chinese populations. It is worth noting that the mutation p.Ile386Hisfs*56 observed in one of our four kindreds has been previously described in 21 families, of which 16 are from North Africa. Our data may provide more evidence and information to confirm that p.Ile386Hisfs*56 is a frequent or potential “hotspot” mutation.Citation1,Citation3,Citation5,Citation9 However, we could not exclude the possibility of a founder effect for this mutation because it might have transferred to China by population migration via Silk Road from North Africa and West Asia more than 1000 years ago. This study is the first report of ATP6V1B1 mutations in Chinese.
Hypercalciuria is a characteristic biochemical finding in patients with dRTA, however, four of six patients were normocalciuric at the time of diagnosis in this study. In fact, normocalciuria is not uncommon at diagnosis in young dRTA patients (<1 year).Citation4,Citation14,Citation15 As we all know, many factors can contribute to the differences in urinary calcium excretion, such as dietary, calcium absorption and hormonal control, as well as renal handling of calcium. Three (patient I-1, II-1 and IV-1) of the four patients with normocalciuria had evident signs of dehydration resulted by vomiting, fever, or other causes at diagnosis in this study. Extracellular volume contraction is a well-known stimulus for para-cellular calcium reabsorption in the proximal tubules. Therefore, volume contraction may be an important cause for normocalciuria in them. In addition, dietary sodium is an important modulator of urinary calcium excretion and a close correlation exists between urinary sodium and urinary calcium excretion. Low salt intake is associated with decreased urinary calcium excretion.Citation15,Citation16 All these children were diagnosed before the age of 6 months. As we know, breast milk or infant formula, which has relatively low sodium content, is the principal source of calories for them at this stage.Citation15 Therefore, we suppose that low dietary salt intake may help to explain their normal urinary calcium excretion. Furthermore, dietary sodium restriction may aggravate volume depletion and augment hypocalciuria.
Undoubtedly, hypercalciuria is a major contributor to nephrocalcinosis in dRTA. It may seem paradoxical that nephrocalcinosis was confirmed in all of our four children with normocalciuria at diagnosis. This may demonstrate that nephrocalcinosis can occur in patients with dRTA without hypercalciuria. However, we would rather believe that normocalciuria may merely stand for a status of patients with dRTA in a dynamic changing process. Tsai HY et al. proposed a dynamic mode of calcium excretion in patients with dRTA which well explained this ambivalence.Citation15 At the early stage, metabolic acidosis promotes bone demineralization and hypercalciuria.Citation17 Hypercalciuria, persistently high urine pH and profound hypocitraturia predispose the development of the nephrocalcinosis. With time, polyuria, renal sodium wasting and poor intake result in volume depletion, especially in young babies with low salt intake. Then volume contraction, low salt content in infant formula, and alkaline urine lead to the increased calcium reabsorption and normocalciuria.
Nevertheless, it should not be neglected that ethnicity and genetic polymorphism may be another determinant factor in the difference of urinary calcium excretion.Citation18 It is well known that the normal spot urine calcium/creatinine (UCa/Cr) ratio has a significantly negative correlation with age. Sargent's group proposed the 95th percentile of normal reference values for UCa/Cr was 0.86 mg/mg for Caucasian infants less than 7 moths.Citation19 However, subsequent investigations have shown that the UCa/Cr may vary with ethnicity, geographic area and genetic polymorphism.Citation18 There is evidence that the UCa/Cr ratio in Chinese adolescents is low when compared with published values for Caucasian children.Citation20 Regrettably, there are no accurate reference values of UCa/Cr for Chinese infants. So the possibility of that hypercalciuria was inappropriately labeled as normocalciuria could not be completely excluded.
We found that two (patient I-1 and IV-1) of six patients presented with hypercalcemia. It is known that hypercalcemia is associated with dRTA not only in neonates but also in other ages.Citation21 A combination of increased calcium reabsorption at proximal tubules due to a decrease in glomerular filtration rate (GFR) secondary to severe dehydration, systemic acidosis causing calcium chelation from bones have been postulated as possible mechanisms for hypercalcemia associated with dRTA.Citation22 The fact that the abnormality of hypercalcemia was rapidly corrected with the rectification of systemic metabolic acidosis and dehydration in patient IV-1 may add weight to this hypothesis. However, even after acid–base disturbance and volume depletion was corrected, the patient I-1 presented sustained hypercalcemia in the absence of excessive vitamin D therapy and primary hyperparathyroidism. The hypercalcemia can be explained by a disequilibrium in which the intake of calcium exceeds the internal buffering capacity of the bone and the renal capacity to excrete calcium. However, further studies will be required to better define the mechanisms and possible etiological factors.
In this study, the association of EVA with ATP6V0A4 was observed for the second time, which in conjunction with the evidence from Kcc4−/− mice confirmed such connection. However, the early onset HL is not always strictly related to EVA in patients with ATP6V0A4 mutations, as can be observed in our patients. The exact pathogenic mechanism of EVA remain unclear, a recent study demonstrated that the lack of endocochlear potential and disruption of endolymph pH homeostasis due to the dysfunction of H+-ATPases which expressing on the endolymphatic sac epithelium may play an important role.Citation11 EVA, as an anomaly of the structures of inner ear, which might lead to SNHL or mixed HL, and manifest with normal hearing, congenital total deafness, progressive HL, or fluctuating HL. In addition, air-bone gap in the audiogram, especially at the lower frequencies, is one of the predominant features of EVA. In this investigation, there is no noticeable difference can be seen between the characteristics of EVA in our primary dRTA patients and that of other common causes of EVA. The loss of function of H+-ATPases in the inner ear is undoubtedly the basic reason of EVA and HL; however, EVA may play a modificatory role to the manifestation of HL. More studies are necessary to clarify the relationships between dRTA, SNHL, EVA, and gene mutations.
It is not clear when hearing loss due to an ATP6V1B1 or ATP6V0A4 mutation begins and how much residual hearing remains at birth, because the level of hearing in these patients at birth has not been studied. We found that one out of two HL patients with ATP6V1B1 mutations and both HL patients with ATP6V0A4 variants passed the screening test in both ears at birth. These data suggest that many patients with ATP6V1V1 or ATP6V0A4 mutations have better hearing than the UNHS threshold (∼35 dB) at birth. This result is similar to the investigation on newborns with PDS mutations.Citation23 As to the hereditary HL, the appropriate early intervention service is important to the development of speech and language, especially pre-lingual deafness. We think that it may be wise to implement repetitive screenings at preverbal and preschool ages in children with primary dRTA. However, in the light of our small sample size, investigations based on more subjects are needed to verify our conclusions.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the article. This work was supported by a grant from the National Natural Scientific Foundation (81170653), a Shandong province Science & Technology Development Plan (BS2010YY011), as well as Qingdao Science & Technology Development Plans (12-1-4-2-(21)-jch &13-1-3-40-nsh).
Supplemental material available online
Supplemental Table 1
Supplementary Material
Download PDF (17.8 KB)Acknowledgements
We are grateful to the family members for their participation in this study.
References
- Karet FE, Finberg KE, Nelson RD, et al. Mutations in the gene encoding B1 subunit of H+-ATPase cause renal tubular acidosis with sensorineural deafness. Nat Genet. 1999;21:84–90
- Shao L, Xu Y, Dong Q, Lang Y, Yue S, Miao Z. A novel SLC4A1 variant in an autosomal dominant distal renal tubular acidosis family with a severe phenotype. Endocrine. 2010;37:473–478
- Elhayek D, Perez de Nanclares G, Chouchane S, et al. Molecular diagnosis of distal renal tubular acidosis in Tunisian patients: Proposed algorithm for Northern Africa populations for the ATP6V1B1, ATP6V0A4 and SCL4A1 genes. BMC Med Genet. 2013;14:119
- Smith AN, Skaug J, Choate KA, et al. Mutations in ATP6N1B, encoding a new kidney vacuolar proton pump 116-kD subunit, cause recessive distal renal tubular acidosis with preserved hearing. Nat Genet. 2000;26:71–75
- Stover EH, Borthwick KJ, Bavalia C, et al. Novel ATP6V1B1 and ATP6V0A4 mutations in autosomal recessive distal renal tubular acidosis with new evidence for hearing loss. J Med Genet. 2002;39:796–803
- Subasioglu Uzak A, Cakar N, Comak E, Yalcinkaya F, Tekin M. ATP6V1B1 mutations in distal renal tubular acidosis and sensorineural hearing loss: Clinical and genetic spectrum of five families. Ren Fail. 2013;35:1281–1284
- Joshua B, Kaplan DM, Raveh E, et al. Audiometric and imaging characteristics of distal renal tubular acidosis and deafness. J Laryngol Otol. 2008;122:193–198
- Miura K, Sekine T, Takahashi K, et al. Mutational analyses of the ATP6V1B1 and ATP6V0A4 genes in patients with primary distal renal tubular acidosis. Nephrol Dial Transplant. 2013;28:2123–2130
- Vargas-Poussou R, Houillier P, Le Pottier N, et al. Genetic investigation of autosomal recessive distal renal tubular acidosis: Evidence for early sensorineural hearing loss associated with mutations in the ATP6V0A4 gene. J Am Soc Nephrol. 2006;17:1437–1443
- Andreucci E, Bianchi B, Carboni I, et al. Inner ear abnormalities in four patients with dRTA and SNHL: Clinical and genetic heterogeneity. Pediatr Nephrol. 2009;24:2147–2153
- Lorente-Cánovas B, Ingham N, Norgett EE, Golder ZJ, Karet Frankl FE, Steel KP. Mice deficient in H+-ATPase a4 subunit have severe hearing impairment associated with enlarged endolymphatic compartments within the inner ear. Dis Model Mech. 2013;6:434–442
- Batlle D, Haque SK. Genetic causes and mechanisms of distal renal tubular acidosis. Nephrol Dial Transplant. 2012;27:3691–3704
- Sinha R, Agarwal I, Bawazir WM, Bruce LJ. Distal renal tubular acidosis with hereditary spherocytosis. Indian Pediatr. 2013;50:693–695
- Gil H, Santos F, García E, et al. Distal RTA with nerve deafness: Clinical spectrum and mutational analysis in five children. Pediatr Nephrol. 2007;22:825–828
- Tsai HY, Lin SH, Lin CC, Huang FY, Lee MD, Tsai JD. Why is hypercalciuria absent at diagnosis in some children with ATP6V1B1 mutation? Pediatr Nephrol. 2011;26:1903–1907
- Breslau NA, McGuire JL, Zerwekh JE, Pak CY. The role of dietary sodium on renal excretion and intestinal absorption of calcium and on vitamin D metabolism. J Clin Endocrinol Metab. 1982;55:369–373
- Houillier P, Normand M, Froissart M, Blanchard A, Jungers P, Paillard M. Calciuric response to an acute acid load in healthy subjects and hypercalciuric calcium stone formers. Kidney Int. 1996;50:987–997
- So NP, Osorio AV, Simon SD, Alon US. Normal urinary calcium/creatinine ratios in African-American and Caucasian children. Pediatr Nephrol. 2001;16:133–139
- Sargent JD, Ta S, Kresel J, Klein RZ. Normal values for random urinary calcium to creatinine ratios in infancy. J Pediatr. 1993;123:393–397
- Wong GW, Lam CW, Kwok MY, Mak TW. Urinary calcium excretion in Chinese adolescents. J Pediatr Child Health. 1998;34:226–228
- Pela I, Seracini D, Lavoratti G, Materassi M. Hypercalcemia and distal renal tubular acidosis: An association not only in the newborn. Pediatr Nephrol. 2003;18:850
- Eshraghi P, Karamifar H, Derakhshan A. Association of distal renal tubular acidosis with hypercalcemia. Iran J Pediatr. 2008;18:369–372
- Kim BG, Shin JW, Park HJ, Kim JM, Kim UK, Choi JY. Limitations of hearing screening in newborns with PDS mutations. Int J Pediatr Otorhinolaryngol. 2013;77:833–837