
Abstract
Renal tubular epithelial cell injury is a major pathological event that contributes to the development of diabetic kidney disease (DKD). Uncoupling protein-2 (UCP2), a mitochondrial membrane protein, has been reported to participate in the regulation of reactive oxygen species (ROS) generation, which contributes to tubular cell apoptosis induced by hyperglycemia. In this study, we found that genipin, a UCP2 inhibitor, dramatically boosted oxidative stress, attenuated antioxidative capacity, and exacerbated cell apoptosis accompanied with caspase-3 activation in rat renal proximal tubular cells (NRK-52E) incubated with high glucose. The present study results suggest that manipulation of UCP2 could be important in the prevention of oxidative stress damage in renal tubular epithelial cells induced by hyperglycemia in vitro.
Introduction
Diabetic kidney disease (DKD) is the leading cause of end-stage renal disease (ESRD) in the world. In the United States, DKD accounted for 44% of new ESRD cases in 2007.Citation1 A recently published cross-sectional survey in China has also shown that diabetes is an independent risk factor of kidney damage.Citation2 Vascular and glomerular injuries are considered the main pathological features of DKD; however, tubular atrophy is also a common event observed in the disease.Citation3 Signs of tubular dysfunction, including elevated levels of beta 2-microglobulin in urine and renal tubular acidosis, can be detected early in diabetic patients.Citation4
It has been suggested that oxidative stress is implicated in the pathogenesis of DKD.Citation5 Hyperglycemia triggers the generation of free radicals in various cells of the kidney, including proximal tubular epithelial cells, and consequently proliferation, extracellular matrix deposition, and apoptosis emerge.Citation6 The mechanisms by which hyperglycemia leads to tubular cell apoptosis are not completely understood. Recently, the mitochondrion has emerged as a major source of reactive ROS and UCP2, a mitochondrial membrane protein, has been reported to participate in the regulation of ROS generation.Citation7,Citation8 UCP2 belongs to the UCP family, which are mitochondrial transporters that are capable of dissipating the proton gradient and increasing thermogenesis while reducing the efficiency of ATP synthesis. Furthermore, several other physiological roles of UCPs have recently become a great concern, including regulation of fatty acid, glucose oxidation, and reduction of mitochondrial ROS generation.Citation9 Several genetic studies have investigated the relationship between UCP2 gene polymorphism and susceptibility for the development of renal damage in humans.Citation10,Citation11 Di Castro et al.Citation12 found that UCP2 is a critical protein to prevent oxidative stress damage in renal mesangial cells in the context of hypertension. However, the role and mechanisms of UCP2 in tubular cell injury caused by hyperglycemia remain unknown.
In the present study, we investigated the function of UCP2 in monitoring ROS generation as well as the subsequent apoptosis of tubular cells induced by hyperglycemia, using an in vitro cell culture system. Based on the identification of reduced UCP2 protein expression induced by genipin, a UCP2 inhibitor, our findings provide evidence that UCP2 may exert a protective effect against oxidative stress during DKD.
Materials and methods
Cell culture and experiment groups
NRK-52E cell, a rat renal proximal tubular cell line, was purchased from Chinese Academy of Sciences Library. The cells were cultured in a humidified atmosphere of 5% CO2 at 37 °C in Dulbecco's modified Eagles medium (DMEM, Gibco, Gran Island, NY), which contained 1000 mg/L glucose supplemented with 10% fetal bovine serum (FBS) (containing 100 U/mL penicillin and 100 U/mL streptomycin). The cells were maintained in serum-free media for 24 h before each treatment. Genipin (Zhixin Bio, Linchuan, China) was added to the cell cultures 30 min prior to changing the medium to a high glucose medium. NRK-52E cells were divided into five groups:
Group NG (normal glucose): cells were incubated with 5.5 mmol/L glucose.
Group MG: cells were incubated with 5.5 mmol/L glucose supplemented with 24.5 mmol/L mannitol, which was served as osmotic control in the experiment.
Group HG (high glucose): cells were incubated with 30 mmol/L glucose.
Group Lg (low genipin): cells were incubated with 30 mmol/L of glucose pretreated with 25 μmol/L genipin.
Group Hg (high genipin): cells were incubated with 30 mmol/L of glucose pretreated with 50 μmol/L genipin.
Measurements of oxidative stress and antioxidant capacity
MDA level
Malondialdehyde (MDA) is a secondary product of lipid peroxidation, which is usually used as the index of oxidative stress in cells. In our study, the MDA contents in the NRK-52E cells were measured with lipid peroxidation (MDA) Assay Kit (Beyotime, Shanghai, China), according to the protocols by the manufacturer. In this kit, lipid peroxidation is determined by the reaction of MDA with thiobarbituric acid (TBA) to form a colorimetric (532 nm)/fluorometric (lex = 532/Lem = 553 nm) product, proportional to the MDA present. The concentration of MDA was expressed as µmol of MDA per mg protein.
SOD activity
The superoxide dismutase (SOD) activity in the NRK-52E cells was measured with SOD Determination Kit (Beyotime, Shanghai, China), according to the protocols by the manufacturer. This kit allows very convenient SOD assaying by utilizing Dojindo's highly water-soluble tetrazolium salt, WST-1 (2-(4-iodophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium, monosodium salt) that produces a water-soluble formazan dye upon reduction with a superoxide anion. The rate of the reduction with O2 is linearly related to the xanthine oxidase (XO) activity and is inhibited by SOD. Therefore, the IC50 (50% inhibition activity of SOD or SOD-like materials) can be determined by a colorimetric method. The enzyme activity was expressed as U/mg protein and 1 unit of enzyme is defined as the enzyme activity that inhibits autoxidation of pyrogallol by 50%.
GPx activity
The glutathione peroxidase (GPx) activity in the NRK-52E cells was measured with Glutathione Peroxidase Cellular Activity Assay Kit (Beyotime, Shanghai, China), according to the protocols by the manufacturer. This kit uses an indirect determination method. It is based on the oxidation of glutathione (GSH) to oxidized glutathione (GSSG) catalyzed by GPx, which is then coupled to the recycling of GSSG back to GSH utilizing glutathione reductase (GR) and -nicotinamide adenine dinucleotide phosphate, reduced (NADPH). The decrease in NADPH absorbance measured at 340 nm during the oxidation of NADPH to NADP+ is indicative of GPx activity, since GPx is the rate limiting factor of the coupled reactions. The activity of GPx in the sample was expressed as U/mg protein.
Cell apoptosis assays
Western blot analysis of caspase-3 activation and flow cytometry were used to assess high glucose-induced apoptotic cell death. Apoptosis was measured using the Annexin V detection kit (Sigma-Aldrich, St. Louis, MO) according to the manufacturer's instructions. Flow cytometric analysis was performed to monitor the green fluorescence of the FITC-conjugated Annexin V (530 ± 30 nm) and the red fluorescence of DNA-bound propidium iodide (PI, 630 ± 22 nm). All data were analyzed with Cell Quest software (BD, Becton Dickinson and Company, Loveton Circle Sparks, MD).
RNA extraction and RT-PCR analysis
The NRK-52E cells were seeded into 6-well plates at a density of 2 × 105 cells/well in 2 mL medium. Total RNA was isolated using TRIzol reagent. Oligo (dT)-primed RNA (1 μg) was reverse transcribed with RevertAid™ M-MuLV reverse transcriptase (Fermentas, Amherst, NY) according to the manufacturer's instructions. The obtained cDNA was used to determine the amount of UCP2 and β-actin mRNA using PCR with Taq DNA polymerase (TAKARA). β-Actin was used as an internal control. The primers that were used for amplification of the UCP2 and β-actin transcripts were as follows: UCP2 forward, 5′-CTC TGG AAA GGG ACC TCT CCCA-3′ and reverse, 5′-AGG CAG AAG TGA AGT GGC AAG G-3′; β-actin forward, 5′-GAA GAT CAA GAT CAT TGC TCC T-3′ and reverse, 5′-TAC TCC TGC TTG CTG ATC CAC A-3′. PCR condition is one cycle of 94 °C for 2 min, 45 cycles of 94 °C for 20 s, 55 °C for 20 s, and 72 °C for 30 s and one cycle of 72 °C for 5 min.
Western blotting
The NRK-52E cells were lysed with mammalian cell lysis buffer containing protease and phosphatase inhibitor cocktails. The lysates were resolved in 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and electro blotted. The polyvinylidene difluoride (PVDF) membranes were blocked with 5% skimmed milk in TBST (20 mM Tris–HCl pH 7.5, 0.5 M NaCl, 0.5% Tween 20) at 37 °C for 1 h and probed with primary antibodies against UCP2 (monoclonal, mouse) or β-actin (polyclonal, rabbit) or caspase-3 (monoclonal, mouse) at 1:1000 dilution overnight at 4 °C. After three washes with PBS containing 0.1% BSA, the cells were incubated with the horseradish peroxidase-conjugated goat anti-mouse IgG for 2 h. Detection was performed using SuperSignal West Pico Chemiluminescent Substrate (Thermo, West Palm Beach, FL).
Statistical analysis
All data are expressed as mean±SD. Statistical analysis of the differences between the experimental groups was performed using a Student's t test. p < 0.05 was considered to be statistically significant.
Results
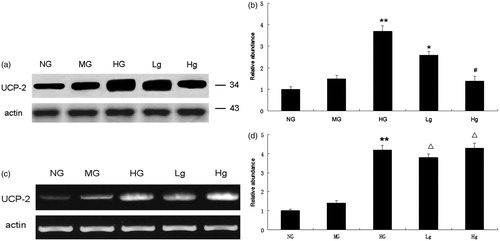
Genipin inhibits UCP2 protein expression induced by high glucose in NRK-52E cells
Both mRNA expression and protein expression of UCP2 were increased in NRK-52E cells after exposure to high glucose for 48 h (p < 0.01). Western blot analysis showed that pretreatment with genipin markedly decreased UCP2 protein expression in a dose-dependent manner (). However, the expression of UCP2 mRNA remained unchanged after genipin administration in RT-PCR analysis (p > 0.05) (). It indicates that genipin down-regulates UCP2 by inhibiting its protein expression.
Figure 1. UCP2 expression in different groups. (a) UCP2 protein expression in western blot analysis. (b) Graphic presentation of relative UCP2 abundance normalized to actin. (c) UCP2 mRNA expression in RT-PCR analysis. (d) Graphic presentation of relative UCP2 mRNA abundance normalized to actin. * p < 0.05 vs. HG; #p < 0.01 vs. HG; △p > 0.05 vs. HG ; **p < 0.01 vs. NG.
Downregulation of UCP2 expression aggravates oxidative stress and reduces antioxidant capacity
shows the MDA levels, SOD, and GPx activities in different groups after the cells were exposed to high glucose for 72 h. The MDA levels were increased significantly in cells incubated with high glucose as compared with the group NG. In addition, antioxidant capacity was diminished by HG. genipin, an inhibitor of UCP2, dramatically boosted MDA levels and reduced SOD and GPx activities as compared with the group HG.
Table 1. MDA levels, SOD, and GPx activities in different groups (mean ± SD).
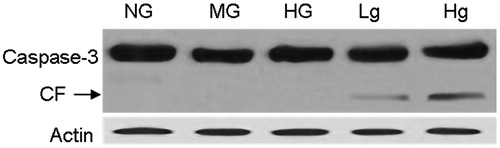
Downregulation of UCP2 expression activates caspase-3
To investigate the role of UCP2 in the genesis of cell injury, we tested the activation of caspase-3 by western blotting. The cells in different groups were lysed to examine caspase-3 cleavage after exposure to high glucose for 72 h. Our data revealed that pretreatment of genipin induced caspase-3 activation, which indicates that downregulation of UCP2 is involved in HG-induced cell apoptosis ().
Figure 2. Downregulation of UCP2 expression in the groups of genipin induced caspase-3 cleavage. CF, cleaved fragments of caspase-3.
Downregulation of UCP2 expression increases cell apoptosis
Cellular apoptosis was assessed by flow cytometry using PI and Annexin V-FITC staining after the cells were exposed to high glucose for 72 h. As shown in , genipin aggravated cell apoptosis induced by high glucose in a dose-dependent manner. Statistical analysis indicated that HG induced significant apoptosis (p < 0.01). Moreover, the preadministration of genipin aggravated the levels of apoptosis compared with the HG group. The apoptosis rates are shown in .
Figure 3. Apoptosis rate. The early apoptotic cells stained by Annexin-V-fluorescein isothiocyanate are located in the lower right quadrant.
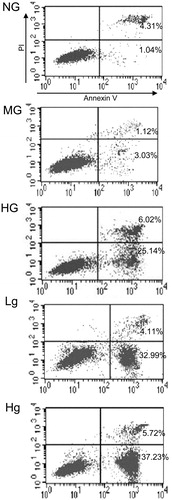
Table 2. Apoptosis rate in different groups (%, mean ± SD).
Discussion
Renal mesangial and podocytes have traditionally attracted the most attention in investigations of DKD; however, tubular apoptosis is one of the major morphologic changes that also contribute to kidney damage. Apoptosis is a form of programmed cell death characterized by cell shrinkage, chromatin condensation, and DNA fragmentation in a variety of cell types, including renal proximal tubular epithelial cells.Citation13,Citation14 The mechanisms by which hyperglycemia promotes apoptosis in renal tubular cells are poorly understood. Previous studies have proved that oxidative stress plays a major role in DKD.Citation15 ROS overproduction in diabetes is a direct consequence of hyperglycemia. Various types of cells in the kidney, including tubular epithelial cells, are capable of producing ROS under hyperglycemic conditions.Citation16 ROS have been shown to function as signaling molecules to activate several cellular stress-sensitive pathways that cause mesangial cell damage.Citation17 Besides, the oxidative stress resulting from increased ROS production can cause apoptosis in proximal tubular epithelial cells through pathways mediated by multiple caspases, such as caspase-3.Citation18 It has been shown that an increase in oxidative stress can induce apoptotic changes in HK2 cells, a novel human renal proximal tubular cell line, after being incubated in a high glucose concentration of 30 mmol/L for 18–48 h.Citation19 In contrast, decreased antioxidant activities are common events in diabetes. Oxidative stress-derived tissue injury occurs when ROS production exceeds local antioxidant capacity. In the present study, we demonstrated that the amount of MDA, which is an index of oxidative stress, dramatically increased in NRK-52E cells exposed to high glucose. Furthermore, the upregulation of oxidative stress is correlative to the severity of cell apoptosis. Simultaneously, a significant downregulation of antioxidant (SOD and GPx) activities was observed. These results are consistent with previous studies.Citation5,Citation20
The mitochondria respiratory system has been reported to be the major source of ROS under normal physiological conditions and hyperglycemia.Citation21 During mitochondrial respiration, a small number of electrons in the electron transport chain impulsively escape to oxygen, following which superoxide is generated. Thus, mitochondria-derived ROS are byproducts of oxidative phosphorylation across the electron transport chain.Citation22 Evidence indicates that mitochondrial ROS in hyperglycemia are the key molecules linking other pathways causing hyperglycemic tissue damage.Citation23 Therefore, inhibition of mitochondrial ROS may serve as a treatment strategy for DKD. Nishikawa et al. have shown that normalizing the levels of mitochondrial ROS prevented several diabetes-induced pathways associated with progressive renal disease.Citation24 In nature, ROS production is decreased by uncoupling of the respiratory chain that reduces the proton electrochemical gradient.Citation25 There are three major isoforms of uncoupling proteins, UCP-1 to -3, which belong to a family of inner mitochondrial membrane proteins that are believed to control several aspects of mitochondrial function, including ROS generation, fatty acid homeostasis, and mitochondrial biogenesis.Citation26 These isoforms have similarities in structure, but different tissue expressions in mammals. UCP1 is expressed primarily in brown adipose tissue, UCP2 is ubiquitous, and UCP3 is present in skeletal muscle.Citation27 UCPs play important roles in decreasing ROS production and regulating mitochondrial apoptosis signals by hyperpolarizing the inner mitochondrial membrane. It has been demonstrated that “mild uncoupling” of the mitochondrial respiratory chain due to UCP2 activity allows a more rapid flux of electrons through the mitochondrial inner membrane, reducing membrane potential and decreasing ROS production.Citation28 Hence, UCP2 may play a protective role in the pathogenesis of DM and its complications. Chuang et al.Citation29 show that upregulation of mitochondrial UCP2 expression decreased ROS overproduction, leading to amelioration of apoptotic neuronal cell death in the hippocampus following status epilepticus. Similarly, peroxisome proliferator-activated receptor alpha induction of uncoupling protein 2 has been shown to protect against elevated ROS generated during drug-induced hepatotoxicity.Citation30 Moreover, the protective role of UCP2 in the pathogenesis of diabetic retinopathy has been identified.Citation31 In the present study, we demonstrated that UCP2 was induced in the tubular epithelial cells under the condition of hyperglycemia as a compensative reaction for ROS overproduction. To test the hypothesis that UCP2 functions to limit ROS production, we used genipin, a UCP2 inhibitor, to inhibit uncoupling protein expression. After pretreatment with genipin, NRK-52E cells were implicated in more serious oxidative stress, which is demonstrated by an increase of MDA and downregulation of antioxidant activities. Furthermore, we assessed cell injury by detecting caspase-3 activation and measuring the percentage of apoptotic cells. Caspase-3 activation plays a central role in the execution phase of cell apoptosis and is traditionally considered as an apoptosis indicator. We observed that pretreatment of NRK-52E cells with genipin dramatically promoted caspase-3 activation and aggravated hyperglycemia-induced cell apoptosis in a dose-dependent manner, which showed a positive correlation with MDA amount.
Except the role in regulation of mitochondrial ROS production, the increased UCP2 expression in the diabetic kidney was detected to result in increased cellular O2 consumption. And the subsequently reduced O2 availability may contribute to the development of progressive kidney damage.Citation32 These results indicate that UCP2 may act as a double-edged sword for tubular cell metabolism. However, the balancing levels of UCP2 for optimal function are still poorly understood. Persson et al.Citation33 have found in their study that siRNA-mediated knockdown of UCP2 for 48 h in the diabetic kidney did not reverse the defect on kidney function in vivo.Citation33 Therefore, the role of UCP-2 in diabetes needs to be further evaluated.
In conclusion, the results of the present study indicate that UCP2 upregulation is a compensatory mechanism of tubular epithelial cells for the high glucose-induced ROS production. UCP2 is a strong candidate protein for investigations into treatments of DKD. However, the mechanisms by which hyperglycemia regulates UCP2 gene expression in renal tubular cells are poorly understood. Further studies are required to elucidate the roles of UCP2 in the cellular injuries caused by hyperglycemia.
Declaration of interest
The authors report no conflicts of interest.
References
- Centers for Disease Control and Prevention (CDC). Incidence of end-stage renal disease attributed to diabetes among persons with diagnosed diabetes—United States and Puerto Rico, 1996–2007. MMWR Morb Mortal Wkly Rep. 2010;59:1361–1366
- Zhang L, Wang F, Wang L, et al. Prevalence of chronic kidney disease in China: a cross-sectional survey. Lancet. 2012;379:815–822
- Barnes DJ, Pinto JR, Davison AM, et al. The patient with diabetes mellitus. In: Oxford Textbook of Clinical Nephrology. 2nd ed. Oxford, United Kingdom: Oxford University Press; 1998:723–775
- Ginevri F, Piccotti E, Alinovi R, et al. Reversible tubular proteinuria precedes microalbuminuria and correlates with the metabolic status in diabetic children. Pediatr Nephrol. 1993;7:23–26
- Zhang W, Miao J, Wang S, Zhang Y. The protective effects of beta-casomorphin-7 against glucose-induced renal oxidative stress in vivo and vitro. PLoS One. 2013;8:e63472
- Habib SL. Diabetes and renal tubular cell apoptosis. World J Diabetes. 2013;4:27–30
- Puddu P, Puddu GM, Cravero E, De Pascalis S, Muscari A. The putative role of mitochondrial dysfunction in hypertension. Clin Exp Hypertens. 2007;29:427–434
- Mattiasson G, Sullivan PG. The emerging functions of UCP2 in health, disease, and therapeutics. Antioxid Redox Signal. 2006;8:1–38
- Nakatani T, Tsuboyama-Kasaoka N, Takahashi M, Miura S, Ezaki O. Mechanism for peroxisome proliferator-activated receptor-alpha activator-induced up-regulation of UCP2 mRNA in rodent hepatocytes. J Biol Chem. 2002;277:9562–9569
- Yoshida T, Kato K, Fujimaki T, et al. Association of genetic variants with chronic kidney disease in Japanese individuals. Clin J Am Soc Nephrol. 2009;4:883–890
- Tripathi G, Sharma RK, Baburaj VP, Sankhwar SN, Jafar T, Agrawal S. Genetic risk factors for renal failure among north Indian ESRD patients. Clin Biochem. 2008;41:525–531
- Di Castro S, Scarpino S, Marchitti S, et al. Differential modulation of uncoupling protein 2 in kidneys of stroke-prone spontaneously hypertensive rats under high-salt/low-potassium diet. Hypertension. 2013;61:534–541
- Habib SL. Diabetes and renal tubular cell apoptosis. World J Diabetes. 2013;4:27–30
- Kumar D, Robertson S, Burns KD. Evidence of apoptosis in human diabetic kidney. Mol Cell Biochem. 2004;259:67–70
- Forbes JM, Coughlan MT, Cooper ME. Oxidative stress as a major culprit in kidney disease in diabetes. Diabetes. 2008;57:1446–1454
- Brezniceanu ML, Liu F, Wei CC, et al. Catalase overexpression attenuates angiotensinogen expression and apoptosis in diabetic mice. Kidney Int. 2007;71:912–923
- Ha H, Lee HB. Reactive oxygen species as glucose signaling molecules in mesangial cells cultured under high glucose. Kidney Int Suppl. 2000;77:S19–S25
- Allen DA, Harwood S, Varagunam M, Raftery MJ, Yaqoob MM. High glucose-induced oxidative stress causes apoptosis in proximal tubular epithelial cells and is mediated by multiple caspases. FASEB J. 2003;17:908–910
- Verzola D, Bertolotto MB, Villaggio B, et al. Oxidative stress mediates apoptotic changes induced by hyperglycemia in human tubular kidney cells. J Am Soc Nephrol. 2004;15(Suppl 1):S85–S87
- Gui D, Guo Y, Wang F, et al. Astragaloside IV, a novel antioxidant, prevents glucose-induced podocyte apoptosis in vitro and in vivo. PLoS One. 2012;7:e39824
- Nishikawa T, Araki E. Impact of mitochondrial ROS production in the pathogenesis of diabetes mellitus and its complications. Antioxid Redox Signal. 2007;9:343–353
- Sinha K, Das J, Pal PB, Sil PC. Oxidative stress: the mitochondria-dependent and mitochondria-independent pathways of apoptosis. Arch Toxicol. 2013;87:1157–1180
- Noh H, Ha H. Reactive oxygen species and oxidative stress. Contrib Nephrol. 2011;170:102–112
- Nishikawa T, Edelstein D, Du XL, et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature. 2000;404:787–790
- Saleh MC, Wheeler MB, Chan CB. Uncoupling protein-2: evidence for its function as a metabolic regulator. Diabetologia. 2002;45:174–187
- Rocha M, Apostolova N, Herance JR, Rovira-Llopis S, Hernandez-Mijares A, Victor VM. Perspectives and potential applications of mitochondria-targeted antioxidants in cardiometabolic diseases and type 2 diabetes. Med Res Rev. 2014;34:160–189
- Hernandez-Mijares A, Rocha M, Apostolova N, et al. Mitochondrial complex I impairment in leukocytes from type 2 diabetic patients. Free Radic Biol Med. 2011;50:1215–1221
- Souza BM, Assmann TS, Kliemann LM, Gross JL, Canani LH, Crispim D. The role of uncoupling protein 2 (UCP2) on the development of type 2 diabetes mellitus and its chronic complications. Arq Bras Endocrinol Metabol. 2011;55:239–248
- Chuang YC, Lin TK, Huang HY, et al. Peroxisome proliferator-activated receptors γ/mitochondrial uncoupling protein 2 signaling protects against seizure-induced neuronal cell death in the hippocampus following experimental status epilepticus. J Neuroinflammation. 2012;9:184–201
- Patterson AD, Shah YM, Matsubara T, Krausz KW, Gonzalez FJ. Peroxisome proliferator-activated receptor alpha induction of uncoupling protein 2 protects against acetaminophen-induced liver toxicity. Hepatology. 2012;56:281–290
- Cui Y, Xu X, Bi H, et al. Expression modification of uncoupling proteins and MnSOD in retinal endothelial cells and pericytes induced by high glucose: the role of reactive oxygen species in diabetic retinopathy. Exp Eye Res. 2006;83:807–816
- Friederich M, Fasching A, Hansell P, Nordquist L, Palm F. Diabetes-induced up-regulation of uncoupling protein-2 results in increased mitochondrial uncoupling in kidney proximal tubular cells. Biochim Biophys Acta. 2008;1777(7 and 8):935–940
- Persson MF, Welch WJ, Wilcox CS, Palm F. Kidney function after in vivo gene silencing of uncoupling protein-2 in streptozotocin-induced diabetic rats. Adv Exp Med Biol. 2013;765:217–223