
Abstract
Genetic factors have been implicated in the pathogenesis of certain cases of MPGN. Familial cases of all three histological subtypes have been described. Genetic defects in the control of complement pathways appear to be at the root of many hereditary forms of MPGN. Here we describe a series of three families with familial MPGN. We have identified 3 individuals with a diagnosis of idiopathic MPGN, each with a sibling with the same diagnosis. Data collected included age at presentation, histological findings and age at commencement of renal replacement therapy. A family pedigree was generated for each family as well as a description of the long-term clinical course and transplant outcomes of affected individuals. We have identified male and female affected individuals in this series of three families. The progression to end-stage kidney disease was universal amongst affected individuals. The majority of cases were successfully transplanted. Recurrence in a transplanted kidney occurred in only one individual. This series of familial MPGN provides further evidence for a genetic basis for the disease. Additional studies on these three families will further our knowledge of the underlying mutations in hereditary MPGN and contribute to the understanding of complement-mediated renal disease.
Introduction
Membranoproliferative glomerulonephritis (MPGN) describes a histological lesion, characterized by mesangial hypercellularity and thickening of the glomerular capillary wall on light microscopy. It usually causes progressive chronic kidney disease (CKD), although the clinical expression is variable. Case reports over the last 40 years have speculated about the role of genetic factors in the pathogenesis of MPGN.
MPGN has traditionally been classified as either primary or secondary. In secondary MPGN, the underlying etiology is known, e.g. hepatitis C infection or systemic lupus erythematosus. Primary, or idiopathic MPGN, is further sub-classified into three types (I–III) according to the glomerular ultrastructural appearance and the location of electron dense deposits. A familial clustering of cases of idiopathic MPGN has been observed. Familial cases of types I, II and III MPGN have been describedCitation1–6 and genetic defects have been identified in a proportion of cases.
The first study to establish a genetic locus for MPGN type III was carried out by Neary et al.Citation7 Affected individuals were identified in three generations of the one family. The mode of inheritance was autosomal dominant. Linkage of this familial MPGN was successfully demonstrated to chromosome 1q. The area identified encompasses the regulators of complement cluster, which code for proteins that regulate C3 convertase activity. This landmark linkage study gave support to the potential genetic basis for certain forms of MPGN.
Another important study in the area of inherited glomerulonephritis described two families of Cypriot origin with an inherited form of renal disease.Citation8 The disease was characterized by the presence of microscopic hematuria and an MPGN lesion in the majority of cases. A mutation in the complement factor H-related protein 5 gene was identified in affected individuals. This gene appears to play a pivotal role in the regulation of the ACP.
Both of these familial studies provide evidence to support the concept that complement pathway dysregulation is integral in the pathogenesis of this renal disease. Genetic defects affecting ACP regulation have been identified in all 3 histological subtypes of MPGN.Citation5–10 It seems probable that defects in the regulation or function of the alternative complement pathway (ACP) are pivotal in the pathogenesis of many (if not all) hereditary forms of MPGN.
Methods
We had identified three individuals with a diagnosis of idiopathic MPGN, each with a sibling with the same diagnosis. Data collected included age at presentation, histological findings and age at commencement of renal replacement therapy. A family pedigree was generated for each family as well as a description of the long-term clinical course and transplant outcomes of affected individuals.
Case series
Family 1
The index case (case 1A) in family 1 () presented with renal impairment at the age of 16 years. She was previously healthy. She proceeded to undergo a renal biopsy, which revealed a membranoproliferative pattern of injury with immune complex deposition. She progressed to end-stage kidney disease (ESKD) and required dialysis within 5 years of her initial presentation. She received her first cadaveric kidney transplant at the age of 22 years. The graft failed after 5 years and she received two subsequent cadaveric transplants. Her first two grafts failed due to chronic allograft nephropathy, with no evidence of recurrence in either. The third graft continues to function well with no clinical evidence of recurrence.
Figure 1. Family Pedigree of Family 1.
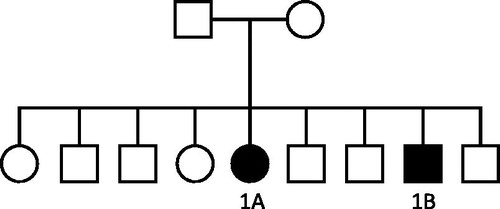
Several years later, it emerged that her brother (case 1B) had been diagnosed with kidney disease at the age of 18 years. His biopsy had evidence of MPGN with immune complex deposition. He commenced hemodialysis at the age of 21 years and received a cadaveric kidney transplant 1 year later. He received a second renal transplant 9 years later. Graft loss in both cases occurred due to chronic allograft nephropathy in the setting of poor adherence with immunosuppression. There was no histological evidence of disease recurrence in either graft.
Family 2
The index case (case 2A, ) presented at the age of 22 years with a severe headache and was found to be hypertensive with impaired renal excretory function. Urinalysis was positive for blood and protein and his serum complement levels were low. His renal biopsy revealed MPGN with a moderate amount of immunoglobulin and complement deposition. He progressed to end-stage renal failure within 12 months of presentation, necessitating initiation of hemodialysis. He received a living related (sibling) kidney transplant 1 year later.
Figure 2. Family Pedigree of Family 2.
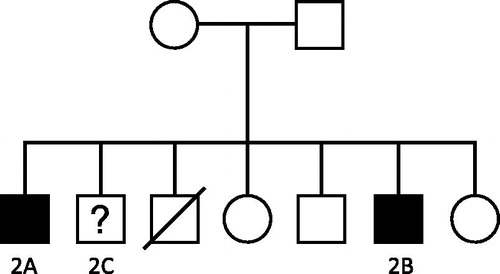
His younger brother (case 2B) was found to have proteinuria on routine examination. A renal biopsy performed at the age of 15 years, revealed MPGN with deposition of immunoglobulin and complement in all glomerulus. He received a pre-emptive, sibling transplant at age 17 years. Graft failure occurred within 6 years due to histologically confirmed recurrence of MPGN. He received a second sibling transplant. Biopsy findings raised a concern regarding early recurrence of MPGN in this graft; however, this was deemed unlikely given the absence of any clinical indication of recurrence. The patient in question enjoys well-preserved graft function at 31 years post-transplant. At the time, electron microscopy was not performed on the specimen.
Interestingly, a third brother (case 2C) underwent a renal biopsy at age 31, to investigate the presence of persistent microscopic hematuria. The histological findings were consistent with a diagnosis of IgA Nephropathy. He did not develop progressive renal disease. The death of another sibling in this family was not related to renal disease.
Family 3
Patient 3A () was noted to have proteinuria at the age of 20 years and was given a clinical diagnosis of an unspecified nephritis at that time. When he developed hypertension and renal impairment 14 years later, he was referred for specialist evaluation. A renal biopsy specimen was obtained and revealed MPGN of the linear dense deposit disease type. Serum complement levels were normal. He became dialysis dependent at the age of 42 years.
Figure 3. Family Pedigree of Family 3. “?” denotes suspected, but unconfirmed, kidney disease in this individual. A histological diagnosis was not obtainable.
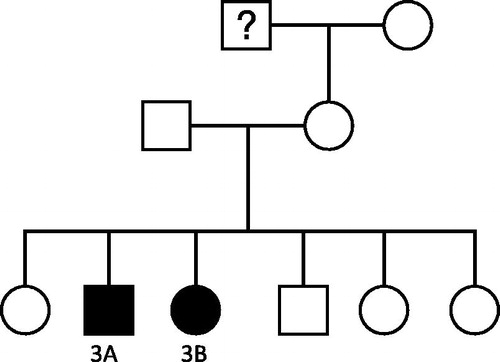
His sister (patient 3b) was also diagnosed with MPGN at the age of 53 years. She received a pre-emptive kidney transplant 4 years later, which is functioning well 6 years post-transplant. In addition, there was a history of “kidney failure” in a maternal grandfather. It was not possible to determine the etiology of this.
Discussion
Familial cases of all three histological types of idiopathic MPGN have previously been described but this is the largest series published to date. It further strengthens the concept of a genetic basis for certain cases of this renal lesion. Hereditary MPGN remains a rare and heterogeneous disorder. However, as the number of affected families described increases and the spectrum of causative genetic defects expands, so too does our understanding of this disease entity.
We have described three families, each with two members who have a histological diagnosis of MPGN (). Families 1 and 2 both have MPGN type I with family 3 affected by MPGN type II. The majority of the affected individuals developed clinically detectable renal disease in their 2nd or 3rd decade. Family 3 were diagnosed with renal disease at an older age and appear to have had a slower progression to ESKD. We have described both male and female affected subjects. The mode of inheritance in Families 1 and 3 appears to be autosomal recessive. In the case of Family 2, the inheritance is likely to be either X-linked recessive or autosomal recessive.
Table 1. Clinical characteristics of affected individuals.
The progression to ESKD was universal in our series, although “unaffected” family members were not screened for possible renal disease. Although histological confirmation of recurrence of MPGN in a kidney transplant can be difficult, we have evidence to suggest that only one affected individual had a recurrence of immune complex MPGN in his transplant. We have demonstrated that patients with hereditary MPGN can be successfully transplanted, although we accept that the underlying genetic defect is likely to determine the probability of disease recurrence.
A more comprehensive and evolving knowledge of complement functions have contributed to the understanding of the pathogenesis of MPGN and other renal diseases. Genetic defects in the control of complement pathways appear to be at the root of many hereditary forms of MPGN. Mutations in C3, complement factor H and complement control proteins have been identified in familial cases of Type II MPGN or dense deposit disease (DDD).Citation5,Citation8–10 Neary et al. demonstrated linkage of MPGN III to chromosome 1q, a locus containing genes that code for a number of important regulators of the complement pathway.Citation7 Mutations in CFH and CFI have been identified in 10.4% and 6.2%, respectively, of a cohort of patients with MPGN type I.Citation10 These patients had positive staining for both complement and immunoglobulin on immunofluorescence. This suggests that complement dysregulation also plays a role in the pathogenesis of immune complex MPGN.
One member of family 2 (subject 2C) was diagnosed with IgA Nephropathy but did not develop progressive renal disease. Whether this represents a different renal phenotype in a family member with the same underlying genetic abnormality as his siblings, or a mere coincidence, is unclear. It is evident that differing renal phenotypes can occur with mutations affecting the regulation of complement pathways. Indeed, there are reports of Types I and III MPGN occurring within the same family.Citation11,Citation12 Walker et al. identified that only a minority of cases of DDD had a membranoproliferative pattern of injury.Citation13 A total of five different histological lesions may be caused by DDD. What determines the renal phenotype in an individual with a genetic mutation affecting ACP control has yet to be comprehensively described?
It is clear that certain forms of MPGN have a genetic basis. Early identification of these familial cases is important. At present, the treatment options for patients with idiopathic MPGN are based on general supportive strategies, rather than targeted therapy. However, with the emergence of new agents that target complement-mediated endothelial and glomerular attack, there is the potential to delay or halt progressive renal injury. As many familial cases of MPGN have been found to have an inherited dysregulation of complement pathways, these individuals may respond to such treatment. In the future, the identification of underlying genetic mutations will allow a targeted approach to treatment and will hopefully promote the development of novel treatment options. Not only will the identification of genetic causes of MPGN allow us to tailor therapies for such patients, but also allows us to offer genetic counseling to affected individuals and to gather information on the disease trajectory depending on the underlying defect.
Conclusion
This series of familial MPGN provides further evidence for a genetic basis for the disease. There was a progression to ESKD in all affected individuals but the majority of our patients were successfully transplanted without disease recurrence. Additional studies on these three families will further our knowledge of the underlying mutations in hereditary MPGN and contribute to the understanding of complement-mediated renal disease.
Declaration of interest
The authors declare that there is no conflict of interest.
References
- Neary J, Dorman A, Campbell E, Keogan M, Conlon P. Familial membranoproliferative glomerulonephritis type III. Am J Kidney Dis. 2002;40(1):E1
- Motoyama O, Sakai K, Ohashi Y, et al. Membranoproliferative glomerulonephritis in a girl and her mother. Clin Exp Nephrol. 2009;13(1):77–80
- Ueda H, Isimura E, Okuno S, et al. Sibling cases of nephritis resembling membranoproliferative glomerulonephritis. Nihon Jinzo Gakkai Shi. 2002;44(4):420–426
- Robles NR, Barquilla JF, Arrobas M, et al. Familial membranoproliferative glomerulonephritis. An Med Interna. 1998;15(7):373–375
- Martínez-Barricarte R, Heurich M, Valdes-Cañedo F, et al. Human C3 mutation reveals a mechanism of dense deposit disease pathogenesis and provides insights into complement activation and regulation. J Clin Invest. 2010;120(10):3702–3712
- Levy M, Halbwachs-Mecarelli L, Gubler MC, et al. H deficiency in two brothers with atypical dense intramembranous deposit disease. Kidney Int. 1986;30(6):949–956
- Neary JJ, Conlon PJ, Croke D, et al. Linkage of a gene causing familial membranoproliferative glomerulonephritis type III to chromosome 1. J Am Soc Nephrol. 2002;13(8):2052–2057
- Gale DP, de Jorge EG, Cook HT, et al. Identification of a mutation in complement factor H-related protein 5 in patients of Cypriot origin with glomerulonephritis. Lancet. 2010;376(9743):794–801
- Linshaw MA, Stapleton FB, Cuppage FE, et al. Hypocomplementemic glomerulonephritis in an infant and mother. Evidence for an abnormal form of C3. Am J Nephrol. 1987;7(6):470–477
- Servais A, Noël LH, Roumenina LT, et al. Acquired and genetic complement abnormalities play a critical role in dense deposit disease and other C3 glomerulopathies. Kidney Int. 2012;82(4):454–464
- Berry PL, McEnery PT, McAdams AJ, West CD. Membranoproliferative glomerulonephritis in two sibships. Clin Nephrol. 1981;16(2):101–106
- Bogdanović RM, Dimitrijević JZ, Nikolić VN, Ognjanović MV, Rodić BD, Slavković BV. Membranoproliferative glomerulonephritis in two siblings: report and literature review. Pediatr Nephrol. 2000;14(5):400–405
- Walker PD, Ferrario F, Joh K, Bonsib SM. Dense deposit disease is not a membranoproliferative glomerulonephritis. Mod Pathol. 2007;20(6):605–616