
Abstract
Background: Mature podocytes are in cell cycle arrest and their inability to proliferate successfully is a consequence of negative cell-cycle regulators’ expression, such as p57. Phosphorylated smad2/smad3 (pSmad2/3) is an intracellular heteromeric mediator of transforming growth factor beta (TGF-β) signals and, together with co-activators such as P300, regulates gene transcription, including cell cycle regulator proteins. Methods: In order to investigate Smad pathway activation and podocyte cell cycle regulation in glomerular injury, we studied the glomerular immunohistochemical expression of p57, pSmad2/3 and P300 in samples from 67 patients with various types of glomerulonephritis (GN) and 10 normal kidney tissue specimens. Results: pSmad2/3 and p300 expression were found significantly increased in all glomerular cell types in both proliferative and nonproliferative GN, while a significant reduction in p57-positive podocytes was observed when compared to controls. Staining for p57 was found to inversely correlate to pSmad2/3 suggesting that glomerular Smad pathway activation is related to down-regulation of p57 expression in proliferative glomerulonephritis. To our knowledge, this is the first study that indicates a relation between the TGF-beta/Smad signalling pathway and the cell cycle regulatory protein p57 in human GN. Conclusion: The increased pSmad2/3 staining together with the reduced p57 expression found in biopsy specimens with intense interstitial inflammation, indicate a possible relation between interstitial inflammation, glomerular Smad pathway activation and podocyte cell-cycle deregulation.
Introduction
Podocyte injury and glomerulosclerosis are the hallmarks of progressive glomerular damage. Podocytes are terminally differentiated epithelial cells with specialized morphology and molecular architecture that are main contributors to the glomerular filtration barrier. Mature podocytes are in cell cycle arrest and their inability to proliferate successfully is most likely the consequence of robust expression of Cyclin-dependent Kinase inhibitors (CKIs), which are negative regulators of cell-cycle progression.Citation1 p57 is a member of the CKIs family associated with G1 cell cycle arrest.Citation2 In glomerular diseases, p57 expression is affected and cell-cycle deregulation can occur. Specifically, in focal segmental glomerulosclerosis (FSGS) cellular lesions, damaged podocytes have reduced p57 protein expression that is related to unsuccessful podocyte proliferation.Citation3 This podocyte cell-cycle deregulation is thought to be involved in the development of cellular FSGS and even the formation of crescents, although it has been argued that proliferating cells in these lesions mainly originate from parietal epithelial cells.Citation4–7
It is well established that TGF-β has effects on proliferation, hypertrophy, and apoptosis in renal cells.Citation8–10 The expression of TGF-β mRNA and protein in podocytes has been found to be increased in a variety of glomerulopathies, such as FSGSCitation11 and Membranous Nephropathy (MN).Citation12 In TGF-β1 transgenic mice, an acute and massive increase in plasma levels of TGF-β1 results in severe GN with crescent formation.Citation13,Citation14 TGF-β is also strongly expressed in the cellular crescents in the early stage of experimental crescentic GN.Citation15 Furthermore, podocytes covering the cellular lesion of FSGS exhibit increased expression of TGF-β1 protein in glomerular diseases.Citation11,Citation16,Citation17 TGF-β activates the Smad, Ras/extracellular signal-regulated kinase (ERK) and phosphatidyl inositol-3-kinase (PI3K) pathways in podocytes, of which the major TGF-β/Smad signalling pathway appears to override the minor TGF-β -induced Ras/ERK/PI3K pathways.Citation18 The TGF-β/Smad signalling pathway is initiated by ligand-binding induced signal transduction through the TGF-β receptor complex which results in the intracellular mediator Phosphorylated smad2/smad3 (pSmad2/3).Citation19 The activated pSmad2/3 heteromer binds with Smad4, translocates into the nucleus and, together with co-activators such as P300, induces transcription of genes such as the cell-cycle regulators p15 and p21.Citation20–23 This process is negatively regulated by the inhibitory Smad7.Citation24 P300 is a transcriptional co-activator that facilitates pSmad2/3 and Smad4 complexes binding to DNA and enables specific sequence targeting.Citation25–27 Increased expression of Smad2/3 and their activated forms pSmad2/3 in glomeruli of both experimental and human glomerulonephritis has been reported in the literature.Citation11,Citation28 Moreover, it has been shown that Smad2/3 and Smad4 require the co-activator p300 to activate transcription.Citation23,Citation26,Citation27 P300 is also involved in TGF-β/Smad mediated type I collagen gene transcription.Citation29–31
Limited data exist on the glomerular expression patterns of p57, p300 and phosphorylated Smads in human glomerulonephritis (GN). It appears that there is interplay between the TGF-β/Smad signaling pathway and podocyte cell cycle regulation that could be involved in the pathogenesis and disease progression of GN. In order to investigate glomerular Smad pathway activation and p57 expression in podocytes, we studied the glomerular immunohistochemical expression of p57, pSmad2/3 and P300 in various types of human GN.
Material and methods
Patients
Percutaneous renal biopsies were obtained from 67 patients (28 male/39 female) with various types of GN. Renal biopsies were performed for diagnostic purposes and informed consent for the use of excess renal tissue was obtained from the patients. GNs were divided into proliferative (n = 40) and non-proliferative (n = 27). The non-proliferative group of primary GNs included Minimal Change Disease (MCD, n = 3), MN (n = 9), FSGS (n = 5), and cases with WHO class II and V Lupus Nephritis (LN, n = 10). The proliferative group of GNs included IgA nephropathy (IGAN, n = 10), pauci-immune GN (Pauci-Im GN, n = 10) as well as cases with WHO class III and IV LN (n = 20). The summary of the classification and the clinical parameters of patients with GN are given in . Ten samples of normal kidney tissue that were surgically removed from patients with renal cell carcinoma served as controls.
Table 1. Summary of the classification and the clinical parameters of patients with glomerulonephritis.
Immunohistochemistry
Immunohistochemical staining was performed on 4 mm thick formalin-fixed paraffin embedded sections, using a biotin free, two-step, HRP-labelled detection system (DACO EnVision™, Glostrup, Denmark).
Sections were deparaffinised in xylene and rehydrated through graded alcohols. After quenching of endogenous peroxidase activity using a methanol/hydrogen peroxide solution (0.3% in TBS for 30 min), we proceeded to microwave heat-mediated antigen retrieval in 10 mM, pH = 6 citrate buffer and blockage of non-specific binding by incubation in 10% normal horse serum in TBS (Vector Lab, Burlingame, CA) for 30 min at room temperature. Subsequently, sections were incubated for 40 min at 4 °C with the primary antibodies.
The primary antibodies used in our study were: (1) anti-p57 rabbit polyclonal antibody(C20/sc-1040) (Santa Cruz Biotechnology Inc., Dallas, TX) at a dilution 1:80, (2) anti-pSmad2/3 rabbit polyclonal antibody (#3101) (Cell Signalling Technology Inc., Danvers, MA) at a dilution 1:500 and (3) anti-p300 rabbit polyclonal antibody (sc-585) (Santa Cruz Biotechnology Inc.) at a dilution 1:500.
Sections were incubated with a biotin free, two-step, HRP-labelled detection system (DACO EnVision™) for 30 min at room temperature followed by the addition of 3,3′-diaminobenzidine tetrachlorohydrate (DAB), in order to achieve visualization of the requested antigens. The biotin-avidin/streptavidin system is often used as an amplification step to increase sensitivity. However, in some tissues such as kidney, “non-specific” interactions may be a problem due to high levels of endogenous biotin-containing proteins.Citation32 Thus, a biotin free detection system was used in our experiments. Finally, sections were rinsed, counterstained and mounted.
Quantification of pSmad2/3 and p300 immunostaining and histologic injury in human renal biopsies
Serial sections were immunostained for each molecule. The intensity of glomerular staining of pSmad2/3 p300 and p57 was evaluated according to the following 0 to 3 scale: 0: <10% cells with nuclear staining; (1) weak nuclear staining intensity or 11–50% of cells with nuclear staining; (2) moderate nuclear staining intensity and >50% of cells with nuclear staining and (3) strong nuclear staining intensity and >50% of cells with nuclear staining. The staining status of glomerular lesions was also assessed, with immunopositivity being recorded when some degree of immunostaining was noticeable. When no staining was observed, the respective lesions were characterized as negative. Sections were scored independently by two investigators (A.N. and L.N.) blinded to the patients’ clinical profile. The human renal biopsy specimens have been reviewed and analyzed by an independent pathologist, who was blinded to the quantification of immunostaining. The severity of glomerulosclerosis was expressed as the percentage of globally sclerosed glomeruli out of the total number of glomeruli in the renal biopsy. The non-globally sclerosed glomeruli were analyzed for the presence of proliferative segmental lesions, crescents, microadhesions, and segmental sclerotic lesions. The degree of interstitial inflammation, interstitial fibrosis and tubular atrophy was scored between 0 and 3 according to the following: 0, 0%, 1, 1 to 25%; 2, 26 to 50%; and 3, >50% of the biopsy area demonstrating inflammation, fibrosis or atrophy, respectively.
Data are presented as mean ±1 standard deviation. Comparisons between the groups were made with the Kruskal–Wallis test. Mann–Whitney U test was used to compare the molecules’ expression between controls and every form of GN. The Spearman correlation analysis was used to detect possible correlations between staining scores, clinical data and indices of histologic injury. Statistical significance was set at 5%.
Results
Normal human kidney
Within normal human kidney tissue, pSmad2/3 was expressed in the nuclei of a few glomerular cells (less than 10%) (), whereas less that 30% of glomerular cells exhibited nuclear positivity for p300 ().Citation35 The expression of both molecules was not restricted in any particular glomerular cell type. p57 immunostaining in the glomerulus was mainly restricted to normal podocytes and was not present in glomerular mesangial or endothelial cells ().
Figure 1. Expression pSmad2/3, p300 and p57 in normal human kidney: A few glomerular cells, including all cell types, were pSmad2/3 positive (a) (arrows). Less than 30% of glomerular cells, including all cell types, were p300 positive (b). Podocyte staining for p57 in the normal kidney was abundant with a nuclear staining pattern (c). Immunopositivity for psmad2/3 and p300 (d, e,) but not for p57 (f) in a cellular crescent (arrows) (Wegener granulomatosis). Immunopositivity for psmad2/3 and p300 (g, h) but not for p57 (i) in hyperplastic lesions and microadhesions (arrows) (proliferative lupus nephritis, WHO class IV). ( are reproduced from Kassimatis et al.Citation35). Original magnification ×400.
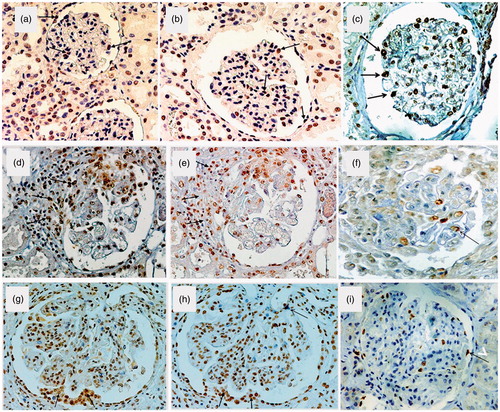
Glomerular pSmad2/3, p300 and p57 expression in human glomerulonephritis
pSmad2/3 and p300 expression was significantly increased (p < 0.001) in all glomerular cell types in both proliferative and nonproliferative GN, when compared to controls. In contrast, a significant reduction in p57-positive podocytes was observed in samples from patients with GN (p = 0.006). In proliferative GN there was a significant general increase in pSmad2/3 (p < 0.001) and p300 (p < 0.001) expression and a decrease in the intensity of p57 podocyte staining (p < 0.001). In areas of proliferation, there was a segmental absence of podocyte immunostaining for p57. The absence was not due to cell loss, as nuclei were easily detected by the hematoxylin counterstain. In these areas, on the other hand, an increase in staining for pSmad2/3 and p300 was observed. In proliferative lupus nephritis, pSmad2/3 and p300, but not p57, were overexpressed in hyperplastic lesions and microadhesions and the same pattern of expression was observed in cellular crescents (). Comparing the non-proliferative GN group with the control group resulted in similar results regarding pSmad2/3 (p = 0.011) and p300 (p = 0.003), while podocyte expression of p57 was found to be preserved (p = 0.418).
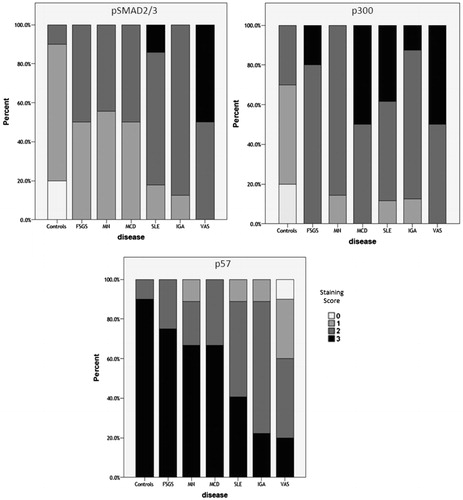
Comparisons between the different types of GN showed a statistically significant difference in the glomerular expression of pSmad2/3, p300 and p57 (). In particular, when compared to controls, a significant decrease in p57 expression was found in pauci-immune GN, SLE and IGA (p = 0.004, p = 0.019, p = 0.01, respectively), but not in FSGS, MN and MCD (p = 0.733, p = 0.4, p = 0.573, respectively). Similarly, increased pSmad2/3 staining was found in pauci- immune GN, SLE and IGA (p < 0.001, p < 0.001, p = 0.03, respectively) but not in FSGS, MN and MCD (p = 0.198, p = 0.095, p = 0.364, respectively). P300 staining was found to be increased in all GN, compared to controls.
Figure 2. Staining scores for the glomerular expression of pSmad2/3, p300 and p57 in different types of GN. (% patients). (FSGS: Focal segmental glomerulosclerosis, MN: Membranous nephropathy, MCD: Minimal Change Disease, pauci-IM-GN: pauci-immune glomerulonephritis, LN: Lupus nephritis, IGAN: IgA nephropathy.)
Correlation of p57 with pSmad2/3 and p300 expression – Correlations between immunohistochemical, clinical, and histologic parameters
Taking into account glomerular staining in all GN, pSmad2/3 expression correlated positively to P300 (r = 0.603, p < 0.01), while an inverse correlation between p57 expression, and pSmad2/3 (r = −0.4, p < 0.01), as well as P300 (r = −0.45, p < 0.01) staining was observed. The expression of pSmad2/3 in diseased glomeruli correlated positively with serum creatinine values (r = 0.36, p < 0.05) and interstitial inflammation scores (r = 0.29, p < 0.05). A negative correlation between p57 and interstitial inflammation was also found. (r = −0.3, p < 0.05), while p300 was found to be inversely correlated to glomerulosclerosis (r = 0.35, p < 0.01). No correlation was found between proteinuria or interstitial fibrosis and the expression of the studied proteins.
Stratification of the GN biopsy samples to proliferative and non-proliferative revealed that the correlations between pSmad2/3, p300 and p57 were significant only for the proliferative GN group. In particular, in the proliferative GN group pSmad2/3 expression correlated positively to P300 (r = 0.35, p = 0.04) and inversely to p57 (r = −0.37, p = 0.03) while in the non-proliferative there were no significant correlations. It should be noted that in the non-proliferative group the significant negative correlation between p300 and glomerulosclerosis persisted (r = −0.49, p < 0.05).
Discussion
To our knowledge, this is the first study that indicates a relation between the TGF-beta/Smad signalling pathway and the cell cycle regulatory protein p57 in human GN.
In contrast to the findings of the present study, no expression of pSmad2/3 in normal human renal tissue was reported by another group of researchers.Citation11 This divergence most likely reflects differences in the immunostaining protocol or the usage of a different primary antibody.Citation33,Citation34 Indeed, in a previous study from our laboratory, similar results of positive immunostaining in normal human renal tissue were found using the same biotin free, two-step, HRP-labelled detection system for antibody localization with increased sensitivity.Citation35 Moreover, Smad2 and 3 have been found to be weakly expressed in the glomeruli and tubules of the mouse embryo and adult kidney.Citation29,Citation36,Citation37 p300 integrates transcriptional signals thus regulating various cellular processes such as DNA repair, cell growth, differentiation, and apoptosis.Citation38 It is therefore possible that its expression in normal glomeruli, detected in our study, implies a role for this molecule in maintaining normal glomerular function. Podocyte staining for p57 in the normal kidney was abundant with a nuclear staining pattern, and reduced in samples from patients with GN, as expected.Citation39 It is known that p57 and p27 are decreased in damaged podocytes from patients with FSGS in comparison with controls.Citation3,Citation40 Our findings are also in agreement with previous studies in experimental models in mice and rat showing that p57 is suppressed in glomerular diseases.Citation41
The reduced glomerular p57 expression together with the increased pSmad2/3 and P300 staining that we observed in the glomeruli of patients with GN indicate that these proteins may be involved in the progression of glomerular injury. The finding that staining for p57 inversely correlated to pSmad2/3 could suggest that glomerular Smad pathway activation is related to down-regulation of p57 expression. Our findings, which are based solely on immunohistochemistry, cannot illustrate a causal relationship between Smad signalling pathway activation and cell cycle deregulation, but there are data in the literature suggesting that this relation might exist. It has been suggested that TGF-β, which is overexpressed by hyperplastic podocytes, may be involved in the regulation of podocyte proliferation.Citation18 Moreover, p57 is thought to be involved in the control of TGF-β-induced proliferation and differentiation of osteoblastic cells.Citation42 In osteoblastic cells on cell cycle arrest, p57 is rapidly degraded upon TGF-β stimulation.Citation43 One proposed mechanism of this p57 degradation is that it is mediated through TGFβ1-activated, Smad-dependent transcription of the gene for the F-box protein FBL12, a protein expressed in the limb bud of developing embryosCitation42,Citation44 FBL12 forms a complex that binds to and ubiquitinates phosphorylated mouse p57. Inhibition of FBL12 suppresses TGFβ-induced degradation of p57, increases the steady-state level of p57, and promotes the differentiation of primary osteoblasts.Citation44 In addition; the interaction between TGF-beta and p57 has been demonstrated to affect proliferative processes of both renal and non-renal cells. TGF-β-induced cell cycle arrest of hematopoietic cells requires p57 up-regulation,Citation45 while the targeted inhibition of p57 blocks TGF-β-inhibited proliferation of primary cultured human limbal epithelial cells.Citation46 In an animal model that involved unilaterally nephrectomised male Sprague-Dawley rats, TGF-β-induced compensatory tubular cell hypertrophy that was found to be regulated by p57.Citation47 Liu et al. demonstrated that TGF-β-mediated hypertrophy in rat renal epithelial cells is regulated by p57, and does not involve regulation by the CKIs p21 and/or p27.Citation48 The finding that staining for p57 inversely correlated to P300 expression suggests that glomerular p300 could also serve as a mediator of podocyte proliferation. Research in cancer cells has shown that p300 plays a crucial role in cell cycle progression and its activity is required for G1/S transition, while p300 inhibition induces block of progression into the S-phase of the cell cycle and apoptosis.Citation49–51 Our finding suggest that p300 over-expression accompanied by p57 down-regulation might induce podocyte cell cycle deregulation and be involved in disease progression, especially in proliferative GNs.
The increased expression of pSmad2/3 and p300 in the glomeruli of proliferative GNs may be related to the inflammation that accompanies disease progression. This is supported by the positive correlation of pSmad2/3 and p300 glomerular immunostaining with interstitial inflammation. It is well accepted that TGF-beta/Smad signalling is a major pathway for renal fibrosis. In experimental and human kidney diseases such as diabetic nephropathy, obstructive kidney disease, 5/6 nephrectomy, and hypertensive nephropathy, Smad2 and Smad3 are strongly activated, in the context of renal fibrosis.Citation52,Citation53 It should be noted, though, that except for its pro-fibrotic role, TGF-β also exerts anti-inflammatory effects and possesses immunoregulatory properties. Deletion of TGF-β1 in mice results in lethal multi-organ inflammation and death at 3 weeks of age. Similarly, conditional deletion of theTGF-β1 gene from T cells in mice develops autoimmune diseases.Citation54,Citation55 These findings suggest a vital role for TGF-β as an anti-inflammatory mediator. This is confirmed further by the findings that application of TGF-β improves autoimmune diseases, including allergic encephalomyelitis, arthritis, and experimental colitis.Citation56–59 In the normal kidney, TGF-β induces Smad7 expression to exert its negative feedback mechanism by causing degradation, and prevent phosphorylation of Smad2 and Smad3.Citation60,Citation61 Recent data have shown that in progressive kidney disease, renal Smad7 protein is reduced, resulting in over activation of TGF-β signalling and progressive renal fibrosis as shown in various rodent models of kidney disease, such as an anti-Thy1 model of glomerulonephritis, obstructive kidney disease, remnant kidney disease, and autoimmune crescentic kidney disease.Citation62–65 The loss of renal Smad7 not only enhances TGF-β Smad3-mediated renal fibrosis, but also enhances renal inflammation through Smad2 and Smad3 phosphorylation and activation of the NF-kB–dependent inflammatory response.Citation66,Citation67 This mechanism could explain our findings of pSmad2/3 and p300 over-expression in the glomeruli of proliferative GNs with an increased inflammation score.
Our results indicate an interaction between the TGF-β/SMAD signaling pathway, the co-activator p300 and the cell cycle regulatory protein p57 in the glomerulus that could be involved in the pathogenesis and disease progression of GN. The increased pSmad2/3 staining together with the reduced p57 expression found in biopsy specimens with intense interstitial inflammation, indicating a possible relation between interstitial inflammation, glomerular Smad pathway activation and podocyte cell-cycle deregulation.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
References
- Vidal A, Koff A. Cell-cycle inhibitors: Three families united by a common cause. Gene. 2000;247:1–15
- Yan Y, Frisen J, Lee MH, Massague J, Barbacid M. Ablation of the CDK inhibitor p57Kip2 results in increased apoptosis and delayed differentiation during mouse development. Genes Dev. 1997;11:973–983
- Wang S, Kim JH, Moon KC, Hong HK, Lee HS. Cell cycle mechanisms involved in podocyte proliferation in cellular lesion of focal segmental glomerulosclerosis. Am J Kidney Dis. 2004;43:19–27
- Bariety J, Bruneval P, Meyrier A, Mandet C, Hill G, Jacquot C. Podocyte involvement in human immune crescentic glomerulonephritis. Kidney Int. 2005;68:1109–1119
- Ding M, Cui S, Li C, Jothy S, et al. Loss of the tumor suppressor Vhlh leads to upregulation of Cxcr4 and rapidly progressive glomerulonephritis in mice. Nat Med. 2006;12:1081–1087
- Thorner PS, Ho M, Eremina V, Sado Y, Quaggin S. Podocytes contribute to the formation of glomerular crescents. J Am Soc Nephrol. 2008;19:495–502
- Smeets B, Uhlig S, Fuss A, et al. Tracing the origin of glomerular extracapillary lesions from parietal epithelial cells. J Am Soc Nephrol. 2009;20:2604–2615
- Bottinger EP, Bitzer M. TGF-beta signaling in renal disease. J Am Soc Nephrol. 2002;13:2600–2610
- Schnaper HW, Hayashida T, Poncelet AC. It’s a Smad world: Regulation of TGF-beta signaling in the kidney. J Am Soc Nephrol. 2002;13:1126–1128
- Liu Y. Renal fibrosis: New insights into the pathogenesis and therapeutics. Kidney Int. 2006;69:213–217
- Kim JH, Kim BK, Moon KC, Hong HK, Lee HS. Activation of the TGF-β/Smad signaling pathway in focal segmental glomerulosclerosis. Kidney Int. 2003;64:1715–1721
- Kim TS, Kim JY, Hong HK, Lee HS. mRNA expression of glomerular basement membrane proteins and TGF-β1 in human membranous nephropathy. J Pathol. 1999;189:425–430
- Kopp JB, Factor VM, Mozes M, et al. Transgenic mice with increased plasma levels of TGF-b1 develop progressive renal disease. Lab Invest. 1996;74:991–1003
- Sanderson N, Factor V, Nagy P, et al. Hepatic expression of mature transforming growth factor b1 in transgenic mice results in multiple tissue lesions. Proc Natl Acad Sci USA. 1995;92:2572–2576
- Shimizu M, Kondo S, Urushihara M, et al. Role of integrin-linked kinase in epithelial mesenchymal transition in crescent formation of experimental glomerulonephritis. Nephrol Dial Transplant. 2006;21:2380–2390
- Kim HW, Moon KC, Park SY, Hong HK, Lee HS. Differential expression of platelet-derived growth factor and transforming growth factor-b in relation to progression of IgA nephropathy. Nephrology. 2002;7:S131–S139
- Patek CE, Fleming S, Miles CG, et al. Murine Denys-Drash syndrome: Evidence of podocyte dedifferentiation and systemic mediation of glomerulosclerosis. Hum Mol Genet. 2003;12:2379–2394
- Lee HS, Song CY. Effects of TGF-β on podocyte growth and disease progression in proliferative podocytopathies. Kidney Blood Press Res. 2010;33:24–29
- Franzen P, Ten Dijke P, Ichijo H, et al. Cloning of a TGF beta type I receptor that forms a heteromeric complex with the TGF beta type II receptor. Cell. 1993;75:681–692
- Massague J, Blain SW, Lo RS. TGF-β signalling in growth control, cancer, and heritable disorders. Cell. 2000;103:295–309
- Nakao A, Imamura T, Souchelnytskyi S, et al. TGF-bJreceptor-mediated signalling through Smad2, Smad3 and Smad4. EMBO J. 1997;16:5353–5362
- Zawel L, Dai JL, Buckhaults P, et al. Human Smad3 and Smad4 are sequence-specific transcription activators. Mol Cell. 1998;1:611–617
- Massague J, Wotton D. Transcriptional control by TGF-bJ/Smad signaling. EMBO J. 2000;19:1745–1754
- Nakao A, Afrakhte M, Moren A, et al. Identification of Smad7, aTGFbeta-inducible antagonist of TGF-beta signalling. Nature. 1997;389:631–635
- Shi Y, Massague J. Mechanisms of TGF-b signaling from cell membrane to the nucleus. Cell. 2003;113:685–700
- de Caestecker MP, Yahata T, Wang D, et al. The Smad4 activation domain (SAD) is a proline-rich, p300-dependent transcriptional activation domain. J Biol Chem. 2000;275:2115–2122
- Shen X, Hu PP, Liberati NT, Datto MB, Frederick JP, Wang XF. TGF-beta-induced phosphorylation of Smad3 regulates its interaction with co-activator p300/CREBbinding protein. Mol Biol Cell. 1998;9:3309–3319
- Hong SW, Isono M, Chen S, et al. Increased glomerular and tubular expression of transforming growth factor-beta1, its type II receptor, and activation of the Smad signaling pathway in the db/db mouse. Am J Pathol. 2001;158:1653–1663
- Kanamaru Y, Nakao A, Tanaka Y, et al. Involvement of p300 in TGF-beta/Smad-pathway mediated alpha2(I) collagen expression in mouse mesangial cells. Nephron Exp Nephrol. 2003;95:e36–e42
- Li JH, Zhu HJ, Huang XR, Lai KN, Johnson RJ, Lan HY. Smad7 inhibits fibrotic effect of TGF-Beta on renal tubular epithelial cells by blocking Smad2 activation. J Am Soc Nephrol. 2002;13:1464–1472
- Yamashita S. Ontogenic expression of estrogen receptor co-activators in the reproductive tract of female mice neonatally exposed to diethylstilbestrol. Reprod Toxicol. 2004;18:275–284
- Bussolati G, Gugliotta P, Volante M, Pace M, Papotti M. Retrieved endogenous biotin: A novel marker and a potential pitfall in diagnostic immunohistochemistry. Histopathology. 1997;31:400–407
- Vyberg M, Nielsen S. Dextran polymer conjugate two step visualization system for immunohistochemistry. A comparison of EnVision+ with two three step avidinbiotin techniques. Appl Immunohistochem. 1998;61:3–10
- Sabattini E, Bisgaard K, Ascani S, et al. The EnVisionTM+ system: A new immunohistochemical method for diagnostics and research. Critical comparison with APAAP, ChemMateTM, CSA, LABC, and SABC techniques. J Clin Pathol. 1998;51:506–511
- Kassimatis TI, Giannopoulou I, Koumoundourou D, Theodorakopoulou E, Varakis I, Nakopoulou L. Immunohistochemical evaluation of phosphorylated SMAD2/SMAD3 and the co-activator P300 in human glomerulonephritis: Correlation with renal injury. J Cell Mol Med. 2006;10(4):908–921
- Flanders K, Kim E, Roberts A. Immunohistochemical expression of Smads 1–6 in the 15-day gestation mouse embryo: Signaling by BMPs and TGF-betas. Dev Dyn. 2001;220:141–154
- Vrljicak P, Myburgh D, Ryan AK. Smad expression during kidney development. Am J Physiol Renal Physiol. 2004;286:F625–F633
- Goodman RH, Smolik S. CBP/p300 in cell growth, transformation and development. Genes Dev. 2000;14:1553–1577
- Shankland SJ, Eitner F, Hudkins KL, Goodpaster T, D'Agati V, Alpers CE. Differential expression of cyclin-dependent kinase inhibitors in human glomerular disease: Role in podocyte proliferation and maturation. Kidney Int. 2000;58(2):674–683
- Srivastava T, Garola RE, Singh HK. Cell-cycle regulatory proteins in the podocyte in collapsing glomerulopathy in children. Kidney Int. 2006;70(3):529–533
- Hiromura K, Haseley LA, Zhang P, et al. Podocyte expression of the CDK inhibitor p57 during development and disease. Kidney Int. 2001;60(6):2235–2246
- Nishimori S, Tanaka Y, Chiba T, et al. Smad-mediated transcription is regulated for transforming growth factor-beta 1-induced p57(kip2) proteolysis in osteoblastic cells. J Biol Chem. 2001;276:10700–10705
- Urano T, Yashiroda H, Muraoka M, et al. p57(Kip2) is degraded through the proteasome in osteoblasts stimulated to proliferation by transforming growth factor beta1. J Biol Chem. 1999;274:12197–12200
- Kim M, Nakamoto T, Nishimori S, Tanaka K, Chiba T. A new ubiquitin ligase involved in p57KIP2 proteolysis regulates osteoblast cell differentiation. EMBO Rep. 2008;9:878–884
- Scandura JM, Boccuni P, Massaque J, et al. Transforming growth factor β-induced cell cycle arrest of hematopoietic cells requires p57kip2 up-regulation. Proc Natl Acad Sci. 2004;101:15231–15236
- Chen Z, Li DQ, Tong L, et al. Targeted inhibition of p57 and p15 blocks transforming growth factor beta-inhibited proliferation of primary cultured human limbal epithelial cells. Mol Vis. 2006;23:983–994
- Sinuani I, Weissgarten J, Beberashvili I, et al. The cyclin kinase inhibitor p57kip2 regulates TGF-beta-induced compensatory tubular hypertrophy: Effect of the immunomodulator AS101. Nephrol Dial Transplant. 2009;24(8):2328–2338
- Liu B, Preisig PA. TGF-β-mediated hypertrophy in rat renal epithelial cells involves inhibiting phosphorylation by preventing activation of cdk2/cyclin E kinase. Am J Physiol. 1999;277:186–194
- Bowers EM, Yan G, Mukherjee C, et al. Virtual ligand screening of the p300/CBP histone acetyltransferase: Identification of a selective small molecule inhibitor. Chem Biol. 2010;17:471–482
- Iyer NG, Xian J, Chin SF, et al. p300 is required for orderly G1/S transition in human cancer cells. Oncogene. 2007;26:21–29
- Heemers HV, Debes JD, Tindall DJ. The role of the transcriptional coactivator p300 in prostate cancer progression. Adv Exp Med Biol. 2008;617:535–540
- Wang W, Koka V, Lan HY. Transforming growth factor-beta and Smad signalling in kidney diseases. Nephrology (Carlton). 2005;10:48–56
- Roberts AB. Molecular and cell biology of TGF-beta. Miner Electrolyte Metab. 1998;24:111–119
- Li MO, Wan YY, Flavell RA. T cell-produced transforming growth factor-beta1 controls T cell tolerance and regulates Th1-and Th17-cell differentiation. Immunity. 2007;26:579–591
- Li MO, Sanjabi S, Flavell RA. Transforming growth factor-beta controls development, homeostasis, and tolerance of T cells by regulatory T cell-dependent and -independent mechanisms. Immunity. 2006;25:455–471
- Chernajovsky Y, Adams G, Triantaphyllopoulos K, Ledda MF, Podhajcer OL. Pathogenic lymphoid cells engineered to express TGF beta 1 ameliorate disease in a collagen-induced arthritis model. Gene Ther. 1997;4:553–1539
- Subra JF, Cautain B, Xystrakis E, et al. The balance between CD45RChigh and CD45RClow CD4 T cells in rats is intrinsic to bone marrow-derived cells and is genetically controlled. J Immunol. 2001;166:2944–2952
- Fuss IJ, Boirivant M, Lacy B, Strober W. The interrelated roles of TGF-beta and IL-10 in the regulation of experimental colitis. J Immunol. 2002;168:900–908
- Hansen G, McIntire JJ, Yeung VP, et al. CD4 T helper cells engineered to produce latent TGF-beta1 reverse allergen-induced airway hyperreactivity and inflammation. J Clin Invest. 2000;105:61–70
- Afrakhte M, Moren A, Jossan S, et al. Induction of inhibitory Smad6 and Smad7 mRNA by TGF-beta family members. Biochem Biophys Res Commun. 1998;249:505–511
- Zhu HJ, Iaria J, Sizeland AM. Smad7 differentially regulates transforming growth factor betamediated signaling pathways. J Biol Chem. 1999;274:32258–32264
- Lan HY, Mu W, Tomita N, et al. Inhibition of renal fibrosis by gene transfer of inducible Smad7 using ultrasound-microbubble system in rat UUO model. J Am Soc Nephrol. 2003;14:1535–1548
- Hou CC, Wang W, Huang XR, et al. Ultrasound-microbubblemediated gene transfer of inducible Smad7 blocks transforming growth factor-beta signaling and fibrosis in rat remnant kidney. Am J Pathol. 2005;166:761–771
- Uchida K, Nitta K, Kobayashi H, et al. Localization of Smad6 and Smad7 in the rat kidney and their regulated expression in the anti-Thy-1 nephritis. Mol Cell Biol Res Commun. 2000;4:98–105
- Fukasawa H, Yamamoto T, Togawa A, et al. Down-regulation of Smad7 expression by ubiquitin-dependent degradation contributes to renal fibrosis in obstructive nephropathy in mice. Proc Natl Acad Sci USA. 2004;101:8687–8692
- Chen H, Huang XR, Wang W, et al. The protective role of Smad7 in diabetic kidney disease: Mechanism and therapeutic potential. Diabetes. 2010;60:590–601
- Wang W, Huang XR, Li AG, et al. Signaling mechanism of TGF-beta1 in prevention of renal inflammation: Role of Smad7. J Am Soc Nephrol. 2005;16:1371–1383