
Abstract
NPHS2 mutations are responsible for autosomal recessive familial steroid-resistant nephrotic syndrome (SRNS) with minor glomerular abnormalities or focal segmental glomerulosclerosis (FSGS), which is characterized by early childhood onset and rapid progression to chronic renal insufficiency. This gene mutation is also responsible for an adolescent onset form of autosomal recessive familial FSGS with heavy proteinuria. Many infants with late steroid-resistant nephrotic syndrome (late SRNS) are prone to an implicated clinical and therapeutic course. The etiopathogenesis and the long-term prognosis of late SRNS remain obscure. Considering the similar steroid resistance between late and initial SRNS, analysis of NPHS2 variation was performed in 70 sporadic Chinese infants with late SRNS and 70 controls in the present study to investigate the possible role of NPHS2 gene in late SRNS. The variation analysis revealed three polymorphisms (288C > T heterozygous in exon 2, 954T > C heterozygous and homozygous, and 1038A > G heterozygous in exon 8) in 22 out of 70 patients studied. In conclusion, NPHS2 gene mutations are not a major cause of chronic renal insufficiency caused by late SRNS in Chinese southern infants.
Introduction
Podocin is an integral membrane protein and is encoded by NPHS2, which is mapped to 1q25-31 and is exclusively expressed in glomerular podocytes. NPHS2 mutations are responsible for autosomal recessive familial steroid-resistant nephrotic syndrome (SRNS) with minor glomerular abnormalities or focal segmental glomerulosclerosis (FSGS), which is characterized by early childhood onset and rapid progression to chronic renal insufficiency. This gene mutation is also responsible for an adolescent/adult-onset form of autosomal recessive familial FSGS with heavy proteinuria without NS.Citation1 It is possible that sporadic SRNS and heavy proteinuria without NS are also due to NPHS2 gene mutations. Several groups from European and Middle Eastern countries recently showed that in 10–30% of patients with sporadic SRNS, the SRNS was caused by NPHS2 gene mutations.Citation2,Citation3 Many of them are prone to a complicated clinical and therapeutic course, however, the etiopathogenesis of late SRNS remains unclear. There is now growing evidence that genetic variation is one of the causes of SRNS. Mutations of NPHS2 gene have been reported in 26–38% of familial SRNS and 10.5–23% of sporadic cases in infants in a large European survey.Citation4–6 In Japan, most children with NS are sporadic and approximately 20% of sporadic cases show steroid resistance, which is similar to the percentage in other countries. We, therefore, isolated genomic DNA from 70 sporadic Chinese infants with late SRNS to investigate the possible role of NPHS2 gene on late SRNS.
Materials and methods
Patients and controls
Inclusion criteria were the following: (1) identified with a clinical diagnosis of SSNS; (2) responded to the initial standard steroid treatment (a daily corticosteroid (prednisone or prednisolone) at a 2 mg/kg/d (maximum 60 mg/d) for 4 weeks and were then switched to alternate day therapy) and then developed a steroid resistance later in the course of treatment; (3) from unrelated families. Seventy eligible patients (38 male and 32 female) were followed by two regional centers for pediatric diseases in China for 1–36 months (mean 1.2 years). The median onset age of the disease was 1.8 years old (3 months–3 years). Patients with congenital NS (NS occurring before 1 month of age) and secondary NS were excluded. Controls included 70 healthy infants with age and ethnic background matched and without personal or family history of a kidney disease. The study was approved by The Ethic Committee of the two regional centers for pediatric diseases. Written informed consent was obtained from each subject or their parents. The definitions and criteria were the following: NS was defined as massive proteinuria (≥40 mg/m2/h), hypoalbuminemia (serum albumin<25 g/L edema), and hyperlipidemia. A partial response was defined as disappearance of edema, an increase in the serum albumin concentration to >35 g/L, and the persistence of proteinuria of >4 mg/m2/h. Steroid-resistant was defined as the persistence of proteinuria after initial daily corticosteroid (prednisone) treatment at 2.0 mg/kg/d for at least 6 weeks. Patients were categorized as being steroid-sensitive if at least a partial response to steroids was observed. Late SRNS was defined as no response of a relapsing proteinuria to steroids in a patient who had initially responded to steroids.Citation7
Variation analysis
Genomic DNA was directly isolated from peripheral blood samples, and subjected in touchdown PCR amplification in a total volume of 25 μL, which contained 50 ng templates, 5 pmol of each primer, 2 × Taq platinum PCR master Mix 12.5 μL (Qiagen, Hilden, Germany), and ddH2O 9.5 μL. DNA was denatured at 94 °C for 5 min. Then the annealing temperature was lowered 1 °C every 2 cycles from 64 to 59 °C, followed by the annealing temperature of 58 °C for 26 cycles. At last, the extension temperature stayed constantly at 72 °C for 7 min. The sequences of the oligonucleotide primers are given in . Variation analysis was performed with direct sequencing of one strand of each exon with an automated sequencer (ABI PRISM 3735, Applied Biosystems, Foster City, CA) after cycle-sequencing reactions and analyzed using Chromas 2.23 software (Applied Biosystems, Foster City, CA). When the results were in doubts, the complementary strand was also sequenced. All variants were confirmed with sequencing of the complementary strand.
Table 1. Sequences of primers.
Statistical analysis
All the data were presented as mean ± standard deviation or as percentage. Chi-squared test was used to analyze the difference between genotypic and allelic frequencies between patients and controls by SPSS software (SPSS Inc., Chicago, IL). A p value of less than 0.05 was viewed as statistically significant.
Results
During the follow-up period, 52 patients were treated with cyclophosphamide and prednisone; eight patients achieved complete remission with prolonged steroid therapy. A large number (38 patients) again became steroid sensitive. Fifty patients maintained stable renal function: 16 remained in remission (20 received cyclophosphamide, six received cyclosporine A, and 6 received both drugs subsequently), six showed recurrence of proteinuria and six experienced partial response. Eight patients with abnormal renal function did not show response to intravenous cyclophosphamide, and one progressed to ESRD followed dialysis (no. 2). One was dead from serious infections. Renal histologic examination, which was available for 24 patients, revealed minimal change lesions (MC) in 12 patients, focal segmental glomerulosclerosis (FSGS) in six patients (nos. 2 and 4), mesangioproliferative glomerulonephritis in two patients, and four patients showed a transition from MC to FSGS in repeated biopsies. No mutation was found in all patients and controls (p < 0.05). Three sequence variations known as single nucleotide polymorphism (SNP) (288C > T heterozygous in exon 2, 954T > C heterozygous, 954T > C homozygous, and 1038A > G heterozygous in exon 8) were observed, as summarized in . To examine the significance of the variations, the same exons were also sequenced from 50 control subjects, and the results were compared with the published data and database (dbSNP and SNPper). These SNPs were found in 70 patients and 70 controls, and none of them caused an amino acid substitution. Three SNPs in NPHS2 gene were identified in Chinese infants with late SRNS, we could not find any causative NPHS2 gene mutations, suggesting that NPHS2 gene mutations are not a major cause of heavy proteinuria in Chinese infants. Although NPHS2 gene mutations were not found in this study, gene mutations of other podocyte-specific proteins, especially those related to podocyte cytoskeleton, including α-actinin-4, could be responsible for the development of SRNS or heavy proteinuria in Chinese infants. The clinical and NPHS2 variations of our patients carrying NPHS2 gene polymorphisms are summarized in .
Figure 1. Direct-sequencing exon in NPHS2. The arrows indicate (A) NPHS2 288C > T heterozygous variation in exon 2; (B) 954T > C heterozygous in exon 8; (C) 1038A > G heterozygous in exon 8.
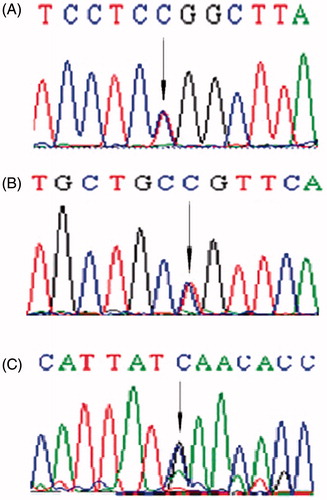
Table 2. The clinical and NPHS2 variations of 22 patients with SNPs.
Discussions
Although NPHS2 gene mutations cause autosomal recessive familial SRNS or familial heavy proteinuria without NS, it is also possible that the gene mutations cause sporadic, non-familial, SRNS, or heavy proteinuria without NS. Several groups from European and Middle Eastern countries, including Italy, France, Germany, and Israel, recently reported that in 10–30% of patients with sporadic SRNS, the SRNS was caused by NPHS2 gene mutations. FSGS with SRNS or heavy proteinuria is a major cause of chronic renal insufficiency in Japanese children. Patients with FSGS were sometimes diagnosed as having minor glomerular abnormalities or diffuse mesangial proliferation in the early stages of the disease.Citation8 The variations in the frequency and the type of NPHS2 mutations among different populations partially explain the inter-ethnic difference in the prevalence as well as the outcome of SRNS. Large-scale mutation studies of SRNS of various ethnic origins revealed that the NPHS2 mutations appear to be very uncommon in Far East Asian Countries (China, Japan, and Korea).Citation9–11 Conversely, the incidence of NPHS2 mutations was 25% in cases of sporadic SRNS in an Egyptian study,Citation12 4% in a Turkish study,Citation13 and 10.5–23% in European studies.Citation4–6 To our knowledge, there is little study on correlation between NPHS2 variations and late SRNS in Chinese infants. In the present study, three SNPs (288C > T, 954T > C, and 1038A > G) were observed in exon 2 and exon 8 of NPHS2 gene in 70 patients and 70 controls, which have been also reported in Chinese infants with SRNS.Citation10,Citation14 Although the three patients (two with FSGS) carrying compound heterozygous variants (288C > T, 954T > C, and 1038A > G) were all suffered more relapses until late resistance than others, the differences were statistically not significant. Weber et al.Citation4 reported that R138Q appeared to be associated with early onset SRNS (12 ± 3 months in 15 patients), whereas V180M and R238S were associated with late-onset SRNS (129 ± 12 months in seven patients). Schwaderer et al.Citation2 reported that uncommon variant of NPHS2 gene (IVS3 + 10insA) in late-onset patients which had no major influence on splicing mechanisms. However, none of the late SRNS patients with those variations, including R229Q which was discussed to play a role as disease modifier for glomerular disorders,Citation15 was observed in this study, similar to the data of Cho et al.Citation11
The German study revealed that six of the seven patients with sporadic SRNS and NPHS2 mutations had homozygous mutations, suggesting a common founder in these families. Children of Israeli-Arab descent with SRNS-related NPHS2 mutations all had the same homozygous R138X mutation, suggesting a common founder in these families with familial SRNS as well as sporadic cases. The Italian study also demonstrated that the 419delG mutation, the most frequently observed mutation in patients with non-familial SRNS and FSGS, was probably due to a founder effect.Citation16,Citation17 These findings might explain a higher incidence of sporadic cases in some populations.Citation3,Citation18 Despite such similarity of clinical features, we found a substantial difference in the time interval from disease onset to late resistance development. The intervals of older patients with more relapses were shorter than younger ones, which suggest that the age at onset was a highly significant factor, and older infants may have a higher tendency to develop late SRNS. In our patient group, 52 patients received cyclophosphamide and steroid therapy, whereas cyclosporine was applied in eight patients who could not go into remission with the therapeutic alliance. Thirty-two patients achieved complete remission followed therapy and only one patient developed ESRD. Data from the present and precious studies showed that complete remission was more likely to occur in patients with late than with initial resistance (22.2%).Citation19–21 So a first treatment attempt with cytostatic drugs might be justifiable. Renal biopsies showed that MCD was the commonest pathology feature in late-onset SRNS. Five patients with MCD experienced steroid sensitive relapse after becoming steroid resistant. Two patients with FSGS did not consent to cyclotate drugs. Hence we suppose that there may be a possible association between histology and response to CsA in late resistance,Citation22 which is observed in this study. Two patients showed MCD in early biopsy but FSGS in subsequent biopsy. Although complete remission was achieved in a higher proportion of patients with MCD compared with FSGS and MC, the differences were not significant.
In conclusion, three SNPs in NPHS2 gene were identified in Chinese infants with late SRNS, we could not find any causative NPHS2 gene mutations, suggesting that NPHS2 gene mutations are not a major cause of heavy proteinuria in Chinese infants. Although NPHS2 gene mutations were not found in this study, gene mutations of other podocyte-specific proteins, especially those related to podocyte cytoskeleton, including α-actinin-4, could be responsible for the development of SRNS or heavy proteinuria in Chinese infants. Further studies are required to elucidate these possibilities.
Declaration of interest
The authors report that they have no conflicts of interest. The authors alone are responsible for the content and writing of this article.
References
- Eddy AA, Symons JM. Nephrotic syndrome in childhood. Lancet. 2003;362:629–639
- Schwaderer P, Knüppel T, Konrad M, et al. Clinical course and NPHS2 analysis in patients with late steroid-resistant nephrotic syndrome. Pediatr Nephrol. 2008;23:251–256
- Kim JS, Bellew CA, Silverstein DM, et al. High incidence of initial and late steroid resistance in childhood nephrotic syndrome. Kidney Int. 2005;68:1275–1281
- Weber S, Gribouval O, Esquivel EL, et al. NPHS2 mutation analysis shows genetic heterogeneity of steroid-resistant nephrotic syndrome and low post-transplant recurrence. Kidney Int. 2004;66:571–579
- Ruf RG, Lichtenberger A, Karle SM, et al. Patients with mutations in NPHS2 (podocin) do not respond to standard steroid treatment of nephrotic syndrome. J Am Soc Nephrol. 2004;15:722–732
- Caridi G, Bertelli R, Di Duca M, et al. Broadening the spectrum of diseases related to podocin mutations. J Am Soc Nephrol. 2003;14:1278–1286
- Qi R, Li W. FK506 inhibits the mice glomerular mesangial cells proliferation by affecting the transforming growth factor β and Smads signal pathways. Ren Fail. 2014;36:589–592
- Tory K, Menyhárd DK, Woerner S, et al. Mutation-dependent recessive inheritance of NPHS2-associated steroid-resistant nephrotic syndrome. Nat Genet. 2014;46:299–304
- Ren-Qi, Zhao H. Losartan reverses glomerular podocytes injury induced by AngII via stabilizing the expression of GLUT1. Mol Biol Reports. 2013;40:6295–6301
- Yu M, Ren Q. Role of nephrin phosphorylation inducted by dexamethasone and angiotensin II in podocytes. Mol Biol Reports. 2014;41:3591–3595
- Bouchireb K, Boyer O, Gribouval O, et al. NPHS2 mutations in steroid-resistant nephrotic syndrome: A mutation update and the associated phenotypic spectrum. Hum Mutat. 2014;35:178–186
- Koziell A, Grech V, Hussain S, et al. Genotype/phenotype correlations of NPHS1 and NPHS2 mutations in nephrotic syndrome advocate a functional inter-relationship in glomerular filtration. Hum Mol Genet. 2002;11:379–388
- Bouchireb K, Boyer O, Gribouval O, et al. NPHS2 mutations in steroid-resistant nephrotic syndrome: A mutation update and the associated phenotypic spectrum. Hum Mutat. 2014;35:178–186
- Ren-Qi. Role of bad in podocyte apoptosis induced by puromycin aminonucleoside. Transplant Proc. 2013;45:569–573
- McKenzie LM, Hendrickson SL, Briggs WA, et al. NPHS2 variation in sporadic focal segmental glomerulosclerosis. J Am Soc Nephrol. 2007;18:2987–2995
- Wang G, Lai FM, Lai KB, et al. Intra-renal and urinary mRNA expression of podocyte-associated molecules for the estimation of glomerular podocyte loss. Ren Fail. 2010;32:372–379
- Komatsuda A, Wakui H, Maki N, et al. Analysis of mutations in alpha-actinin 4 and podocin genes of patients with chronic renal failure due to sporadic focal segmental glomerulosclerosis. Ren Fail. 2003;25:87–93
- Gbadegesin RA, Winn MP, Smoyer WE. Genetic testing in nephrotic syndrome—challenges and opportunities. Nat Rev Nephrol. 2013;9:179–184
- Efstratiadis G, Memmos D, Tsiaousis G, et al. Strumpell's disease in a family with hereditary focal segmental glomerulosclerosis. Ren Fail. 2006;28:351–354
- Kerti A, Csohány R, Szabó A, et al. NPHS2 p.V290M mutation in late-onset steroid-resistant nephritic syndrome. Pediatr Nephrol. 2013;28:751–757
- Bouchireb K, Boyer O, Gribouval O, et al. NPHS2 mutations in steroid-resistant nephrotic syndrome: A mutation update and the associated phenotypic spectrum. Hum Mutat. 2014;35:178–186
- Stojnev S, Pejcic M, Dolicanin Z, et al. Challenges of genomics and proteomics in nephrology. Ren Fail. 2009;31:765–772