
Abstract
Recent studies have identified that high glucose-induced renal tubular cell damage. We previously demonstrated that high glucose treatment induced oxidative stress in human renal proximal tubular epithelial cells (RPTECs), and angiotensin II type 1 (AT1) receptor blockers reduce high glucose-induced oxidative stress in RPTEC possibly via blockade of intracellular as well as extracellular AT1 receptor. However, exact roles of tumor necrosis factor (TNF)-α and AT1 receptor on high glucose-induced renal tubular function remain unclear. N-acetyl-beta-glucosaminidase (NAG), concentrations of TNF-α/angiotensin II and p22phox protein levels after high glucose treatment with or without AT1 receptor blocker or thalidomide, an inhibitor of TNF-α protein synthesis, were measured in immortalized human renal proximal tubular epithelial cells (HK2 cells). AT1 receptor knockdown was performed with AT1 receptor small interfering RNA (siRNA). High glucose treatment (30 mM) significantly increased NAG release, TNF-α/angiotensin II concentrations in cell media and p22phox protein levels compared with those in regular glucose medium (5.6 mM). Candesartan, an AT1R blocker, showed a significant reduction on high glucose-induced NAG release, TNF-α concentrations and p22phox protein levels in HK2 cells. In addition, significant decreases of NAG release, TNF-α concentrations and p22phox protein levels in HK2 cells were observed in high glucose-treated group with thalidomide. AT1R knockdown with siRNA markedly reversed high glucose, angiotensin II or TNF-α-induced p22phox protein levels in HK2 cells. TNF-α may be involved in high glucose-induced renal tubular damage in HK2 cells possibly via AT1 receptor signaling.
Introduction
Renal proximal tubular cells (RPTCs) play several important physiological roles including glucose reabsorptionCitation1,Citation2. It is now well accepted that there is a local intrarenal renin–angiotensin system (RAS),Citation3 and all components of the RAS are expressed in human, rat and murine RPTCs.Citation4–8 Angiotensin II concentrations, angiotensinogen and renin mRNA expression in the kidney are increased in early diabetes,Citation8–10 indicating that hyperglycemia or augmented intrarenal angiotensin II, or both, may be directly or indirectly responsible for renal proximal tubular hypertrophy and tubulointerstitial fibrosis in diabetes. In addition, our previous study demonstrated that angiotensin II type 1 (AT1) receptor blockers (candesartan, olmesartan or valsartan) showed a significant reduction in high glucose-induced N-acetyl-beta-glucosaminidase (NAG), 8-hydroxy-2′-deoxyguanosine and p22phox protein levels in renal proximal tubular epithelial cells (RPTECs).Citation11 Therefore, RAS in proximal tubular cells have been suggested to contribute to diabetic nephropathy.Citation12 Cytokines including tumor necrosis factor (TNF)-α involved in the inflammatory process and endothelial damage have emerged as potential markers of progressive diabetic nephropathy.Citation13 TNF-α is expressed, synthesized and released by infiltrating macrophages and by intrinsic kidney cells, namely, endothelial, mesangial, glomerular and tubular epithelial cells.Citation14,Citation15 Moreover, a recent study demonstrated that aldosterone-enhanced TNF-α expression and secretion in renal proximal tubular cells.Citation16
It is well known that a multicomponent phagocyte-type nicotinamide adenine dinucleotide phosphate [NAD(P)H] oxidase is a major source of reactive oxygen species (ROS) production in many nonspherocytic cells, including renal mesangial cells and tubular cells. TNF-α as well as angiotensin II activates increases various ROS in the mammalian cells.Citation17,Citation18 A recent study suggested that ΑΤ1 receptor blocker preferentially restored increased free radical production in diabetic kidney.Citation19 We previously demonstrated that significant increases of p22phox protein [a component of NAD(P)H oxidase] levels were observed in TNF-α-treated group in human RPTCs, and ΑΤ1 receptor blocker significantly decreased TNF-α-induced p22phox protein levels.Citation20 However, precise mechanism between AT1 receptor and TNF-α on high glucose-induced renal tubular damage is not fully elucidated. In order to investigate the roles of AT1 receptor and TNF-α on high glucose-induced cell damage in immortalized human renal proximal tubular epithelial cells (HK2 cells), we examined the effects of candesartan or thalidomide, an inhibitor of TNF-α protein synthesis on high glucose-induced NAG release, TNF-α levels and the p22phox protein levels. In addition, we utilized AT1R small interfering RNA (siRNA) to further explore the roles of AT1R on high glucose-induced cell damage.
Materials and methods
Cell preparation
HK2 cell line derived from normal kidney was purchased from ATCC (Manassas, VA). These cells were cultured in growth medium containing DMEM/F-12HAM, 10% FBS, penicillin (100 units/mL), streptomycin (100 µg/mL). HK2 cells between passages 8 and 10 were used in this study. HK2 cells were made quiescent by incubation in serum-free DMEM medium with 5.6 mM glucose for 24–48 h. All the following experiments were performed in quiescent HK2 cells.
High glucose and AT1 receptor blockers/TNF-α inhibitor treatment
HK2 cells were plated on 96-well plate for NAG studies and 6-well plate for Western blotting. On the experimental day, the media in plates or chambers were washed and replaced with fresh DMEM/F-12 media without FBS. Various doses of candesartan (Takeda Pharmaceutical Co., Ltd., Osaka, Japan) and/or TNF-α inhibitor thalidomide (Enzo Life Science, Plymouth Meeting, PA) were added to the media alone or in combination with glucose (30 mM as high glucose treatment) at 24 h prior to the various measurements.
NAG assay
NAG as a marker of lysosomal damage was assessed using the following modified commercial procedure (PPR Diagnostics, London, UK) as previously described.Citation21
TNF-α and angiotensin II enzyme immunoassay
TNF-α and angiotensin II concentrations in cell media were assayed with the commercially available ELISA kits (Human TNF-α ELISA kit, Thermo Fisher Scientific Inc., Yokohama, Japan, and Human/Rat Angiotensin II ELISA, BioVendor, Candler, NC) following the manufacturer’s protocol.
Western blotting
The whole cells were collected and mixed in electrophoresis sample buffer and were incubated at 100 °C for 5 min and electrophoresed by SDS-PAGE. After electrophoresis, separated proteins were transferred to polyvinylidene difluoride membranes followed by blocking with 5% skim milk in TBS solution (0.15 M NaCl/20 mM Tris-HCl, pH 7.6) for 60 min at room temperature. The membrane was incubated with anti-p22phox (sc-20781; Santa Cruz Biotechnology Inc., Santa Cruz, CA) or anti-α tubulin (sc-5286; Santa Cruz Biotechnology Inc.) at a final concentration of 1:200–1000 for anti- anti-p22phox or 1:10,000 for anti-α tubulin in 5% skim milk in 1 × TBS solution overnight at 4°C. Subsequently, the membranes were washed for 5 min (× 5) and incubated with biotinylated F (ab')2 fragment of polyclonal swine anti-rabbit immunoglobulin for anti-p22phox or polyclonal rabbit anti-mouse immunoglobulin for anti-α tubulin (Dako A/S, Glostrup, Denmark) as second antibodies for 30 min at room temperature and washed. The membranes were incubated with streptavidin horseradish peroxidase conjugate (Chemicon, Boronia, VIC, Australia) for 20 min at room temperature and washed in TBS + Tween 20 for 5 min (×3) and TBS for 10 min (× 2). The bands were visualized by Chemi-Lumi One Super (Nakalai Tesque Inc., Kyoto, Japan) and quantitated using a computer-assisted image analyzer (Kodak 1D Image Analysis Software, Scientific Imaging Systems, New Haven, CT).
Gene silencing by siRNA
AT1 receptor siRNA was purchased from Ambion Inc. (Silencer Select pre-designed: ID# s135416). In addition, Silencer select negative control #1 siRNA (Ambion Inc., Carlsbad, CA) was utilized as a negative control siRNA. The transfection was conducted using Lipofectamine 2000 transfection reagent (Invitrogen, Carlsbad, CA). Transfection efficiency was assessed by measuring AT1 receptor protein expression by Western blot analysis. The AT1 receptor antibody was purchased from Santa Cruz Biotechnology Inc. (sc-1173).
Data analysis
All results are expressed as mean ± SEM. All analyses were performed with the program SPSS ver.20 statistical package (IBM, Armonk, NY). Data were analyzed by Mann–Whitney U test or ANOVA followed by Fisher’s protected least squares difference test. A level of p < 0.05 was considered to be statistically significant.
Results
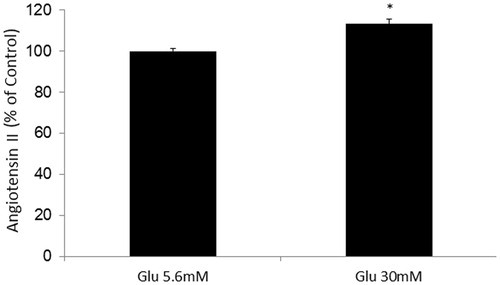
Effects of AT1 receptor blocker or thalidomide treatment on high glucose-induced NAG release in HK2 cells
Glucose treatment (30 mM) increased NAG release in HK2 cells compared with glucose treatment (5.6 mM). Candesartan (1 × 10–7 mol/L) or thalidomide (4 × 10–5 mol/L) treatment significantly decreased high glucose-induced NAG release in HK2 cells ().
Figure 1. Effects of AT1 receptor blocker or thalidomide treatment on high glucose-induced NAG release in HK2 cells. *Denotes significant alterations at p < 0.05, when compared to control group (5.6 mM glucose) and +denotes significant alterations at p < 0.05, when compared to high glucose treatment (30 mM) group. Notes: Glu, glucose; HG, high glucose treatment (30 mM); Cand, candesartan; and Thal, thalidomide.
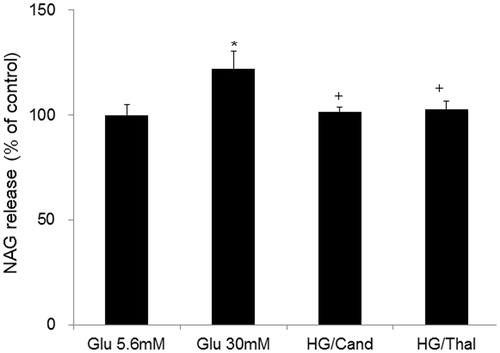
Effects of AT1 receptor blocker or thalidomide treatment on high glucose-induced p22phox protein levels in HK2 cells
Glucose treatment (30 mM) showed significant increases of p22phox protein levels in HK2 cells compared with glucose treatment (5.6 mM) group. Candesartan (1 × 10–7 mol/L) or thalidomide (4 × 10–5 mol/L) treatment significantly reduced high glucose-induced p22phox protein levels ().
Figure 2. Effects of AT1 receptor blocker or thalidomide treatment on high glucose-induced p22phox protein levels in HK2 cells. A representative example of Western blotting was shown above. *Denotes significant alterations at p < 0.05 when compared to control group (5.6 mM glucose). +Denotes significant alterations at p < 0.05, when compared to high glucose treatment (30 mM) group. Notes: Glu, glucose; HG, high glucose treatment (30 mM); Cand, candesartan; and Thal, thalidomide.
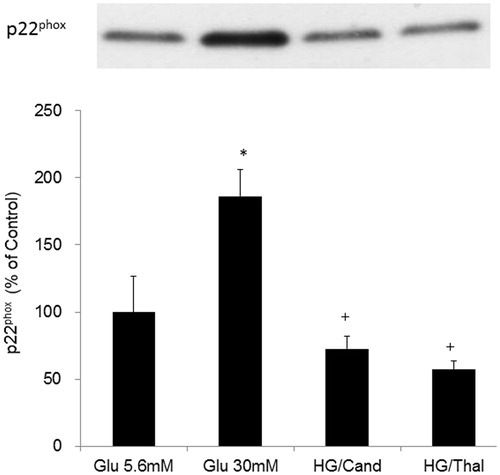
Effects of high glucose on TNFα concentrations in the cell media
Glucose treatment (30 mM) significantly increased TNF-α concentrations in cell media. Candesartan (1 × 10–7 mol/L) or thalidomide (4 × 10–5 mol/L) treatment significantly decreased high glucose-induced TNF-α concentrations in cell media ().
Figure 3. Effects of high glucose on TNF-α concentrations in the cell media in HK2 cells. *Denotes significant alterations at p < 0.05 when compared to control group (5.6 mM glucose) and +Denotes significant alterations at p < 0.05 when compared to high glucose treatment (30 mM) group. Notes: Glu, glucose; HG, high glucose treatment (30 mM); Cand, candesartan; and Thal, thalidomide.
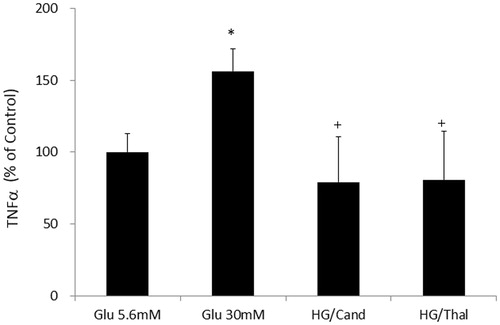
Effects of high glucose on angiotensin II concentrations in the cell media
Glucose treatment (30 mM) induced slight but significant increase of angiotensin II concentrations in cell media ().
Figure 4. Effects of high glucose on angiotensin II concentrations in the cell media in HK2 cells. *Denotes significant alterations at p < 0.05 when compared to control group (5.6 mM glucose). Glu, glucose.
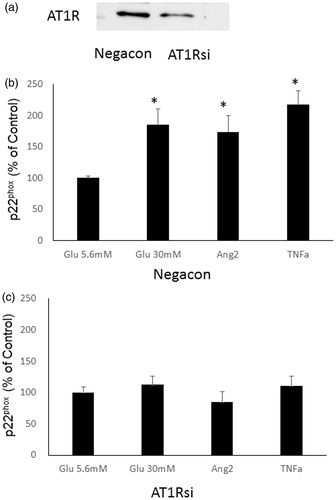
Effects of AT1 siRNA treatment on high glucose, angiotensin II- or TNF-α-induced p22phox protein in HK2 cells
AT1 siRNA significantly reduced AT1 receptor protein compared with negative control siRNA treatment in HK2 cells (). Glucose treatment (30 mM), angiotensin II (1 × 10–7 mol/L) or TNF-α (10 ng/mL) significantly induced p22phox protein in HK2 cells with negative control siRNA (). In contrast, the increment of p22phox protein by high glucose, angiotensin II or TNF-α was markedly attenuated by AT1 siRNA ().
Figure 5. Effects of AT1 siRNA treatment on high glucose, angiotensin II- or TNF-α-induced p22phox protein in HK2 cells. (a) A representative example of knockdown of the AT1R protein using siRNA. Effects of glucose treatment (30 mM), angiotensin II- or TNFα-induced p22phox protein in HK2 cells with negative control siRNA (b) and with AT1 siRNA (c). *Denotes significant alterations at p < 0.05 when compared to control group (5.6 mM glucose). Glu, glucose and Ang, angiotensin.
Discussion
In this study, high glucose significantly increased NAG release, angiotensin II and TNF-α concentrations. These results are agreement with that high glucose increased angiotensinogen, angiotensin-converting enzyme and AT1R mRNA levels and angiotensin II concentrations in the renal tubular cell culture medium.Citation22 Moreover, a recent study investigated the role of TNF-α/TNF receptors in the development of early and late diabetic nephropathy,Citation23 suggesting that angiotensin II/AT1R and TNF-α may be involved in high glucose-induced renal tubular damage.
High glucose (25 mM) evoked ROS generation and p38 MAPK phosphorylation as well as stimulated immunoreactive rat angiotensin II secretion and angiotensin II mRNA expression in rat immortalized renal proximal tubular cells.Citation24 While, TNF-α markedly induced NAD(P)H oxidase-derived ROS generation in rat embryonic-heart derived H9c2 cells.Citation25 In addition, TNF can induce activation of NAD(P)H oxidase leading to enhanced oxidative stress and decreased bioavailability of NO.Citation26 We also demonstrated that high glucose and TNF-α markedly induced p22phox protein levels,Citation11,Citation20 suggesting that angiotensin II/AT1R and TNF-α may be involved in high glucose-induced ROS generation in RPTECs.
The mechanisms on interaction between TNF-α and RAS in high glucose-induced renal tubular damage were investigated. Both in vitro and in vivo studies have demonstrated a cross-talk between angiotensin II and TNF.Citation27,Citation28 Angiotensin II can act as a potent proinflammatory agent and stimulate the production of chemokines such as MCP-1, and PICs, such as TNF, interleukin (IL)-6 and IL-1β in the brain.Citation29,Citation30 Olmesartan, an AT1 receptor blocker, has been investigated and found to inhibit the expression of TNF-α in vascular smooth muscle cells.Citation31 In addition, olmesartan resulted in decreased levels of TNF-α in male Wistar albino rats,Citation32 suggesting that the stimulatory role of AT1R on TNF-α production. In this study, the siRNA-mediated AT1 receptor knockdown abolished high glucose, angiotensin II and TNF-α-induced p22phox protein levels in HK2 cells, supporting a role for AT1R in the development of ROS generation. These results are keeping with the report that the AT1 receptor blocker decreased the TNF-α-dependent increase of 8-isoprostane, phosphorylation of I kappa B alpha and cell death in cultured human umbilical vein endothelial cells.Citation33 Taken together, the results that high glucose treatment induced slight but significant increase of angiotensin II concentrations and thalidomide treatment significantly reduced high glucose-induced p22phox protein levels in HK2 cells, high glucose may induce angiotensin II, which stimulates TNF-α through AT1 receptor, and the increased TNF-α induces NAD(P)H oxidase, which causes the renal tubular cells damage. Further studies using other kidney injury markers such as KIM-1 or NGAL are required to clarify the mechanisms on interaction between TNF-α and RAS in high glucose-induced renal tubular damage.
In conclusion, high glucose-induced increases in NAG and p22phox protein were accompanied by increased secretion of angiotensin II and TNF-α in HK2 cells. The increment of p22phox protein by high glucose, angiotensin II or TNF-α was markedly attenuated by AT1 siRNA. This work shows that AT1 receptor blockade may be useful in preventing TNF-α-induced additional renal tubular damage in diabetic nephropathy.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Mather A, Pollock C. Glucose handling by the kidney. Kidney Int Suppl. 2011;(120):S1–S6
- Vallon V, Platt KA, Cunard R, et al. SGLT2 mediates glucose reabsorption in the early proximal tubule. J Am Soc Nephrol. 2011;22(1):104–112
- Dzau VJ, Paul M, Nakamura N, Pratt RE, Ingelfinger JR. Role of molecular biology in hypertension research. State of the art lecture. Hypertension. 1989;13(6 Pt 2):731–740
- Ingelfinger JR, Zuo WM, Fon EA, Ellison KE, Dzau VJ. In situ hybridization evidence for angiotensinogen messenger RNA in the rat proximal tubule. An hypothesis for the intrarenal renin angiotensin system. J Clin Invest. 1990;85(2):417–423
- Wolf G, Neilson EG. Effects of angiotensin II on proximal tubular cells stably transfected with the c-mas oncogene. Am J Physiol. 1992;263(5 Pt 2):F931–F938
- Loghman-Adham M, Rohrwasser A, Helin C, et al. A conditionally immortalized cell line from murine proximal tubule. Kidney Int. 1997;52(1):229–239
- Burns KD, Inagami T, Harris RC. Cloning of a rabbit kidney cortex AT1 angiotensin II receptor that is present in proximal tubule epithelium. Am J Physiol. 1993;264(4 Pt 2):F645–F654
- Lai KN, Leung JC, Lai KB, To WY, Yeung VT, Lai FM. Gene expression of the renin-angiotensin system in human kidney. J Hypertens. 1998;16(1):91–102
- Andersen S, Tarnow L, Rossing P, Hansen BV, Parving HH. Renoprotective effects of angiotensin II receptor blockade in type 1 diabetic patients with diabetic nephropathy. Kidney Int. 2000;57(2):601–606
- Zimpelmann J, Kumar D, Levine DZ, et al. Early diabetes mellitus stimulates proximal tubule renin mRNA expression in the rat. Kidney Int. 2000;58(6):2320–2330
- Takao T, Horino T, Kagawa T, et al. Possible involvement of intracellular angiotensin II receptor in high-glucose-induced damage in renal proximal tubular cells. J Nephrol. 2011;24(2):218–224
- Zhang SL, To C, Chen X, et al. Essential role(s) of the intrarenal renin-angiotensin system in transforming growth factor-beta1 gene expression and induction of hypertrophy of rat kidney proximal tubular cells in high glucose. J Am Soc Nephrol. 2002;13(2):302–312
- Moresco RN, Sangoi MB, De Carvalho JA, Tatsch E, Bochi GV. Diabetic nephropathy: Traditional to proteomic markers. Clin Chim Acta. 2013;421:17–30
- Ortiz A, Egido J. Is there a role for specific anti-TNF strategies in glomerular diseases? Nephrol Dial Transpl. 1995;10(3):309–311
- Zhang B, Ramesh G, Norbury CC, Reeves WB. Cisplatin-induced nephrotoxicity is mediated by tumor necrosis factor-alpha produced by renal parenchymal cells. Kidney Int. 2007;72(1):37–44
- Kitada K, Nakano D, Hitomi H, et al. Aldosterone induces p21-regulated apoptosis via increased synthesis and secretion of tumour necrosis factor-alpha in human proximal tubular cells. Clin Exp Pharmacol Physiol. 2012;39(10):858–863
- Sachse A, Wolf G. Angiotensin II-induced reactive oxygen species and the kidney. J Am Soc Nephrol. 2007;18(9):2439–2446
- Hennet T, Richter C, Peterhans E. Tumour necrosis factor-alpha induces superoxide anion generation in mitochondria of L929 cells. Biochem J. 1993;289(Pt 2):587–592
- Sonta T, Inoguchi T, Matsumoto S, et al. In vivo imaging of oxidative stress in the kidney of diabetic mice and its normalization by angiotensin II type 1 receptor blocker. Biochem Biophys Res Commun. 2005;330(2):415–422
- Kagawa T, Takao T, Horino T, et al. Angiotensin II receptor blocker inhibits tumour necrosis factor-alpha-induced cell damage in human renal proximal tubular epithelial cells. Nephrology (Carlton). 2008;13(4):309–315
- Takao T, Horino T, Kagawa T, et al. Effects of angiotensin II type 1 receptor blocker on albumin-induced cell damage in human renal proximal tubular epithelial cells. Am J Nephrol. 2009;29(2):102–108
- Zhou L, Xue H, Yuan P, et al. Angiotensin AT1 receptor activation mediates high glucose-induced epithelial-mesenchymal transition in renal proximal tubular cells. Clin Exp Pharmacol Physiol. 2010;37(9):e152–e157
- Speeckaert MM, Speeckaert R, Laute M, Vanholder R, Delanghe JR. Tumor necrosis factor receptors: Biology and therapeutic potential in kidney diseases. Am J Nephrol. 2012;36(3):261–270
- Hsieh TJ, Zhang SL, Filep JG, Tang SS, Ingelfinger JR, Chan JS. High glucose stimulates angiotensinogen gene expression via reactive oxygen species generation in rat kidney proximal tubular cells. Endocrinology. 2002;143(8):2975–2985
- Yang CM, Lee IT, Hsu RC, Chi PL, Hsiao LD. NADPH oxidase/ROS-dependent PYK2 activation is involved in TNF-alpha-induced matrix metalloproteinase-9 expression in rat heart-derived H9c2 cells. Toxicol Appl Pharmacol. 2013;272(2):431–442
- Zhang C, Hein TW, Wang W, Ren Y, Shipley RD, Kuo L. Activation of JNK and xanthine oxidase by TNF-alpha impairs nitric oxide-mediated dilation of coronary arterioles. J Mol Cell Cardiol. 2006;40(2):247–257
- Arenas IA, Xu Y, Lopez-Jaramillo P, Davidge ST. Angiotensin II-induced MMP-2 release from endothelial cells is mediated by TNF-alpha. Am J Physiol Cell Physiol. 2004;286(4):C779–C784
- Sasamura H, Nakazato Y, Hayashida T, Kitamura Y, Hayashi M, Saruta T. Regulation of vascular type 1 angiotensin receptors by cytokines. Hypertension. 1997;30(1 Pt 1):35–41
- Shi P, Diez-Freire C, Jun JY, et al. Brain microglial cytokines in neurogenic hypertension. Hypertension. 2010;56(2):297–303
- Lu Y, Chen J, Yin X, Zhao H. Angiotensin II receptor 1 involved in the central pressor response induced by interleukin-1 beta in the paraventricular nucleus. Neurol Res. 2009;31(4):420–424
- Li Z, Chen XD, Ni SK, Li JW, Lin MS. Olmesartan inhibits the expression of monocyte chemoattractant protein-1 and tumor necrosis factor-alpha and improves vascular remodeling after vascular injury in mouse. Chin J Traumatol. 2004;7(1):56–61
- Araujo AA, Lopes de Souza G, Souza TO, et al. Olmesartan decreases IL-1beta and TNF-alpha levels; downregulates MMP-2, MMP-9, COX-2, and RANKL; and upregulates OPG in experimental periodontitis. Naunyn Schmiedeberg's Arch Pharmacol. 2013;386(10):875–884
- Kataoka H, Murakami R, Numaguchi Y, Okumura K, Murohara T. Angiotensin II type 1 receptor blockers prevent tumor necrosis factor-alpha-mediated endothelial nitric oxide synthase reduction and superoxide production in human umbilical vein endothelial cells. Eur J Pharmacol. 2010;636(1–3):36–41