
Abstract
Background: Overwhelming evidences suggest epithelial to mesenchymal transition (EMT) of tubular epithelial cells contributes to renal fibrosis of chronic kidney disease (CKD). Connective tissue growth factor (CTGF) plays an important role in the pathogenesis of EMT. However, the molecular mechanisms that regulate cell behaviors are not clear. Objective: The purpose of this study was to investigate whether CTGF induces EMT via activation of canonical Wnt signaling in renal tubular epithelial cells. Methods: Human renal proximal tubular epithelial cells (HK-2) were divided into control group, CTGF group and dickkopf (Dkk)-1 plus CTGF group. We assessed the biological changes of canonical Wnt signaling, including phosphorylation of low-density lipoprotein receptor-related protein (LRP6) and glycogen synthase kinase-3β (GSK-3β) and accumulation and nuclear localization of β-catenin. Meanwhile, morphological changes of the three groups were observed and tubular EMT was further confirmed by detecting the expression of α-SMA and E-cadherin. Results: The phosphorylation levels of LRP6 and GSK-3β and the expression of β-catenin in CTGF group were higher than control group (p < 0.05). The accumulation and nuclear localization of β-catenin was induced in CTGF group. Meanwhile, CTGF group cells showed a mesenchymal morphological phenotype and exhibited increased expressions of E-cadherin and decreased expressions of α-SMA compared to control group (p < 0.05), suggesting tubular EMT. Furthermore, we also found that Dkk-1 blocked the above CTGF’s effects by binding with LRP6. Conclusion: CTGF induces EMT via activation of canonical Wnt signaling in HK-2 cells in vitro, which may play an important role in the renal fibrosis of CKD.
Introduction
A common pathologic change of all forms of chronic kidney disease (CKD) is renal fibrosis, although a huge range of diseases, such as glomerulonephritis; metabolic diseases, including diabetes mellitus and atherosclerosis; and interstitial nephritis can be major causes of CKD.Citation1 Renal fibrosis includes glomerulosclerosis and tubulointerstitial fibrosis (TIF). However, TIF has been considered as the most reliable indication of an irreversible loss of renal function. Recent evidences have showed that injured renal tubular epithelial cells induce accumulation of fibroblasts or myofibroblasts and the deposition of abundant extracellular matrix (ECM) through epithelial to mesenchymal transition (EMT), ultimately leading to TIF and the progression of CKD.Citation2
Connective tissue growth factor (CTGF), a number of the CCN (Cyr61/CTGF/Nov) family of proteins, regulates cell adhesion, migration, growth, chemotaxis, and the synthesis and accumulation of ECM during pathologic conditions.Citation3 Mnay evidences stated that expression of CTGF increases in a variety of progressive CKDs, and CTGF is strongly involved in the pathogenesis of fibrotic disorders.Citation4,Citation5 It was reported that CTGF represents a mediator of tubular EMT by interacting with downstream protein of the actions of advanced glycation end products or transforming growth factor-1, inducing the expression of ILK protein and effecting on the activity of BMP-7 and HGF.Citation6–8 However, a detailed understanding of the complex signal pathway has not been well understood.
Recently, a number of reports have highlighted the role of Wnt signaling in the pathogenesis of EMT.Citation9–12 For example, lymphoid enhancer factor-1 promotes EMT when its activity is regulated by Wnt signaling.Citation9 Importantly, it was reported that in Xenopus laevis embryos and in mesangial cells, CTGF modulated Wnt signaling by binding to lipoprotein receptor-related protein (LRP6).Citation13,Citation14 On the basis of the previous studies, we hypothesized that canonical Wnt signaling pathway can be activated by CTGF, and tubular EMT can be induced by activation of canonical Wnt signaling pathway in human renal proximal tubular epithelial cells (HK-2) in vitro. In this study, Dickkopf (Dkk)-1, the endogenous LRP6 receptor antagonist, was used to investigate the mechanism of CTGF-induced EMT, and we examined whether CTGF-LRP6 interactions activate canonical Wnt signaling pathway, which contributes indirectly to EMT in tubular epithelial cells in vitro. Simultaneously, we investigated the underlying mechanisms, which may provide new insight into pathogenesis and treatment of tubular EMT and renal tubular interstitial fibrosis of CKD.
Materials and methods
Cell culture
HK-2 cells used for experiments, human proximal tubular epithelial cells immortalized by transduction with human papillomavirus 16 E6/E7 genes, was purchased from China Center for Type Culture Collection (Wuhan, China). In our studies, HK-2 cells were cultured in Dulbecco’s Modified Eagle Medium (Gibco, Gaithersburg, MD) supplemented with 10% heat-inactivated fetal calf serum (Gibco), 1000 mg/L glucose, 100 U/mL penicillin, 100 U/mL streptomycin, 2 mmol/L supplemental glutamine, and 10 ng/mL epidermal growth factor at 37 °C in an atmosphere of 5% CO2. The culture medium was changed every 24–48 h until the cells reached approximately 70–80% confluence, at which point cells were passaged with trypsin-contained ethylene diamine tetraacetic acid (Gibco) on six-well laminin-coated culture plates (Henry Marroum & Sons, Amman, Jordan), where each well had 1.5 × 105 cells and subjected to incubate for 24 h in serum-free medium, and all treatments were carried out in serum-free conditions. HK-2 cells were divided into three goups: control group, CTGF group and Dkk-1 plus CTGF group. In CTGF group, HK-2 cells were stimulated by 50 ng/mL recombinant human CTGF (rhCTGF; PeproTech, Rocky Hill, NJ) for 0.5, 6 and 24 h. In Dkk-1 plus CTGF group, cells were pre-treated with 20, 200, 300 ng/mL of recombinant human Dkk-1 (rhDkk-1; PeproTech) for 1 h prior to stimulation of rhCTGF. CTGF group was subdivided into three subgroups: 50 ng/mL rhCTGF for 0.5 h group, 50 ng/mL rhCTGF for 6 h group, 50 ng/mL rhCTGF for 24 h group. Dkk-1 group was also subdivided into three subgroups: 20 ng/mL, 200 ng/mL and 300 ng/mL groups. The morphological changes of HK-2 cells cultured under different conditions were analyzed by inverted phase contrast microscope (Nikon, Tokyo, Japan).
Western blotting analysis
Total protein of HK-2 cells was extracted by total protein extraction kit (DBI Bioscience, Newark, DE), and the protein concentration was quantified using the Bradford method (Thermo Scientific, Waltham, MA). Whole-cell lysates were subjected to 8–10% SDS-PAGE and transferred onto polyvinylidene fluoride membranes (Millipore, Temecula, CA). The membranes were blocked with 5% skim milk for 1 h at room temperature. After blocking, the membranes were incubated for 16 h at 4 °C with primary antibodies to p-LRP6 (Ser1490; 1:1000, Cell Signaling Technologies, Danvers, MA), total LRP6 (1:1000, Cell Signaling Technologies), p-glycogen synthase kinase-3β (GSK-3β; Ser 9; 1:1000, Cell Signaling Technologies), total GSK-3β (1:1000, Cell Signaling Technologies), β-catenin (1:1000, Cell Signaling Technologies), α-SMA (1:1000; Abcam, Burlingame, CA), E-cadherin (1:1000, Santa Cruz Biotechnology, Santa Cruz, CA) and β-actin (1:1000, Bioworld Technology, St. Louis Park, MN) and then incubated with horseradish peroxidase-conjugated secondary antibody for 1.5 h at room temperature. The protein was detected using the electrochemiluminescence detection kit (Millipore) via enhanced chemiluminescence, which was captured on X-ray film. Band intensity was quantified from scanned membrane images by ImageJ software 1.42q (National Institutes of Health, Bethesda, MD).
Real-time polymerase chain reaction
To measure the mRNA levels of α-SMA and E-cadherin, total RNA from HK-2 cells was extracted using Trizol reagent (Invitrogen, Carlsbad, CA) and reverse transcribed using PrimeScript RT reagent Kit (TaKaRa Biotechnology, Dalian, China) according to the manufacturer’s instructions. Real-time polymerase chain reaction (PCR) was performed using the SYBR Green real-time PCR Master Mix (Roche, Pudong, China) routinely by a sequence detection system (Eppendorf, Hamburg, Germany) for quantification of mRNA expression level. Glyceraldehyde phosphate dehydrogenase (GAPDH) was employed as internal reference for gene evaluation. The following gene-specific primers were used: E-cadherin, forward 5′-AAATCTGAAAGCGGCTGATACTG-3′, reverse 5′-CGGAACCGCTTCCTTCATAG-3′; α-SMA, forward 5′-GACAATGGCTCTGGGCTCTGTAA-3′, reverse 5′-ATGCCATGTTCTATCGGGTACTTCA-3′; GAPDH, forward 5′-CCTCAAGATCATCAGCAAT-3′, reverse 5′-CCATCCACAGTCTTCTGGGT-3′. All primers used in the experiments were designed and generated by Beijing Genomics Institute. The threshold cycle (Ct) was determined for each sample and the relative expression quantity of mRNA was calculated by 2−ΔΔCt. PCR of each sample was performed in triplicate to obtain a mean value.
Immunofluorescence staining
HK-2 cells grown on coverslips were fixed in 4% formaldehyde for 30 minutes, permeabilized in 0.1%Triton X-100 for 10 minutes and blocked in 1:20 normal sheep serum for 30 minutes at 37 °C to block nonspecific binding. After washing three times with PBS, cells were incubated with primary antibodies used for immunofluorescence: E-cadherin (Santa Cruz Biotechnology), α-SMA (Abcam) and β-catenin (Cell Signaling), followed by three washes and incubation with fluorescein-conjugated affinipure goat anti-rabbit IgG (ZSGB-Bio, Beijing, China) for 2 h at room temperature. 4,6-Diamidino-2-phenylindole dihydrochloride hydrate was used for nuclear staining. After mounting, cells were visualized under a Leica TCD laser confocal microscope (Leitz, Wetzlar, Germany) using a 400× objective.
Statistical analysis
Statistical analyses were carried out by SPSS software, version 16.0 (Chicago, IL). Mean ± standard deviation was applied to express data. Differences of quantitative parameters among different groups were assessed by one-way analysis of variance followed by the Student–Newman–Keuls tests. A value of p < 0.05 was considered to manifest significant.
Results
Modulation of canonical Wnt signaling pathway components under the influence of CTGF in HK-2 cells in vitro
We investigated how CTGF altered Wnt/β-catenin signaling. Our studies found that Wnt co-receptor LRP6 was expressed in HK-2 cell in vitro (). The phosphorylation of LRP6 and GSK-3β and the expression of β-catenin showed by western blotting increased gradually as rhCTGF-treated time extends (p < 0.05; ). Immunofluorescence staining showed nuclear accumulation of β-catenin (, middle). Western blotting () and immunofluorescence staining (, right) further showed that pretreatment of cells with the Dkk-1, Wnt signaling antagonist, blocked phosphorylation of GSK3-β, and rhCTGF-mediated accumulation and nuclear localization of β-catenin. These findings indicated that HK-2 cells responded to rhCTGF via combining with LRP6, suppressing GSK-3β and activating Wnt/β-catenin signaling. All of the above were consistent with activation of CTGF-induced canonical Wnt signaling.
Figure 1. CTGF activates canonical Wnt signaling in HK-2 cells. The cells were cultured with recombinant human CTGF (50 ng/mL) for various duration when grown to 80% confluency and serum starved for 24 h. (A) All canonical Wnt signaling pathway components were measured by western blotting. (B) Quantitative analysis of protein expression as measured by western blotting. *p < 0.05 versus control group, **p < 0.01 versus control group. All results are representative of at least three individual experiments.
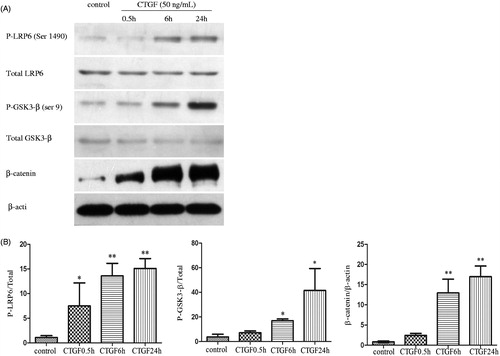
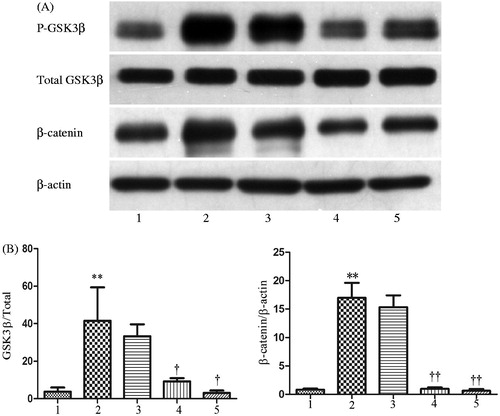
Figure 2. Effects of CTGF and Dkk-1 on β-catenin in HK-2 cells were showed by immunofluorescence staining (original magnification ×400). Left: normal control; middle: HK-2 cells were treated with CTGF (50 ng/mL) for 24 h; right: HK-2 cells were pretreated with Dkk-1 (200 ng/mL) for 1 h and then treated with CTGF (50 ng/mL) for 24 h. β-catenin expression is shown in green and cell nucleuses are shown in blue.
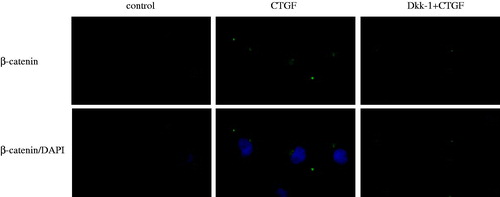
Figure 3. Dkk-1 blocked CTGF-mediated activation of canonical Wnt signaling in HK-2 cells. The cells were pretreated with various concentrations of Dkk-1 for 1 h prior to stimulation with rhCTGF (50 ng/mL). Lane 1: control group; lane 2: 50 ng/mL rhCTGF for 24 h group; lane 3: rhDkk-1 (20 ng/mL) pretreated cells prior to rhCTGF; lane 4: rhDkk-1 (200 ng/mL) pretreated cells prior to rhCTGF; lane 5: rhDkk-1 (300 ng/mL) pretreated cells prior to rhCTGF. (A) Phosphorylation of GSK3-β and protein expression of β-catenin were measured by western blotting. (B) Quantitative analysis of protein expression as measured by western blotting. **p < 0.01 versus control group, †p < 0.05 versus 50 ng/mL rhCTGF for 24 h group, ††p < 0.01 versus 50 ng/mL rhCTGF for 24 h group. All results are representative of at least three individual experiments. All results are representative of at least three individual experiments.
Morphological and phenotypic changes of HK-2 cells induced by CTGF as a result of activation of canonical Wnt signaling
As shown in inverted phase contrast microscopy, normal cultured HK-2 cells in control group produced a sub-confluent with typical epithelial cobblestone morphology (, A1). After being exposed to 50 ng/mL rhCTGF for 24 h, the cells became more elongated, less adhered and lost their apical-to-basal polarity (, A2); however, the morphological changes inducted by rhCTGF were inhibited by pretreatment of 200 ng/mL rhDkk-1, with cells retaining epithelial polarity and a cobblestone growth pattern (, A3).
Figure 4. Morphological and phenotypic changes of HK-2 cells cultured under different conditions were determined by immunofluorescence staining under a inverted microscope. The cellular morphology (A1–A3; original magnification ×200) and the expression of α-SMA (B1–B3; original magnification ×400) and E-cadherin (C1–C3; original magnification ×400) are shown, respectively. Left: normal control and middle: HK-2 cells were treated with rhCTGF (50 ng/mL) for 24 h; right: HK-2 cells were pretreated with rhDkk-1 (200 ng/mL) for 1 h and then treated with rhCTGF (50 ng/mL) for 24 h. E-cadherin and α-SMA expression is shown in green. Cells were counterstained with 4,6-diamidino-2-phenylindole dihydrochloride hydrate (DIPA) to demonstrate the nuclei (blue).
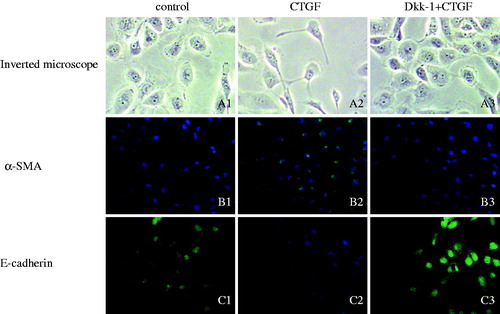
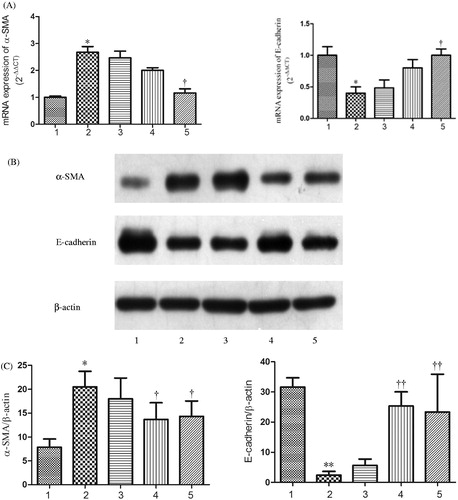
Since α-SMA and E-cadherin were the typical markers of tubular EMT, mRNA and protein levels of α-SMA and E-cadherin were detected by real-time PCR () and western blotting (). Real-time PCR demonstrated that the mRNA expression of E-cadherin was reduced and α-SMA was aggrandized gradually by the induction of rhCTGF from 0.5 h to 24 h compared with the control group, which were greatly improved by rhDkk-1 (). The protein expression level of E-cadherin and α-SMA detected by western blotting was consistent with real-time PCR analysis (). By immunofluorescence staining, HK-2 cells of control group exhibited a normal epithelial staining of E-cadherin (, C1). HK-2 cells treated with rhCTGF for 24 h had lost quite a bit positive staining of E-cadherin (, C2). By the pretreatment of rhDkk-1 (200 ng/mL), the positive staining of E-cadherin in HK-2 cells was significantly retrieved (, C3). Immunofluorescence staining also demonstrated that cells exposed to rhCTGF for 24 h had increased expression of α-SMA versus control group, which were not observed in Dkk-1 plus CTGF group cells (, B1 and B2).
Figure 5. CTGF activates tubular EMT in HK-2 cells. The cells were cultured with rhCTGF (50 ng/mL) for various duration, when grown to 80% confluency and serum starved for 24 h. (A) The mRNA expressions of α-SMA and E-cadherin were measured by real-time PCR. (B) The protein expression of α-SMA and E-cadherin were measured by western blotting. (C) Quantitative analysis of protein expression as measured by western blotting. *p < 0.05 versus control group and **p < 0.01 versus control group. All results are representative of at least three individual experiments.
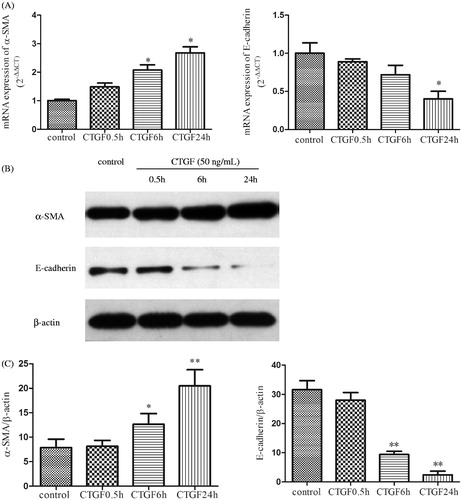
Figure 6. Dkk-1 suppressed CTGF-mediated EMT in HK-2 cells. The cells were pretreated with various concentrations of Dkk-1 for 1 h prior to stimulation with rhCTGF (50 ng/mL). Lane 1: control group; Lane 2: 50 ng/mL rhCTGF for 24 h group; Lane 3: rhDkk-1 (20 ng/mL) pretreated cells prior to rhCTGF; Lane 4: rhDkk-1 (200 ng/mL) pretreated cells prior to rhCTGF; Lane5: rhDkk-1(300 ng/mL) pretreated cells prior to rhCTGF. (A) The mRNA expressions of α-SMA and E-cadherin were measured by real-time PCR. (B) The protein expression of α-SMA and E-cadherin were measured by western blotting. (C) Quantitative analysis of protein expression as measured by western blotting. *p < 0.05 versus control group, **p < 0.01 versus control group; †p < 0.05 versus 50 ng/mL rhCTGF for 24 h group; and ††p < 0.01 versus 50 ng/mL rhCTGF for 24 h group. All results are representative of at least three individual experiments.
Discussion
Our study elaborates that CTGF activates canonical Wnt signaling pathway, which contributes indirectly to EMT of HK-2 cells in vitro, and extends a potential stimuli for renal fibrosis of CKD. The major and novel findings of our study include that HK-2 cells expressed LRP6, and canonical Wnt signaling pathway was activated by CTGF-LRP6 interaction, which led to EMT of HK-2 cells in vitro. Inhibitory experiments based on the use of specific endogenous LRP6 receptor antagonist, Dkk-1, additionally suggested that the mechanism of CTGF-induced tubular EMT was in vitro. Therefore, the covalent attachment and interaction of CTGF with Wnt co-receptor LRP6 seems to be an important mediator of tubular EMT.
Excessive accumulation of matrix protein in the glomerular mesangium and tubular interstitium intensively contribute to progressive renal dysfunction in CKDs and tubulointerstitial lesions were more significant in prognosis of CKD rather than glomerular lesions.Citation15,Citation16 CTGF is an important pro-fibrogenic factor associated with CKD, but the mechanism is not fully understood. The experiment has revealed that CTGF expression was increased in tubular epithelial cells in multiple forms of CKD.Citation17,Citation18 However, CTGF, a 36–38-kDa small molecule, can filter across glomerular basement membrane and be present in the tubular fluid in pharmacologically active concentrations. Therefore, tubular epithelial cells not only express and secrete CTGF but are also exposed to CTGF from the glomerulus.Citation19 In our experiments, to explore how CTGF affects tubular epithelial cells, the CTGF group and Dkk-1 plus CTGF group HK-2 cells were cultured with ectogenic rhCTGF.
It is widely recognized that EMT plays a significant role in the renal fibrosis of CKD.Citation2,Citation12,Citation16,Citation20 In addition, the interaction of CTGF and tubular epithelial cell has been proven to contribute to the mechanisms underlying tubular EMT.Citation21,Citation22 However, although the role of CTGF in tubular EMT of CKD is established, new study highlights the significance of understanding how CTGF interacts with other molecules to EMT. Recently, increasing evidence has showed that Wnt/β-catenin signaling contributes to the tubular EMT in CKD.Citation9–12 Our study demonstrated that the Wnt co-receptor LRP6 was expressed in HK-2 cell in vitro. Meanwhile, corresponding with the studies in embryos of X. laevis and primary human mesangial cells,Citation13,Citation14 we also found rhCTGF initiated the phosphorylation of LRP6 (Ser 1490), which suggests that CTGF may function as an LRP6 ligand in HK-2 cells. In canonical Wnt signaling pathway, the LRP6 intracellular domain can directly promote phosphorylation of GSK-3β.Citation23–25 An increase in phosphorylation of Ser9 in GSK-3β is known to reduce GSK-3β activity and intensified β-catenin translocation into the nucleus.Citation26 Our studies found that in HK-2 cells, the phosphorylation of LRP6 and GSK-3β, and the accumulation and nuclear localization of β-catenin increased gradually when CTGF-treated time extends.
Tubular EMT is a phenotype change where cells lose epithelial phenotype and acquire a new mesenchymal one along with morphological changes. In general, the techniques provided a consistent pattern; that is, upregulation of α-SMA or vimentin and downregulation of E-cadherin or bone morphogenic protein 7 in renal tubular epithelial cells. In this study, Wnt/β-catenin signaling activated by rhCTGF initiated increase of α-SMA and decrease of E-cadherin in HK-2 cells. Dkk-1, a secretory protein, binds to the Wnt co-receptor LRP6 and blocks canonical Wnt signaling by inhibiting the formation of Wnt-Frizzled-LRP6/arrow receptor complexes.Citation27 Our results in this study demonstrated pre-treatment with rhDkk-1-blocked CTGF-induced tubular EMT. These findings provide a novel mechanism for CTGF-mediated renal tubular EMT and renal fibrosis associated with complement system in CKD. For further study, our next work will be based on this cell experiment in vitro to conduct animal experiment in vivo so as to better explore this effect of CTGF in EMT.
In conclusion, our study demonstrated that CTGF can induce the transdifferentiation of normal tubular epithelial cells into mesenchymal cells via activating Wnt/β-catenin signaling pathway in HK-2 cells in vitro. Then, we propose CTGF antagonism, like Dkk-1, may be considered as a potential therapeutic intervention at the center of multiple pathways, including Wnt, to block the progression of renal TIF associated with deterioration of renal function.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
This research was supported by the Twelfth Five-Year National Science and Technology Support Programme project (no. 2011BAI10B00).
References
- Cho MH. Renal fibrosis. Korean J Pediatr. 2010;53:735–740
- Liu Y. New insights into epithelial-mesenchymal transition in kidney fibrosis. J Am Soc Nephrol. 2010;12(2):212–222
- Takigawa M. CTGF/Hcs24 as a multifunctional growth factor for fibroblasts, chondrocytes and vascular endothelial cells. Drug News Perspect. 2003;16:11–21
- Phanish MK, Winn SK, Dockrell ME. Connective tissue growth factor-(CTGF, CCN2)—A marker, mediator and therapeutic target for renal fibrosis. Nephron Exp Nephrol. 2010;114(3):e83–e92
- Chen XM, Qi W, Pollock CA. CTGF and chronic kidney fibrosis. Front Biosci. 2009;1:132–141
- Burns WC, Twigg SM, Forbes JM, et al. Connective tissue growth factor plays an important role in advanced glycation end product-induced tubular epithelial-to-mesenchymal transition: Implications for diabetic renal disease. J Am Soc Nephrol. 2006;17:2484–2494
- Liu XC, Liu BC, Zhang XL, Li MX, Zhang JD. Role of ERK1/2 and PI3-K in the regulation of CTGF-induced ILK expression in HK-2 cells. Clin Chim Acta. 2007;382(1–2):89–94
- Wahab NA, Mason RM. A critical look at growth factors and epithelial-to-mesenchymal transition in the adult kidney. Interrelationships between growth factors that regulate EMT in the adult kidney. Nephron Exp Nephrol. 2006;104(4):e129–e134
- Kim K, Lu Z, Hay ED. Direct evidence for a role of beta-catenin/LEF-1 signaling pathway in induction of EMT. Cell Biol Int. 2002;26(5):463–476
- Zhou D, Tan RJ, Zhou L, Li Y, Liu Y. Kidney tubular β-catenin signaling controls interstitial fibroblast fate via epithelial-mesenchymal communication. Sci Rep. 2013;3:1878
- Xiao L, Wang M, Yang S, Liu F, Sun L. A glimpse of the pathogenetic mechanisms of Wnt/β-catenin signaling in diabetic nephropathy. Biomed Res Int. 2013;2013:987064
- Kim MK, Maeng YI, Sung WJ, et al. The differential expression of TGF-β1, ILK and wnt signaling inducing epithelial to mesenchymal transition in human renal fibrogenesis: An immunohistochemical study. Int J Clin Exp Pathol. 2013;6(9):1747–1758
- Mercurio S, Latinkic B, Itasaki N, Krumlauf R, Smith JC. Connective-tissue growth factor modulates WNT signalling and interacts with the WNT receptor complex. Development. 2004;131(9):2137–2147
- Rooney B, O’Donovan H, Gaffney A, et al. CTGF/CCN2 activates canonical Wnt signalling in mesangial cells through LRP6: Implications for the pathogenesis of diabetic nephropathy. FEBS Lett. 2011;585:531–538
- Fioretto P, Mauer M. Histopathology of diabetic nephropathy. Semin Nephrol. 2007;27:195–207
- Liu Y. Epithelial to mesenchymal transition in renal fibrogenesis: Pathologic significance, molecular mechanism, and therapeutic intervention. J Am Soc Nephrol. 2004;15(1):1–12
- Zuehlke J, Ebenau A, Krueger B, Goppelt-Struebe M. Vectorial secretion of CTGF as a cell-type specific response to LPA and TGF-βin human tubular epithelial cells. Cell Commun Signal. 2012;10(1):25
- Boor P, Floege J. Chronic kidney disease growth factors in renal fibrosis. Clin Exp Pharmacol Physiol. 2011;38(7):441–450
- Wang S, DeNichilo M, Brubaker C, Hirschberg R. Connective tissue growth factor in interstitial injury of diabetic nephropathy. Kidney Int. 2001;60:96–105
- Wing MR, Ramezani A, Gill HS, Devaney JM, Raj DS. Epigenetics of progression of chronic kidney disease: Fact or fantasy? Semin Nephrol. 2013;33(4):363–374
- Gupta S, Clarkson MR, Duggan J, Brady HR. Connective tissue growth factor: Potential role in glomerulosclerosis and tubulointerstitial fibrosis. Kidney Int. 2000;58:1389–1399
- Liu BC, Zhang JD, Zhang XL, Wu GQ, Li MX. Role of connective tissue growth factor (CTGF) module 4 in regulating epithelial mesenchymal transition (EMT) in HK-2 cells. Clin Chim Acta. 2006;373(1–2):144–150
- Wu G, Huang H, Garcia Abreu J, He X. Inhibition of GSK3 phosphorylation of beta-catenin via phosphorylated PPPSPXS motifs of Wnt coreceptor LRP6. PLoS One. 2009;4:e4926
- Cselenyi CS, Jernigan KK, Tahinci E, Thorne CA, Lee LA, Lee E. LRP6 transduces a canonical Wnt signal independently of Axin degradation by inhibiting GSK3’s phosphorylation of beta-catenin. Proc Natl Acad Sci USA. 2008;105:8032–8037
- Piao S, Lee SH, Kim H, Yum S, Stamos JL, Xu Y, et al. Direct inhibition of GSK3β by the phosphorylated cytoplasmic domain of LRP6 in Wnt/beta-catenin signaling. PLoS One. 2008;3:e4046
- Akiyama T. Wnt/beta-catenin signaling. Cytokine Growth Factor Rev. 2000;11:273–282
- Bafico A, Liu G, Yaniv A, Gazit A, Aaronson SA. Novel mechanism of Wnt signaling inhibition mediated by Dickkopf-1 interaction with LRP6/Arrow. Nat Cell Biol. 2001;3:683–686