
Abstract
Caspase-1, IL-1α, and IL-1β are known to be activated in the NLRP3 inflammasome. The inflammasome is activated mostly in inflammatory cells. The presence of inflammasome proteins in proximal tubules (PTs) and the effect of cisplatin-treatment or caspase inhibition on inflammasome proteins in PTs are not known. The aim of this study was to investigate the effect of cisplatin on inflammasome proteins in freshly isolated PTs and also to determine the effect of caspase inhibition on inflammasome proteins and PT injury. PTs were isolated using collagenase digestion and Percoll centrifugation. After recovery period, freshly isolated PTs were incubated with vehicle, 50 µM cisplatin or 50 µM cisplatin plus 50 µM pan caspase inhibitor, QVD-OPH. PTs treated with 50 µM cisplatin showed Propidium Iodide staining indicative of necrosis. Necrotic cells (%) were 2.2 in Vehicle-treated, 37.7 in Cisplatin-treated (p < 0.05 vs. Vehicle), and 3.3 in QVD-treated (p < 0.05 vs. Cisplatin). LDH release (%), a marker of cell membrane damage seen in necrosis was 7.1 in Vehicle-treated, 39.7 in Cisplatin-treated (p < 0.05 vs. Vehicle), and 13.5 in QVD-treated (p < 0.05 vs. Cisplatin). Caspase-1 activity and active caspase-1 protein (10 kDa) were significantly increased in Cisplatin-treated PTs. NLRP3 was strongly expressed in PTs, but there were no significant changes between groups. Pro-apoptotic BID (22 kDa) was unchanged between groups. IL-1α and IL-1β activity was increased in Cisplatin-treated PTs. QVD-OPH co-treatment decreased caspase-1, IL-1α, and IL-1β. In summary, caspase inhibition decreases caspase-1, IL-1α, and IL-1β but not NLRP3 or BID protein and protects against necrosis in cisplatin-treated freshly isolated PTs.
Introduction
Mammalian cell death is usually classified into two types: apoptosis and necrosis. Apoptosis is a fundamental type of programmed cell death that can occur by genetically controlled processes.Citation1,Citation2 There is now evidence that necrosis, traditionally considered an accidental form of cell death, can in certain instances be initiated or modulated by programmed control mechanisms.Citation3–6 The effect of caspase inhibition on apoptosis in proximal tubules (PTs) is well known.Citation7,Citation8 However, the effect of caspase inhibition on necrosis in PTs is less well known. Within the cytoplasm, NLRP3 (nucleotide-binding oligomerization domain, leucine-rich repeat and pyrin domain containing protein 3) forms a complex, NLRP3 inflammasome with ASC (apoptosis-associated speck-like protein containing caspase recruitment domain) and inactive caspase-1 (procaspase-1). Pro-inflammatory caspase-1, activated by inflammasome complexes in response to pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs), converts the cytokines interleukin (IL)-1β and IL-18 to their active forms and also participates in apoptosis through the activation of caspase-3.Citation9–11 The NLRP3 inflammasome is present mainly in inflammatory cells. The presence of inflammasome proteins in PTs and the effect of caspase inhibition on inflammasome proteins in PTs are not known.
It is well appreciated that cells in culture undergo a variety of phenotypic changes to survive in vitro. These changes are particularly problematic for studies of the pathogenesis of injury to the PT. PTs change from their normal dependence on oxidative, mitochondrial metabolism to glycolysis under culture conditions.Citation12 There are some studies on the effect of caspase inhibition in cultured renal PTs treated with cisplatin in vitro.Citation13–17 However, the effects of caspase inhibition in freshly isolated PTs, a model that more closely resembles the in vivo situation, are less well known. Thus the effect of caspase inhibition was investigated in freshly isolated PTs.
Materials and methods
Animals and reagents
For all the mouse studies, 8 to 10-week-old C57BL/6 mice (Harlan, Indianapolis, IN and Jackson Labs, Bar Harbor, ME) weighing 20 to 25 g were used. Mice were maintained on a standard diet and water was freely available. Mice were housed five per cage under a 12-hour light and dark schedule for at least 1 week prior to experiment.
Cisplatin was purchased from Sigma-Aldrich (Milwaukee, WI). Pan Caspase inhibitor, Q-Val-Asp-Oph (QVD-OPH) was purchased from MP Biochemicals (Aurora, OH). DMSO was obtained from Aldrich. The vital nuclear dye bisbenzamide (Hoechst 33342) and propidium iodide (PI) were obtained from Molecular Probes (Eugene, OR).
Preparation of renal PTs
PTs were isolated from kidney cortex of male C57BL/6 mice using collagenase digestion and Percoll centrifugation as we have previously described in detail.Citation18,Citation19 Six-milliliter aliquots of tubule suspension (approximately 1 to 2 mg/mL) were placed in siliconized 25 mL Erlenmeyer flasks for a “recovery” period, which included gassing with 95% O2/5% CO2 for 5 min on ice. Flasks were then capped with rubber stoppers and kept at room temperature for 5 min, then placed in a shaking water bath at 37 °C for 10 min. After adding the reagents, the flasks were regassed with 95% O2/5% CO2 for 5 min and then closed and kept in the shaking water bath for 25 min. At the end of the “pre-incubation and experiment” period, 1 mL of tubule suspension was sampled for measurement of lactate dehydrogenase (LDH), 5 mL for caspase activity, ELISA, or immunoblots.
Cisplatin treatment
After “recovery” period, PTs were incubated with cisplatin (final concentration 50 µM). Cisplatin was freshly prepared and dissolved in DMSO (final concentration 0.1%) at the time of the experiment. Control PTs were treated with the vehicle (0.1% DMSO).
Caspase inhibition
To determine the role of inflammasome and caspase-1 in cisplatin-treated PTs, PTs were incubated with 50 µM pan caspase inhibitor, QVD-OPH (catalogue number OPH109, MP Biochemicals, Aurora, OH).
Measurement of LDH release and protein
Leakage of lactate dehydrogenase (LDH) was measured as an index of lethal membrane injury.Citation19 The incubation medium was collected and the remaining pellet was dissolved in 1.5% Triton X-100 to the original volume. The percentage of LDH released from tubules was calculated by determining the ratio of LDH in the supernatant compared to that in the lysed tubule pellet plus the supernatant. Tubule protein was measured by the Lowry method, using bovine serum albumin as standard.
Immunofluorescence study
Nuclear morphology of the PTs was examined to identify the mechanism of cell death. The cells were stained with the DNA-specific dyes Hoechst 33342 and Propidium Iodide (PI). Fluorescent microscopy was used to determine necrosis. Cells were washed in PBS and then stained with Hoechst 33342 (final concentration 5 µM) and PI (final concentration 0.5 µM) for 10 min at 37 °C. Cells were washed again in PBS and then fixed with 3% paraformaldehyde. Fields of cells were photographed (magnification ×400), by using appropriate filters to examine Hoechst 33342 and PI fluorescence staining. Nuclear morphology was assessed with the cell membrane-permeant supravital DNA dye Hoechst 33342 (excitation wavelength 348 nm; emission 479 nm). Hoechst 33342, unlike PI, enters and stains the nucleus of both viable cells and cells that have died by apoptosis or necrosis. Plasma membrane integrity was assessed using the cell membrane-impermeant DNA dye PI (excitation wavelength 535 nm; emission 617 nm). Necrosis was determined on the basis of positive PI staining in red color, indicative of loss of membrane integrity.
Caspase-1 activity
The caspase assay in PTs was measured as we have previously described in detail.Citation7 Proximal tubule extracts for the caspase assay were prepared as follows. After exposure to normoxia or to hypoxia, the tubule pellet was separated from 2 mL of tubule suspension by centrifugation. The tubule pellet was mixed with 1 mL of lysis buffer on ice for 10 min. The lysis buffer used contained 20 mM Hepes, 100 mM NaCl, 10 mM dithiothreitol, 1 mM EDTA, 0.1% CHAPS, 10% sucrose, and 1% Triton X-100, pH 7.2. The lysate was then centrifuged at 5000 g for 10 min. The supernatant was mixed with an equal quantity of 50% glycerol. The caspase assay was then performed on this supernatant as follows: 200 μL of supernatant (200–400 μg protein) and 10 μL of the substrate (final concentration, 50 μM) were added to the caspase assay buffer. The total volume of the assay solution was adjusted to 2 mL using the assay buffer. The assay buffer contained 100 mM Hepes, 100 mM NaCl, 10 mM dithiothreitol, 1 mM EDTA, 10% sucrose, 0.1% CHAPS, and 10% glycerol, pH 7.4. Ac-YVAD-AMC was used as a susceptible substrate for caspase-1. Fluorescence was measured at 380 nm excitation and 460 nm emission was determined using a Hitachi F2000 spectrophotometer. Caspase activity was expressed in pmol AMC released per minute of incubation time per mg of lysate protein. Lysate protein was measured by the Bradford method as described in the Bio-Rad protein assay kit with bovine serum albumin as standard.
Statistical analysis
Experiments were performed in triplicate and repeated at least three times. All values are expressed as mean ± standard error (SE). Data were analyzed for significance using unpaired, two-tailed Student’s t tests. A p value of <0.05 was considered statistically significant.
Results
Nuclear morphology of the PTs after cisplatin treatment
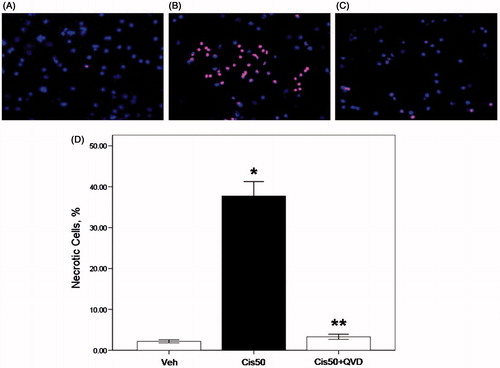
With the color combination, vehicle-treated PTs excluded PI staining and stained mostly with Hoechst 33342 in blue color (). In contrast, 50 µM cisplatin-treated PTs showed extensive PI staining in red color, indicative of loss of membrane integrity and necrosis (). Those nuclei were smaller in size compared with controls which is indicative of homogenous masses of chromatin condensation in globular or crescent-shaped figures. Necrotic cells were quantitated in at least 200 randomly selected cells stained with Hoechst 33342 by fluorescent microscopy. Percentage of necrotic cells was highest in 50 µM cisplatin-treated group (p < 0.05, 37.7 ± 3.5 vs. 2.2 ± 0.4% in vehicle-treated group) (). PTs co-treated with 50 µM cisplatin plus pan caspase inhibitor, QVD-OPH demonstrated significant reduction of necrotic cells to the level of vehicle-treated group (p < 0.05, 3.3 ± 0.6 vs. 37.7 ± 3.5% in 50 µM cisplatin group) ().
Figure 1. Propidium iodide staining of nuclei in freshly isolated proximal tubule cells is increased by cisplatin and decreased by the caspase inhibitor. The cells were stained with color combination of DNA-specific dyes Hoechst 33342 and propidium iodide (PI). Vehicle-treated proximal tubule cells (Veh) demonstrated an intact structure; they excluded PI and stained mostly with Hoechst in blue color (A). Cells treated with 50 µM cisplatin (Cis50) showed extensive PI staining in red color, indicative of loss of membrane integrity and necrosis. Nuclear morphology was disrupted and those cells were more brightly stained indicative of chromatin condensation in globular or crescent-shaped figures (B). Cells treated with 50 µM cisplatin in combination with pan caspase inhibitor, 50 µM QVD-OPH (Cis50+QVD) demonstrated a normal morphology compared with the cells treated with 50 µM cisplatin only (C,D). (*p < 0.05 vs. Veh, **p < 0.05 vs. Cis50).
LDH release from the PTs after cisplatin treatment
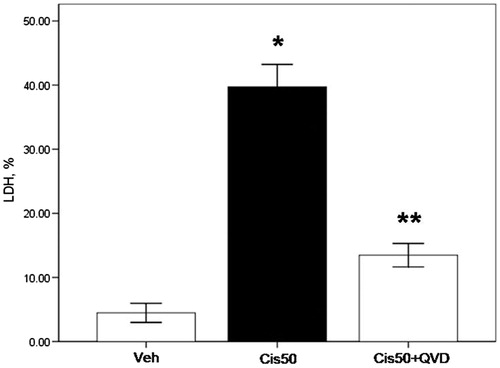
LDH release is a marker of cell membrane damage that occurs in necrosis. LDH release was increased by over 20% in 50 µM cisplatin-treated PTs (p < 0.05, 39.7 ± 3.5 vs. 7.1 ± 0.9 in vehicle-treated group). Addition of QVD-OPH decreased LDH release almost to the levels of controls (p < 0.05, 13.5 ± 1.8 vs. 39.7 ± 3.5% in 50 µM cisplatin-treated group; ).
Figure 2. LDH release (%) from freshly isolated proximal tubule cells is increased by cisplatin and decreased by the caspase inhibitor. LDH release was under 10% in vehicle-treated proximal tubule cells (Veh). Cells treated with 50 µM cisplatin (Cis50) showed increased LDH release. Co-treatment of QVD-OPH (Cis50 + QVD) decreased LDH release significantly. (*p < 0.05 vs. Veh, **p < 0.05 vs. Cis50).
Activation of caspase-1 in vitro in PTs after cisplatin treatment
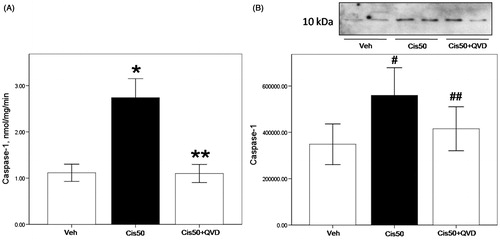
Caspase-1 activity was increased significantly in 50 µM cisplatin-treated PTs compared with vehicle-treated group (p < 0.05, 3.2 ± 0.5 vs. 0.9 ± 0.2 nmol/mg/min in vehicle-treated group). There was a significant reduction in caspase-1 activity of cisplatin-treated cells in combination with QVD-OPH (p < 0.05, 1.1 ± 0.2 vs. 3.2 ± 0.5 nmol/mg/min in 50 µM cisplatin-treated group) (). The active form of caspase-1 (10 kDa) was increased significantly in 50 µM cisplatin-treated groups (p < 0.05 vs. vehicle-treated group), and decreased significantly in combination with QVD-OPH (p < 0.05 vs. 50 µM cisplatin-treated group; ).
Figure 3. Caspase-1 activity and protein of freshly isolated proximal tubule cells treated with cisplatin is reduced by the caspase inhibitor. (A) Caspase-1 activity was increased significantly in 50 µM cisplatin-treated proximal tubule cells (Cis50) compared with vehicle-treated group (Veh) (*p < 0.05 vs. Veh). Co-treatment of QVD-OPH (Cis50 + QVD) decreased caspase-1 activity significantly (**p < 0.05 vs. Cis50). (B) Representative immunoblot of caspase-1 showed active caspase-1 (10 kDa) was increased significantly in Cis50 compared with Veh (#p < 0.05 vs. Veh). Cis50 + QVD decreased the expression of active caspase-1 (##p < 0.05 vs. Cis50 + Veh). Densitometry of caspase-1 immunoblots is shown.
The NLRP3 inflammasome and caspase-1 substrates (IL-1β, IL-1α, BID) in vitro in PTs after cisplatin treatment
NLRP3 was strongly expressed in PTs, however there were no significant changes between groups (). Immunoblotting of BID (22 kDa) showed no difference between the groups (). IL-1α activity was significantly increased in 50 µM cisplatin-treated groups (p < 0.05, 3.1 ± 0.7 vs. 0.1 ± 0.0 nmol/mg/min in vehicle-treated group), and was decreased significantly in combination with QVD-OPH (p < 0.05, 0.8 ± 0.2 vs. 3.1 ± 0.7 nmol/mg/min in 50 µM cisplatin-treated group; ).
Figure 4. Activation of caspase-1 substrates IL-1α and IL-1β in cisplatin-treated freshly isolated proximal tubule cells. (A) On immunoblot, NLRP3 was strongly expressed in PTs with no significant changes between groups on densitometry. (B) On immunoblot, BID (22 kDa) was unchanged between the groups. (C) IL-1α activity was significantly increased in Cis50 (*p < 0.05 vs. Veh), and was decreased significantly in combination with QVD-OPH (**p < 0.05 vs. Cis50). (D) IL-1β activity showed increases in Cis50 (#p < 0.05 vs. Veh), and was decreased significantly in combination with QVD-OPH (##p < 0.05 vs. Cis50).
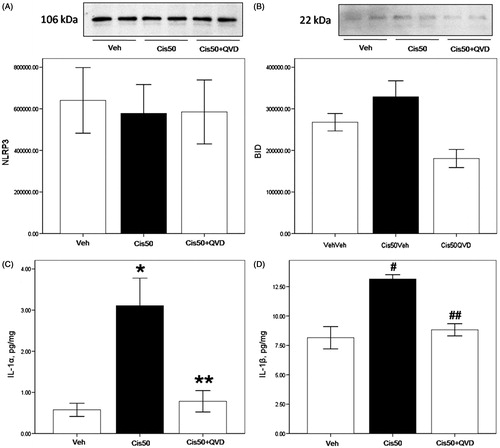
IL-1β activity was significantly increased in 50 µM cisplatin-treated groups (p < 0.05, 13.2 ± 0.4 vs. 7.6 ± 0.9 nmol/mg/min in vehicle-treated group). Co-treatment of QVD-OPH reduced IL-1β activity significantly (p < 0.05, 8.8 ± 0.5 vs. 13.2 ± 0.4 nmol/mg/min in 50 µM cisplatin-treated group; ).
Discussion
Cells in culture undergo a variety of phenotypic changes driven in large part by the adaptations necessary to survive in vitro as well as by a shift to a more proliferative mode that characterizes that setting. Cultured tubular cells also undergo considerable structural changes that include simplification of both their apical and basolateral membrane compartmentsCitation20 that affect cellular characteristics prominently involved in the behavior of the tubules during AKI in vivo. For these reasons, freshly isolated PTs have been favored by a number of investigators for studies of the cellular biology of PT injury.Citation21–27 In vitro preparations of freshly isolated PTs have been valuable for both metabolic and structural investigations, because the PTs retain the biochemical properties of the in vivo state as well as a high degree of structural integrity – highly polarized and fully differentiated features – of the normal proximal tubular epithelium. Thus we chose freshly isolated PTs rather than cultured cells for the present study.
Previous studies in cultured PTsCitation7,Citation8 and the present study in freshly isolated PTs demonstrate that caspase inhibitors prevent cisplatin-induced PT injury. Our published data demonstrate that caspase-1 −/− mice are protected against cisplatin-induced AKICitation28 and ischemic AKI.Citation29 The caspase inhibitor, OPH 001, protects against ischemic AKI in mice.Citation30 However, there are no in vivo studies that demonstrate that caspase inhibitors protect against cisplatin-induced AKI. In view of the dramatic protection against cisplatin-induced PT injury by caspase inhibitors in vitro, it would be expected that caspase inhibitors would protect against cisplatin-induced AKI in vivo. However, a recent study demonstrated that the caspase inhibitor, zVAD-fmk, worsened renal function in cisplatin-induced AKI in vivo.Citation31 The caspase inhibitor prevented cisplatin-induced cleavage of autophagy proteins, but impaired autophagic flux and worsened renal function. Autophagy has been shown to be protective in ischemic AKI.Citation32 Thus differences in autophagy induced by cisplatin treatment in vivo versus in vitro may explain the protection against cisplatin-induced AKI in vitro but not in vivo.
The inflammasome is a molecular complex which contains NLRP (Nucleotide-binding oligomerization domain, Leucine rich Repeat and Pyrin domain containing) proteins and an adaptor protein called ASC (Apoptosis-associated Speck-like protein containing a Caspase recruitment domain).Citation33,Citation34 The most fully characterized inflammasome is the NLRP3 inflammasome which contains the NLRP3 protein.Citation35
Caspase-1 is activated in the inflammasome. The inflammasome is activated mostly in inflammatory cells rather than epithelial cells and plays an important part in the innate immune response.Citation34,Citation35 The inflammasome plays an important role in causing tissue inflammation. In the present study, NLRP3 and inflammasome proteins were determined in isolated PTs, a system devoid of inflammatory cells.
There have been few studies of the inflammasome in PT epithelial cells. In human and mouse primary renal tubular epithelial cells, NLRP3 expression was increased in response to TGF-β1 stimulation. TGF-β1-induced effects were significantly decreased in mouse NLRP3 −/− renal tubular epithelial cells, suggesting a role for NLRP3 in TGF-β-dependent signaling. In response to TGF-β1, the effect of NLRP3 was inflammasome independent, as IL-1β, IL-18, and caspase-1 were not involved.Citation36 In the present study, in cisplatin-treated PTs, there was activation of caspase-1, IL-1β and IL-1α suggesting activation of the inflammasome. However, while NLRP3 protein was abundantly present in PTs, there was no increase in NLRP3 arguing against activation of the NLRP3 inflammasome. In addition to the NLRP3 inflammasome, caspase-1 is also activated in the NLRP1, NLRC4 or AIM2 inflammasomes. Our previous study showed NLRP1 protein was increased in cisplatin-induced AKI suggesting a role of NLRP1 inflammasome in cisplatin-induced AKI.Citation37 In the present study, it is possible that caspase-1 is activated in the NLRP1, NLRC4 or AIM2 inflammasome. In support of the lack of a role of the NLRP3 inflammasome in cisplatin-induced PT injury is our recent study that despite activation of caspase-1 in cisplatin-induced AKI, NLRP3 −/− mice are not protected against cisplatin-induced AKI in vivo.Citation37
Activation of caspase-1 in the inflammasome results in secretion of “leaderless” proteins via direct or indirect physical interaction.Citation38 Active caspase-1 in the inflammasome activates IL-1β, IL-18 and IL-1α. In the present study, there was an increase in caspase-1 protein and activity in cisplatin-treated PTs. IL-1β and IL-1α that are activated by caspase-1 in the inflammasome were also increased in cisplatin-treated PTs. BID, that was originally thought to be activated by caspase-1 but is now thought to be activated by caspase-8,Citation39 was not increased by cisplatin in PTs. NLRP3, the major component of the NLRP3 inflammasome was present in the isolated PTs but unchanged between the groups. The present results demonstrate the novel finding of activation of caspase-1-dependent proteins, IL-1β and IL-1α in isolated PTs. The activation of caspase-1, IL-1β and IL-1α was independent of an increase in NLRP3.
Caspase-1 is a mediator of hypoxic PT injury in vitro and ischemic AKI in vivo. PTs isolated from caspase-1 −/− mice are protected against hypoxic injury.Citation40 Caspase-1 −/− mice are protected against ischemic AKI in vivo.Citation29 Based on these studies, the question arose as to whether caspase-1 activated in the inflammasome was the mediator of the hypoxic and ischemic PT injury. Our recent study demonstrating that NLRP3 −/− mice are protected against ischemic AKI suggests a role for caspase-1 activation in the NLRP3 inflammasome as a mediator of ischemic AKI
While the present study suggests that caspase inhibitors offer promise in the prevention of cisplatin-induced PT injury, a recent study showing that caspase inhibition worsens AKI by preventing autophagy,Citation31 dampens the enthusiasm to use caspase inhibitors to prevent cisplatin-induced AKI. Little is known of the expression of inflammasome-related genes in renal cells or in kidney diseases. The human kidney expresses much lower mRNA levels of most inflammasome-related molecules as compared with the human spleen except for NLRP2 and NLRP10. In contrast, kidneys from C57BL/6 mice express much higher levels of most NLR genes compared with the spleen, with the exception of neuronal apoptosis inhibitory protein (NIAP) and NLRP3.Citation41 Although NLRP1 and NLRP3 expression more or less overlaps in blood immune cells, expression in epithelial cells is quite different. NLRP1 is localized mainly in the nucleus, whereas NLRP3 is predominantly cytoplasmic. A difference in subcellular distribution between NLRP1 and NLRP3 suggests site-specific roles of NLRPs in the inflammation. Which cells express functional inflammasomes in the kidney is largely unknown. However, some studies suggested the potential of tubular epithelial cells to secrete IL-1β and IL-18;Citation42 hence, we should harbor all necessary elements of the inflammasome-caspase-1-IL-1β/-18 axis.
In summary, the present study demonstrates the following novel findings: (1) NLRP3 was expressed in PTs, but not increased by cisplatin. (2) Caspase-1, IL-1α, and IL-1β were activated in PTs. (3) Pan Caspase inhibitor, QVD-OPH protected against necrosis, and the protection was associated with a decrease in caspase-1 as well as IL-1α, and IL-1β.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
References
- Kroemer G, Galluzzi L, Vandenabeele P, et al. Classification of cell death: Recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009;16:3–11
- Majno G, Joris I. Apoptosis, oncosis, and necrosis: An overview of cell death. Am J Pathol. 1995;146:3–15
- Zong WX, Thompson CB. Necrotic death as a cell fate. Genes Dev 2006;20:1–15
- Golstein P, Kroemer G. Cell death by necrosis: Towards a molecular definition. Trends Biochem Sci. 2007;32:37–43
- Luke CJ, Pak SC, Askew YS, et al. An intracellular serpin regulates necrosis by inhibiting the inductionnd sequelae of lysosomal injury. Cell. 2007;130:1108–1119
- Hotchkiss RS, Strasser A, McDunn JE, Swanson PE. Cell death. N Engl J Med. 2009;361(16):1570–1583
- Edelstein CL, Shi Y, Schrier RW. Role of caspases in hypoxia-induced necrosis of rat renal proximal tubules. J Am Soc Nephrol. 1999;10(9):1940–1949
- Yang B, El Nahas AM, Fisher M, et al. Inhibitors directed towards caspase-1 and -3 are less effective than pan caspase inhibition in preventing renal proximal tubular cell apoptosis. Nephron Exp Nephrol. 2004;96(2):e39–e51
- Dinarello CA. Biologic basis for interleukin-1 in disease. Blood. 1996;87:2095–2147
- Fantuzzi G, Puren AJ, Harding MW, Livingston DJ, Dinarello CA. Interleukin-18 regulation of interferon gamma production and cell proliferation as shown in interleukin-1 beta-converting enzyme (caspase-1)-deficient mice. Blood. 1998;91:2118–2125
- Van de Craen M, Declercq W, Van den brande I, Fiers W, Vandenabeele P. The proteolytic procaspase activation network: An in vitro analysis. Cell Death Differ. 1999;6:1117–1124
- Tang MJ, Suresh KR, Tannen RL. Carbohydrate metabolism by primary cultures of rabbit proximal tubules. Am J Physiol Cell Physiol. 1989;256:C535–C539
- Nagothu KK, Bhatt R, Kaushal GP, Portilla D. Fibrate prevents cisplatin-induced proximal tubule cell death. Kidney Int. 2005;68:2680–2693
- Arany I, Megyesi JK, Kaneto H, Price PM, Safirstein RL. Cisplatin-induced cell death is EGFR/src/ERK signaling dependent in mouse proximal tubule cells. Am J Physiol Renal Physiol. 2004;287:F543–F549
- Cummings BS, McHowat J, Schnellmann RG. Role of an endoplasmic reticulum Ca2+-independent phospholipase A2 in cisplatin-induced renal cell apoptosis. J Pharmacol Exp Ther. 2004;308:921–928
- Baek SM, Kwon CH, Kim JH, Woo JS, Jung JS, Kim YK. Differential roles of hydrogen peroxide and hydroxyl radical in cisplatin-induced cell death in renal proximal tubular epithelial cells. J Lab Clin Med. 2003;142:178–186
- Price PM, Safirstein RL, Megyesi J. Protection of renal cells from cisplatin toxicity by cell cycle inhibitors. Am J Physiol Renal Physiol. 2004;286:F378–F384
- Edelstein CL. Calpain activity in rat renal proximal tubules. An in vitro assay. Methods Mol Biol. 2000;144:233–238
- Edelstein CL, Wieder ED, Yaqiib MM, et al. The role of cysteine proteases in hypoxia-induced rat renal proximal tubular injury. Proc Natl Acad Sci USA. 1995;92(17):7662–7666
- Kroshian VM, Sheridan A, Lieberthal W. Functional and cytoskeletal changes induced by sublethal injury in proximal tubular epithelial cells. Am J Physiol Renal Fluid Electrolyte Physiol. 1994;266:F21–F30
- Almeida AR, Wetzels JF, Bunnachak D, et al. Acute phosphate depletion and in vitro proximal tubule injury: Protection by glycine and acidosis. Kidney Int. 1992;41:1494–1500
- Wetzels JFM, Wang X, Gengaro PE, Nemenoff RA, Burke TJ, Schrier RW. Glycine protection against hypoxic but not phospaolipase A2-induced injury in rat proximal tubules. Am J Physiol. 1993;264:F94–F99
- Bunnachak D, Almeida AR, Wetzels JFM, et al. Ca2+ uptake, fatty acid, and LDH release during proximal tubule hypoxia: Effects of mepacrine and dibucaine. Am J Physiol. 1994;266:F196–F201
- Yu L, Gengaro PE, Niederberger M, Burke TJ, Schrier RW. Nitric oxide: A mediator in rat tubular hypoxia/reoxygenation injury. Proc Natl Acad Sci USA. 1994;91:1691–1695
- Choi KH, Edelstein CL, Gengaro PE, Schrier RW, Nemenoff RA. Hypoxia induces changes in phospholipase A2 in rat proximal tubules: Evidence for multiple forms. Am J Physiol. 1995;269: F846–F853
- Alkhunaizi AM, Yaqoob MM, Edelstein CL, et al. Arachidonic acid protects against hypoxic injury in rat proximal tubules. Kidney Int. 1996;49:620–625
- Yaqoob MM, Edelstein CL, Wieder ED, Alkhunaizi AM, Gengaro PE, Schrier RW. Nitric oxide kinetics during hypoxia in proximal tubules: Effects of acidosis and glycine. Kidney Int. 1996;49:1314–1319
- Faubel S, Ljubanovic D, Reznikov L, Somerset H, Dinarello CA, Edelstein CL. Caspase-1-deficient mice are protected against cisplatin-induced apoptosis and acute tubular necrosis. Kidney Int. 2004;66:2202–2213
- Melnikov VY, Ecder T, Fantuzzi G, et al. Impaired IL-18 processing protects caspase-1-deficient mice from ischemic acute renal failure. J Clin Invest. 2001;107(9):1145–1152
- Melnikov VY, Faubel S, Siegmund B, Lucia MS, Ljubanovic D, Edelstein CL. Neutrophil-independent mechanisms of caspase-1-and IL-18-mediated ischemic acute tubular necross in mice. J Clin Invest. 2002;110(9):1083–1091
- Herzog C, Yang C, Holmes A, Kaushal GP. zVAD-fmk prevents cisplatin-induced cleavage of autophagy proteins but impairs autophagic flux and worsens renal function. Am J Physiol Renal Physiol. 2012;303(8):F1239–1250
- Pallet N, Livingston M, Dong Z. Emerging functions of autophagy in kidney transplantation. Am J Transplant. 2014;14(1):13–20
- Franchi L, Eigenbrod T, Munoz-Planillo R, Nunez G. The inflammasome: A caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat Immunol. 2009;10(3):241–247
- Schroder K, Tschopp J. The inflammasomes. Cell 2010;140(6):821–832
- Schroder K, Zhou R, Tschopp J. The NLRP3 inflammasome: A sensor for metabolic danger? Science. 2010;327(5963):296–300
- Wang W, Wang X, Chun J, et al. Inflammasome-independent NLRP3 augments TGF-β signaling in kidney epithelium. J Immunol. 2013;190(3):1239–1249
- Kim HJ, Lee DW, Ravichandran K, et al. NLRP3 inflammasome knockout mice are protected against ischemic but not cisplatin-induced acute kidney injury. J Pharmacol Exp Ther. 2013;346(3):465–472
- Keller M, Rüegg A, Werner S, Beer HD. Active caspase-1 is a regulator of unconventional protein secretion. Cell. 2008;132(5):818–831
- Jung EB, Lee CS. Baicalein attenuates proteasome inhibition-induced apoptosis by suppressing the activation of the mitochondrial pathway and the caspase-8- and Bid-dependent pathways. Eur J Pharmacol. 2014;730:116–124
- Edelstein CL, Hoke TS, Somerset H, et al. Proximal tubules from caspase-1-deficient mice are protected against hypoxia-induced membrane injury. Nephrol Dial Transplant. 2007;22(4):1052–1061
- Anders HJ, Muruve DA. The inflammasomes in kidney disease. J Am Soc Nephrol. 2011;22:1007–1018
- Homsi E, Janino P, de Faria JB. Role of caspases on cell death, inflammation, and cell cycle in glycerol-induced acute renal failure. Kidney Int. 2006;69:1385–1392