Abstract
The Hepatitis C virus (HCV) NS4B protein, a multispanning endoplasmic reticulum (ER) membrane protein, generates intracellular rearrangements of ER-derived membranes, essential for HCV replication. In this study, we characterized NS4B elements involved in the process of targeting, association and retention in the ER membrane. We investigated the localization and membrane association of a number of C- or N-terminal NS4B deletions expressed as GFP chimeras by biochemical and fluorescence microscopy techniques. A second set of GFP-NS4B chimeras containing the plasma membrane ecto-ATPase CD39 at the C-terminus of each NS4B deletion mutant was used to further examine the role of N-terminal NS4B sequences in ER retention. Several structural elements, besides the first two transmembrane domains (TMs), within the NS4B N-terminal half (residues 1–130) were found to mediate association of the NS4B-GFP chimeras with ER membranes. Both TM1 and TM2 are required for ER anchoring and retention but are not sufficient for ER retention. Sequences upstream of TM1 are also required. These include two putative amphipathic α-helices and a Leucine Rich Repeat-like motif, a sequence highly conserved in all HCV genotypes. The N-terminal 55peptidic sequence, containing the 1st amphipathic helix, mediates association of the 55N-GFP chimera with cellular membranes including the ER, but is dispensable for ER targeting of the entire NS4B molecule. Importantly, the C-terminal 70peptidic sequence can associate with membranes positive for ER markers in the absence of any predicted TMs. In conclusion, HCV NS4B targeting and retention in the ER results from the concerted action of several NS4B structural elements.
| Abbreviations: | ||
| HCV, | = | Hepatitis C virus |
| ER, | = | endoplasmic reticulum |
| TM, | = | transmembrane domain |
| GFP, | = | green fluorescent protein |
| a.a, | = | amino acid; pAb, polyclonal antibody |
| mAb, | = | monoclonal antibody |
| GST, | = | glutathione transferase |
| MBP, | = | Maltose binding protein; p.t., post transfection |
Introduction
The formation of a membrane associated replication complex consisting of viral proteins, replicating RNA and altered cellular membranes is a characteristic feature of positive-strand RNA viruses [Citation1–4]. This membrane microenvironment is necessary for RNA synthesis and/or facilitates the recruitment of membrane-associated host cell proteins involved in viral replication.
Hepatitis C virus (HCV), a leading cause of chronic liver disease [Citation5,Citation6], is a positive strand RNA virus of the genus, Hepacivirus, in the Flaviridae family [Citation7]. The HCV genome contains a single open reading frame encoding a polyprotein of about 3,000 residues that is co- and post-translationally cleaved by host and viral proteases. This gives rise to the structural proteins Core (C), E1, E2, the membrane associated protein p7, and the non-structural (NS) proteins NS2, NS3, NS4A, NS4B, NS5A, and NS5B in the stated order [Citation8,Citation9–11]. A second open reading frame overlapping the core protein has been identified to express additional protein(s) of unknown function [Citation12]. The HCV replicon system [Citation9,Citation13,Citation14] and the HCV infectious system [Citation15–17] constitute now important cell-culture based systems that enable HCV RNA replication and production of infectious virus particles. Several reports on intracellular membrane rearrangements, caused by non-structural proteins of other RNA positive viruses, have shown the importance of membrane traffic and reorganization in the replication of these viral genomes [Citation2,Citation18–22].
The HCV 26-27 kDa non-structural protein 4B (NS4B) contains a central hydrophobic domain predicted to be transmembrane in its large part. Interaction of NS4B with the host cell, when expressed alone or in the context of other HCV non-structural proteins, alters the host cell's ultrastructural morphology by inducing a perinuclear structure consisting of vesicles in a membranous matrix, referred to as the “membranous web” [Citation23]. Consequently, the morphological alterations induced by NS4B, its association with endoplasmic reticulum (ER) membranes as well as its topology in the ER led to the hypothesis that NS4B may play an important role in HCV replication. The latter has been confirmed by studies performed with the HCV replicon system in which specific mutations introduced in the NS4B sequence affect the efficiency of subgenomic replicons [Citation24].
Whether expressed alone or in the presence of the entire HCV polyprotein, NS4B localizes in the ER membrane [Citation25,Citation26] in the absence of a cleavable N-terminal ER signal peptide or ER retention signals identified thus far for membrane proteins, such as di-Lys motifs [Citation27] at the C-terminus, or di-Arg motifs at the N-terminus [Citation28,Citation29]. Given the fact that the mechanisms for retaining membrane proteins (particularly multispanning ones) are neither well established nor universal [Citation27,Citation30] NS4B can serve as an interesting model protein for studying novel ER retention structural requirements.
NS4B behaves biochemically as an integral membrane protein inserted in the ER membrane co-translationally [Citation25] and is predicted to contain at least four transmembrane domains (TMs) [Citation26]. However, the precise topology and the boundaries of the first two TMs as well as the signals required for anchoring and retention of the protein in the ER membrane are not yet clearly defined. As far as it concerns signals mediating the initial association of the NS4B protein with the ER membrane, when it is expressed alone and not as part of the HCV polyprotein, it has been shown that an N-terminal amphipathic helix within the first 27 amino acids of NS4B is sufficient to drive a GFP chimeric protein to the ER membrane [Citation31]. The exact position and boundaries of the first predicted TM and its expected role in ER anchoring is however unclear. A truncated NS4B-GFP chimera containing the N-terminal amphipathic helix and the first putative TM (residues 1–93) of NS4B, although localized in the ER, could be easily removed from the membranes after sodium carbonate wash [Citation26]. This result suggested that the N-terminal part of NS4B up to amino acid 93 cannot be anchored to the ER membrane.
Here we present a systematic approach for identification of the ER targeting/anchoring sequences in the NS4B protein and of structural features responsible for retaining the protein in the ER. We studied the subcellular localization of a series of NS4B truncated polypeptides expressed as chimeras with the GFP protein or with the plasma membrane ecto-nucleotidase CD39 and demonstrated that several NS4B structural elements acting in concert mediate targeting and retention of the NS4B protein in the ER membrane.
Materials and methods
Reagents and antibodies
Cycloheximide, saponin and proteolytic inhibitors were from Sigma and Mowiol from Calbiochem. Anti-calnexin pAb (purified IgG) was from Sigma (C-4731). The anti-GTL2 mAb (affinity purified, 1 mg/ml) specific for β-1,4-galactosyl transferase was a gift from Dr T. Suganuma (Miyazaki Medical School, Miyazaki, Japan). Anti-mouse and anti-rabbit antibodies conjugated to Alexa546 were purchased from Molecular Probes (Invitrogen) while goat anti-rabbit or donkey anti-mouse HRP conjugated Abs from Pierce Biotechnology. The mAb against the CD39 ecto-domain (Clone BU61) was from Ancell Corp. (Bayport, MN). The human serum used in this study was obtained from a Greek patient infected with HCV genotype 1b (described by Kalamvoki et al. [Citation32]).
Cloning and expression of recombinant proteins in Ε.coli
NS4B sequence coding for amino acids 1–55 (NS4B-N) was amplified by PCR using the 55Ν sense and antisense (2) oligonucleotides (), cloned by EcoRI/SalI restriction fragment replacement into the pBS SK+ vector (Stratagene) for sequence verification and subcloned into the pmal-C2 (New England Biolabs Inc.) and the pGEX-4T1 (Pharmacia) bacterial expression vectors. The MBP-NS4B-N and the GST-NS4B-N fusion proteins were produced in DH5alpha cells upon induction with 0.15–0.3 mM IPTG (Supplemtary Figure S1, online version only) and were affinity purified according to manufacturers' instructions. The MBP-NS4B-N recombinant protein reacted with human sera obtained from HCV positive patients (Supplemtary Figure S1B, online version only) confirming that the bacterially expressed NS4B-N polypeptide contains antigenic epitopes present in the native HCV NS4B protein. The GFP protein was produced in BL21 DE3 pLysS bacteria transformed with the pET-9d-GFP plasmid (kind gift of Dr J. White [email protected]) containing the GFP coding sequences between the NcoI and BamHI sites. His-GFP produced after induction of bacterial cultures with 0.5 mM IPTG, was purified by affinity chromatography on Ni++ NTA resin (QIAexpressionist, Qiagen).
Table I. Oligonucleotides used for the constructs of this study.
Generation of polyclonal antibodies
Polyclonal antibodies (pAbs) against the MBP-NS4B-N or the GFP protein were generated following protocols provided by Harlow and Lane [Citation33]. Antibodies were affinity purified over CNBr-activated Sepharose 4B matrix (Pharmacia). Antibodies raised against the MBP-NS4B-N protein were affinity purified over matrix containing GST-NS4B-N protein to eliminate any anti-MBP antibodies. The affinity purified antisera reacted in Western Blots with bacterially expressed NS4B-N and with the entire length NS4B protein expressed in eukaryotic cells (Supplemtary Figure S1B, online version only) and in immunofluorescence (, and ).
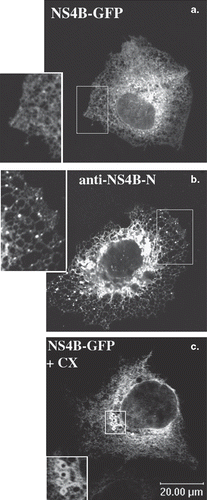
Figure 1. ER localization of the NS4B-GFP chimera in the Huh7 cells. Huh7 cells transiently transfected with the pEGFP-NS4B plasmid were fixed and mounted directly on glass slides (a and c) or fixed and stained with the anti-NS4B-N (1.3 μg/ml) affinity purified pAb followed by incubation with the goat anti-rabbit Alexa546 secondary pAb (b). Treatment with cycloheximide (c) was performed 24 h p.t (50 μg/ml, 5 h at 37°C). Representative confocal images are shown in black and white. The details (insets) represent 2× magnifications of the framed areas. (a) and (c): GFP auto fluorescence; (b): indirect immunofluorescence labelling with the anti-NS4B-N pAb.
Plasmid constructs
Full length NS4B (HCV1a isolate H77) and various fragments of it () were produced by PCR from the pF90/HCV FL-long pU plasmid [Citation34] using oligonucleotide pairs shown in . The PCR products larger than 100 bp were cloned into the pBS SK+ vector by EcoRI/SalI restriction fragment replacement; PCR products smaller than 100 bp were cloned directly into the Topo 2.1 system (Invitrogen). The inserts, after sequence verification were subcloned into the pEGFP-N3 or the pEGFP-C2 vectors (Clontech). For construction of the NS4B-CD39 chimeras, the coding sequences for NS4B deletions () were amplified by PCR using oligonucleotide pairs shown in , and were inserted into the EcoRI site of the pEGFPC1-hpCD39 plasmid [Citation35] between the GFP and CD39 sequences. For generation of the NtCD39-GFP chimera, the N-terminal cytoplasmic and transmembrane domain of the hpCD39 (a.a. 1–44) was amplified with appropriate primers () and subcloned into the pEGFP-N1 expression vector. In all constructs the sequences between the ligation junctions were verified with double stranded DNA sequencing (MWG sequencing services).
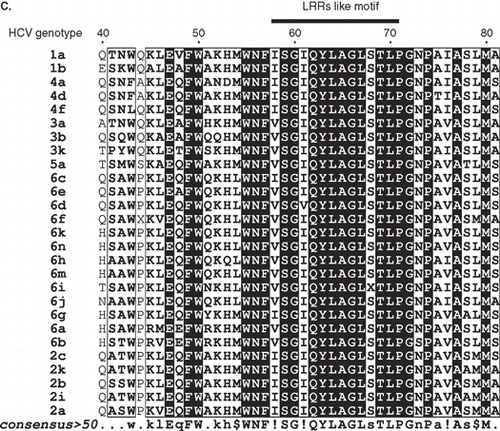
Figure 2. (A) Schematic representation of the NS4B TM topology and the NS4B-GFP chimeras used. The TM NS4B topology model shown at the top is based on the TMHMM trasmembrane prediction method (Supplementary Table S1, online version only). As residue 1 is considered the Ser residue 1712 of the HCV 1aH77 polyprotein (GenBank, accession no. AF009606.1 [Citation34]). White boxes: NS4B sequence; Dark grey boxes: GFP sequence. Dark grey vertical bars: the position of the putative NS4B TM helices. Broken lines: deleted NS4B sequences. Black boxes: the linker arms of 11 (pEGFP-N3) or 12 (pEGFP-C2) residues between the NS4B and GFP sequences. Numbering of the NS4B residues included in the chimeras is given at the beginning and end of the white boxes. The molecular size of the chimeras is indicated on the right. (B) A helical wheel plot of the predicted amphipathic helix between NS4B residues 45-62. The indicated sequence was analysed by the Helical Wheel Custom Images and the Interactive Java Applet (http://kael.net/helical.htm). (C) Sequence alignment of the NS4B LRRs like motif from different HCV isolates. A CLUSTALW multiple sequence alignment was performed for NS4B sequences from representative subtypes of all six major HCV genotypes. The genotypic origin and the subtype of each sequence is indicated on the left. Names of isolates, accession numbers and sequence references in PUBMED are presented in Supplementary Table S2; online version only. The consensus sequence is shown beneath the alignment. Numbering of the sequence presented, and the region including the LRRs are shown at the top. For graphic representation of the aligned sequences the ESPript.2.2 software (http://espript.ibcp.fr/ESPript/ESPript/) was used.
![Figure 2. (A) Schematic representation of the NS4B TM topology and the NS4B-GFP chimeras used. The TM NS4B topology model shown at the top is based on the TMHMM trasmembrane prediction method (Supplementary Table S1, online version only). As residue 1 is considered the Ser residue 1712 of the HCV 1aH77 polyprotein (GenBank, accession no. AF009606.1 [Citation34]). White boxes: NS4B sequence; Dark grey boxes: GFP sequence. Dark grey vertical bars: the position of the putative NS4B TM helices. Broken lines: deleted NS4B sequences. Black boxes: the linker arms of 11 (pEGFP-N3) or 12 (pEGFP-C2) residues between the NS4B and GFP sequences. Numbering of the NS4B residues included in the chimeras is given at the beginning and end of the white boxes. The molecular size of the chimeras is indicated on the right. (B) A helical wheel plot of the predicted amphipathic helix between NS4B residues 45-62. The indicated sequence was analysed by the Helical Wheel Custom Images and the Interactive Java Applet (http://kael.net/helical.htm). (C) Sequence alignment of the NS4B LRRs like motif from different HCV isolates. A CLUSTALW multiple sequence alignment was performed for NS4B sequences from representative subtypes of all six major HCV genotypes. The genotypic origin and the subtype of each sequence is indicated on the left. Names of isolates, accession numbers and sequence references in PUBMED are presented in Supplementary Table S2; online version only. The consensus sequence is shown beneath the alignment. Numbering of the sequence presented, and the region including the LRRs are shown at the top. For graphic representation of the aligned sequences the ESPript.2.2 software (http://espript.ibcp.fr/ESPript/ESPript/) was used.](/cms/asset/4295485c-17c8-4ed3-a94c-205f9a639031/imbc_a_442821_f0002_b.gif)
Secondary structure and transmembrane domain predictions
The algorithms used for secondary structure and transmembrane domain predictions were as follows: (1) Jpred (www.combio.dundee.ac.uk/www-jpred/); (2) the PROF-Secondary Structure Prediction System (http://www.aber.ac.uk/∼phiwww/prof/); (3) PredictProtein PHDhtm (PHDhtm: Protein [Citation36] (http://cubic.bioc.columbia.edu); (4) TMpred (www.ch.embnet.org); (5) TMHMM (www.cbs.dtu.dk); and (6) HMMTop www.enzim.hu/hmmtop/ [Citation37,Citation38].
Tissue culture and transfections
WRL68 (kindly provided by A. Budkowska, Institut Pasteur) and Huh7 hepatocellular carcinoma cells (kindly provided by Dr R. Bartenschlager, University of Heidelberg, Germany) were grown as adherent cultures in DMEM medium + 1% (w/v) glutamate + 10% (v/v) FBS at 37°C in 5% (v/v) CO2 atmosphere. Huh7 cells were grown in the additional presence of 1% (w/v) non-essential amino acids (BIOCHROM, AG). Transfections of Huh7 and WRL68 were performed with the lipofectamine-plus reagent (Invitrogen, Life Technologies) according to the manufacturer's instructions and analyzed for expression of the chimeric proteins 24–48 h post-transfection (p.t.).
Analysis of cell lysates by SDS-PAGE and Western Blot
Total cell lysates were prepared by resuspending the cell pellet in 50 mM Tris-HCl pH 8, 2 mM CaCl2, and solubilizing by the addition of 0.3% (w/v) SDS and 1% (v/v) β-mercaptoethanol and boiling (3 min). The DNA was removed by DNAse (10U-50U) treatment (15 min, 0°C), SDS sample buffer was added and the samples were analyzed by SDS-PAGE (10–12% (w/v) and by Western Blot where the proteins were probed with anti-GFP, anti-NS4B-N or anti-calnexin rabbit pAbs (0.1–0.5 μg/ml) followed by HRP conjugated secondary Abs. Signals were developed by ECL (Amersham) or by the chromogenic reaction using 3,3′ diaminobenzidine tetrahydrochloride (C-8890 Sigma) and H2O2 as substrates.
Triton X-114 extraction of membrane-associated proteins
Cell lysis and phase separation with Triton X-114 were performed as described by Bordier [Citation39]. Briefly, 1–2 × 106 cells treated with Cytochalasin B (20 μg/ml, 1 h, 37°C) were harvested by trypsinization. The trypsin was neutralized (1× wash with 10 volumes DMEM medium + 10% v/v FBS) and cells were further washed with PBS and collected by centrifugation (150 g, 8 min, 4°C). The cell pellet was lysed (15 min, on ice) with 100 μl extraction buffer (1% Triton X-114, 150 mM NaCl, 10 mM Tris-HCl pH 8.0) containing proteolytic inhibitors. Nuclei were pelleted by centrifugation (1000 g, 10 min, 4°C). The post-nuclear supernatant was loaded on the surface of a sucrose cushion (100 μl of 6% (w/v) sucrose, 10 mM Tris-HCl pH 8.0, 150 mM NaCl and 0.06% (v/v) Triton X-114) and incubated at 37°C for 5 min. The detergent phase was centrifuged through the sucrose cushion (10,000 rpm, 3 min, RT), the upper aqueous phase (100 μl) was removed into a different eppendorf tube and Triton X-114 stock solution was added to a 1% (w/v) final concentration. The detergent was dissolved by cooling at 0°C and the solution was reloaded onto the same sucrose cushion used before and incubated at 37°C for 5 min. Centrifugation followed to collect the detergent insoluble-phase as a pellet. The aqueous and detergent phases were collected in separate tubes and were brought up to the same final volume by addition of salt buffer (10 mM Tris-HCl pH 8.0, 150 mM NaCl) and SDS sample buffer. Samples were analyzed by SDS-PAGE and Western Blot analysis. The optical density of the bands in the ECL films was measured using the BIORAD software «Quantity-One».
Isolation of cellular membranes and sodium carbonate treatment
Total membranes enriched in microsomes were prepared by subcellular fractionation and differential centrifugation according to protocols published by Graham (1993) [Citation40]. In brief, transfected cells grown in 10 cm diameter dishes were washed twice with PBS and harvested after 40–45 h p.t. The cells were collected by centrifugation, resuspended in 2 ml isoosmotic medium (0.25 M sucrose, 10 mM triethanolamine-acetic acid pH 7.6, 1 mM MgCl2) and dounce homogenized by 30 strokes in a 3 ml Dounce homogenizer placed on ice. Nuclei and unbroken cells were removed by centrifugation (2700 g, 15min, 4°C) and total membranes were collected by an additional centrifugation (200,000 g in an 80Ti Beckman rotor, 4°C, 1 h). The membrane pellets were resuspended in 2 ml of 100 mM sodium carbonate pH 11.5 and incubated on ice for 30 min [Citation41]. The membrane fraction was recovered by centrifugation as above, washed with dH2O and solubilized in 120 μl, 1× sample buffer. The S/N (sodium carbonate extracted fraction) was subjected to TCA protein precipitation by the addition of 20% w/v final TCA concentration from a fresh 100% (w/v) TCA stock solution, incubation on ice for 10 min and centrifugation at 14,000 rpm for 5 min. The protein pellets were subjected to two acetone washes, dried by heating at 95°C and resuspended in 120 μl 1× sample buffer.
Enzymatic assays
Ecto-nucleotidase enzymatic activity was measured in WRL68 cells transiently expressing CD39 fusion constructs as described before [Citation35]. Briefly, cells grown in 6- or 12-well dishes were washed with HNMC buffer (20 mM Hepes, 150 mM NaCl, 1 mM MgCl2, 1 mM CaCl2) 24 or 44 h p.t. and incubated with 1.0 ml HNMC buffer containing 1 mM ATP for 15 min at 37°C with gentle agitation. The reaction was stopped by the addition of EDTA (10 mM final concentration). The released inorganic phosphate was determined by the malachite green colorimetric assay [Citation42]. A standard curve for free phosphate was generated for each experiment using phosphorus standard solutions (Sigma P3869). The ecto-nucleotidase activity of cells expressing GFP alone was subtracted as background from the activity measured in cells expressing GFP-CD39 or the GFP-NS4B-CD39 chimeras. The released inorganic phosphate was corrected for the percentage of transfected cells in each case and the activity was expressed as the percentage of the enzymatic activity measured in cells expressing GFP-CD39.
Flow cytometry
Transfected cells were washed with PBS-EDTA, left to detach at RT (5–10 min) and analyzed live (5,000–10,000 cells/sample) for GFP fluorescence by the FACS calibur flow cytometer (Becton-Dickinson Immunocytometry System, San Jose, CA, USA). The CELLQuestTM software was used for quantitative analysis.
Indirect Immunofluorescence Microscopy
Cells grown on coverslips were fixed with paraformaldehyde (4% (w/v) in PBS) and neutralized with 50 mM NH4Cl (10 min, RT). They were subsequently mounted onto glass slides (SuperFrost Plus; Menzel-Glaser, Germany) with Mowiol (10% w/v Mowiol, 25% v/v glycerol, 100 mM Tris-HCl, pH 8.5) either immediately or after staining with pAbs or mAbs (1–5 μg/ml) diluted in PBS containing BSA (1 mg/ml) and 0.05% (w/v) saponin or 0.1% (v/v) Triton X-100. As secondary Abs were used, anti-mouse or anti-rabbit pAbs conjugated to Alexa546 fluorochrome (Molecular Probes). For detection of extracellular epitopes, cells were stained live in the absence of detergent and were fixed after removal of the second Ab. Images were acquired with the 63X apochromat lens of the Leica TCS-SP four channels Confocal Microscope equipped with Argon Ion and Helium-Neon Lasers.
Results
Addition of GFP to NS4B does not interfere with its targeting to the ER
The NS4B-GFP chimera, was examined for its association to cellular membranes upon transient expression in the Huh7 cells and WRL68 cells. As revealed by direct fluorescence or by indirect immunofluorescence microscopy (, ) the NS4B-GFP gave a characteristic ER-like reticular localization pattern. At high expression levels, strong perinuclear expression and cytoplasmic foci on the ER network were observed (). This could represent either aggregation due to local protein overexpression or accumulation of the protein in ER exit sites [Citation43]. Treatment with cycloheximide to detect the final destination of the newly synthesized proteins allowed detection of vesicular clusters at the perinuclear region (). These clusters could correspond to the “membranous web” detected by electron microscopy in cells expressing NS4B alone or in the context of the HCV polyprotein [Citation23,Citation44]. The ER localization of NS4B-GFP was confirmed by immunostaining with an anti-calnexin antibody (Supplementary Figure S3, online version only). Correct, full length expression of NS4B-GFP was confirmed by immunoblot analysis using both the anti-NS4B (Supplementary Figure S1B, online version only) and the anti-GFP antibodies (not shown) and membrane association was validated by the Triton X-114 phase separation assay (Supplementary Figure S4B and C, online version only). Thus, in our system, the NS4B-GFP chimera mimicks the ER-localization pattern previously reported for NS4B [Citation25,Citation26].
Generation of NS4B C-terminal and N-terminal deletion mutants in fusion with GFP
To identify the ER targeting/anchoring or retention signals of NS4B, we constructed a series of NS4B N- or C-terminal deletion mutants on the basis of transmembrane topology reported previously [Citation26] or calculated by different methods used in this study (methods, Supplementary Table S1 and Figure S2A, online version only). Eight NS4B-GFP chimeras were generated (methods) either with no putative TMs (55N, 70N and 191-261N) or with one, two or four TMs (95N, 130N, and 43–195N respectively) (). In particular, the 70N deletion was designed to contain an amphipathic helix predicted to be located between residues 45 and 62 () and a Leucine Rich Repeat (LRRs)-like motif (i.e., ISGIQYLAGLSTL). This sequence corresponding to the NS4B residues 58–70 and to residues 1770–1782 of the HCV 1aH77 polyprotein [Citation34], is a highly conserved sequence in all HCV genotypes (). In the design of the NS4B deletions the secondary structure prediction Jpred and PROF (methods, Supplementary Figure S2, online version only) were taken into account so that α-helices or β-strands will not be disrupted.
For the transmembrane topology prediction, we used the TMHMM method, due to its high prediction reliability [Citation45]. However, we also considered results from other transmembrane prediction methods (methods, Supplementary Table S1, online version only) because: (a) The sequence suggested by the TMHMM algorithm as TM1 helix (i.e., residues 66–89) contains two strong helix breaking Pro residues in the middle and is too short for a transmembrane helix ( and Supplementary Figure S2A, online version only); and (b) results from the study by Lundin et al. [Citation26] did not agree with the TMHMM prediction for the boundaries and the topology of the putative TM1 and TM2. Interestingly the four prediction algorithms used in this study to analyze the NS4B sequence showed significant variations in the prediction of the TM1 and TM2 boundaries (Supplementary Table S1).
The NS4B-GFP chimeric mutants are localized in several cellular membrane structures
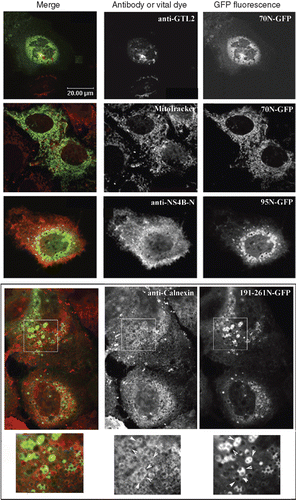
The cellular localization of the truncated NS4B GFP chimeras was examined after transient expression in Huh7 cells by fluorescence microscopy. The 55N-GFP chimera showed a plasma membrane and ER localization (, ), the latter confirmed in immunostaining with the anti-calnexin pAb (Supplementary Figure S3). The green fluorescence detected in the nuclei most probably represents free GFP as shown by immunoblot analysis of extracts from cells expressing 55N-GFP (not shown) due to proteolysis or to additional initiation of translation from the GFP ATG site. The 70N-GFP and 95N-GFP chimeras were partially localized to the ER but as well in distinct clusters of vesicles perinuclearly (–), in the Golgi, (), at the plasma membrane (– and ) and occasionally in mitochondria (). Finally, the 130N-GFP and the 43-195N-GFP chimeras showed distinct ER localization (– and Supplementary Figure S3). The 191-261N-GFP chimera was detected in blebs and protrusions at the plasma membrane (, ) and in clusters of vesicles found positive for calnexin (, inset therein). Fusion of GFP to the 191-261N C-terminus did not alter the protein localization (data not shown). These data indicate that structural elements in the NS4B 70peptidic N- and C-termini other than the predicted TMs target the 70N-GFP and 191-261N-GFP chimeras to the ER membrane or to ER localized structures. The occasional mitochondrial localization observed for these chimeras is rather puzzling. Although these sequences do not have structural characteristics of mitochondria targeting signals [Citation46], they may interact with mitochondria resident proteins.
Figure 3. Predominant localization of the GFP chimeric NS4B mutants in Huh7 cells. Huh7 cells expressing the GFP NS4B chimeric deletions were visualized 24–48 h p.t.either by indirect immunofluorescence (left panels) using the anti-NS4B-N (2.6 μg/ml) (a, c, e, g, j); or the anti-GFP pAbs (4.5 μg/ml) (k, m) and the goat anti-rabbit Alexa546 pAb or by GFP auto fluorescence (right panels). Representative confocal microscopy images are shown in black and white.
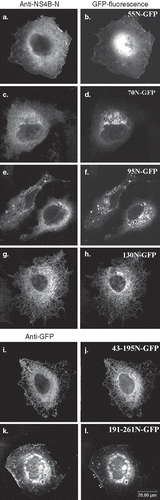
Figure 4. Colocalization of the GFP chimeric NS4B mutants with intracellular markers. Huh7 cells expressing the 70N, 95N or 161-261N GFP chimeras were fixed and mounted directly on coverslips or stained for Golgi with the anti-GTL2 mAb (10 μg/ml), for NS4B with the anti-NS4B-N (2.6 ng/ml), or for ER with the anti-calnexin pAb followed by the anti-mouse Alexa546 or the anti-rabbit Alexa546 pAbs respectively. Mitochondria were labeled by incubation with the MitoTracker Orange CMTMRs (30 min, 37°C, 50 ng/ml). Representative confocal microscopy images are shown in black and white. Details at the bottom represent 2× magnifications of the framed areas shown in panels above. A colour version of this figure with merged images of the two colours shown on the left is published in Molecular Membrane Biology online.
Topological features of the NS4B truncated GFP chimeras were examined by indirect immunofluorescence in non-permeabilized cells using the anti-GFP antibody. GFP was not detected extracellularly in any of the chimeras detected at the plasma membrane (data not shown), suggesting that it faced the cytoplasmic side of the membranes.
The NS4B-GFP chimeric mutants associate with cellular membranes
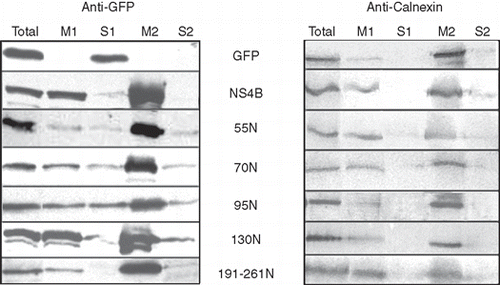
Membrane binding of the NS4B-GFP chimeric mutants was analyzed biochemically by recovering membranes from cells transiently expressing the 55N, 70N, 95N, 130N and 191-261N-GFP chimeras, and submitting them to a carbonate buffer treatment at pH 11.5. This procedure, used to separate integral from peripheral membrane-bound proteins [Citation41], allowed here to examine the nature of association of the NS4B deletions with the cellular membranes. The 130N-GFP was almost completely recovered in the membrane fractions (; M1 and M2) as the NS4B-GFP while the GFP was found exclusively in the soluble fraction of the homogenate (; S1). All the other GFP chimeras including the 55N-GFP, 70N-GFP and 191-261N-GFP which are devoid of TMs, were also recovered in large extent in the M1 membrane fractions, and remained associated with the membranes (, M2 fraction) after pH treatment although to a lesser extent than NS4B-GFP and 130N-GFP. The 55N-GFP, 70N-GFP and 95N-GFP chimeras were also recovered in the S1 and S2 fractions, a result indicating a less tight association with the membranes. Calnexin, a known ER integral membrane protein, was mainly detected in the membrane fractions (, M1 and M2) as expected for an integral membrane protein. In a separate analysis, using a Triton X-114 phase separation assay, we found that all the NS4B-GFP chimeras partitioned, though to different extents, in the detergent phase, a result confirming their lipophilic nature (Supplementary Figure S4A and B, online version only). Thus, the above results confirm that the NS4B truncated polypeptides bind to cellular membranes, as suggested by the localization studies and indicate that this binding involves other mechanisms besides integration to the membranes via transmembrane domains.
Figure 5. Partition of the GFP chimeric NS4B mutants into sodium carbonate extracted membranes. WRL68 cells, expressing GFP chimeric NS4B mutants or NS4B-GFP or GFP, were subjected to subcellular fractionation (methods) 40 h p.t. and the isolated membrane fractions were treated with 100 mM sodium carbonate pH 11.5 (methods). Proteins were analyzed by SDS PAGE (12% w/v) and Western Blot. Nitrocellulose filters were probed with the anti-GFP (0.45 μg/ml) pAb followed by the goat anti-rabbit HRP conjugated pAb and developped by the ECL system (A) and were subsequently stripped of the anti-GFP and anti-rabbit Abs, reprobed with the anti-calnexin pAb (1:2000) and the anti-rabbit HRP conjugated Ab and developped with the diaminobenzidine chromogen as a substrate for the reaction (B). Total: 1/60th of the cell homogenate subjected to centrifugation at 200,000 g, after removal of nuclei and unbroken cells; M1: membrane fraction, 1/60th of the membrane pellet collected by the 200,000 g centrifugation; S1: supernatant of the first 200,000 g centrifugation, 1/60th of the soluble fraction; M2: membranes after sodium carbonate extraction, 1/3rd of the total membrane fraction (20× more concentrated sample from M1); S2: 1/3rd of the proteins extracted with sodium carbonate treatment of the M1 fraction.
The NS4B N-terminal half retains a plasma membrane protein in the ER
To establish whether NS4B ER targeting and retention sequences could redirect other membrane proteins to the ER we attached NS4B N-terminal half sequences to the CD39 human placental ecto-ATP diphosphohydrolase I (), a plasma membrane glycoprotein with an extracellular central enzyme domain flanked by two long hydrophophobic TMs ( [Citation47]). Two more NS4B deletions, the 113N and 85-130N, were constructed for this part of the study, on the basis of recently published data [Citation48,Citation49] suggesting that the NS4B TM1 is located downstream the putative TM1 predicted by the transmembrane prediction methods used in this study and by others [Citation26].
Figure 6. (A) ER Transmembrane topology model of the human placental CD39 protein encoded by the hpCD39 plasmid [Citation35]. Numbering corresponds to extramembrane regions. (B) Schematic representation of the GFP-CD39 chimeric NS4B mutants. Regions of the NS4B N-terminal half were inserted between the GFP and the CD39 coding sequences as described in methods. White boxes: NS4B sequence; Light grey boxes: CD39 sequence; Dark grey boxes: GFP sequence. Dark gray vertical bars: the position of the putative NS4B and CD39 TM helices. The NS4B residues in the chimeras are numbered at the beginning and the end of the white boxes. The molecular size of the chimeras is indicated on the right.
![Figure 6. (A) ER Transmembrane topology model of the human placental CD39 protein encoded by the hpCD39 plasmid [Citation35]. Numbering corresponds to extramembrane regions. (B) Schematic representation of the GFP-CD39 chimeric NS4B mutants. Regions of the NS4B N-terminal half were inserted between the GFP and the CD39 coding sequences as described in methods. White boxes: NS4B sequence; Light grey boxes: CD39 sequence; Dark grey boxes: GFP sequence. Dark gray vertical bars: the position of the putative NS4B and CD39 TM helices. The NS4B residues in the chimeras are numbered at the beginning and the end of the white boxes. The molecular size of the chimeras is indicated on the right.](/cms/asset/f20b2261-bcaa-44ee-925e-59c8bf47d393/imbc_a_442821_f0006_b.gif)
Correct expression and folding of the chimeras was verified by the GFP fluorescence () detection of the NS4B sequences with the anti-NS4B-N antibody (not shown) and detection of the CD39 ecto domain and ATPase activity at the cell surface (, ). In all cells examined, all chimeras except the GFP-130N-CD39 were localized at the plasma membrane, besides the ER and Golgi ( and ). The GFP-130N-CD39 chimera was mostly detected in the ER ( and ) and only in a very small proportion of cells was found at the cell surface, 44 h post transfection (data not shown).
Figure 7. Localization of the GFP-CD39 chimeric NS4B mutants in Huh7 cells. Huh7 cells expressing GFP-CD39 chimeric NS4B mutants or GFP-CD39. Localization of the GFP chimeras was analyzed 24 h p.t. by confocal microscopy. Representative images of the GFP auto fluorescence are shown in black and white
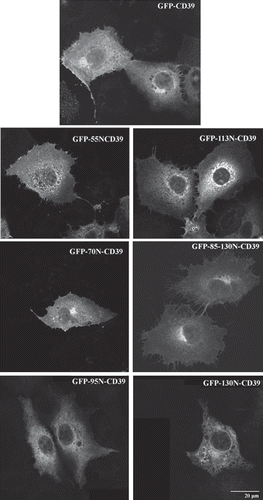
Figure 8. (A) Extracellular detection of the CD39 ecto domain in the GFP-CD39 chimeric NS4B mutants. Huh7 cells expressing GFP-CD39 chimeric NS4B mutants or GFP-CD39 were stained 24 h p.t. without fixation with the mouse anti-CD39 mAb followed by the anti-mouse Alexa546 pAb and were subsequently fixed and mounted on coverslips. Merged images of the green (GFP) and red (Alexa546) fluorescence are presented. The red fluorescence denotes the extracellular localization of the CD39 ecto-domain. (B) Cell surface ATPase activity of the GFP-CD39 chimeric NS4B mutants. ATPase activity of intact WRL68 cells transiently expressing the GFP-CD39 chimeric NS4B mutants, was measured at 24 or 44 h p.t. in the presence of ATP and was expressed as the percentage of the ATPase activity in cells expressing GFP-CD39. The estimated ATPase activity values for each chimera were normalized for the population of transfected cells estimated by FACS (methods). Activity assays were performed in duplicates in at least two independent experiments. Error bars represent standard errors (for two values from independent experiments).
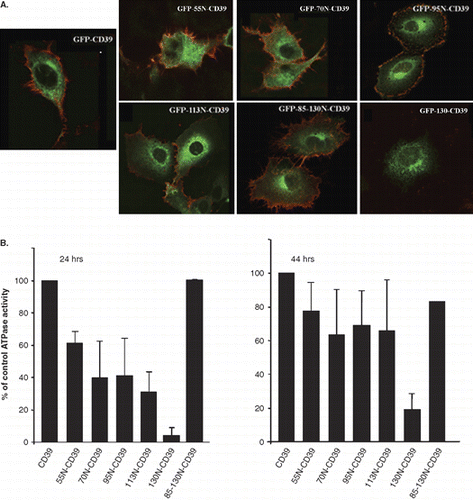
The CD39 chimeras containing the 55N, 70N, 95N and 113N NS4B sequences showed reduced cell surface ATPase activity (35–65%) when compared to the activity of the GFP-CD39 at 24 h p.t. However, this activity returned nearly to 65– > 80% of the control activity 44 h p.t. () indicating that addition of the NS4B sequences delayed trafficking of the chimeras to the cell surface. In contrast, the GFP-130N-CD39 showed dramatically reduced activity with respect to the GFP-CD39 (10% residual activity) which increased only by two-fold (20% activity) 44 h p.t. (). Interestingly, the ATPase activity of GFP-85-130N-CD39 was similar to that of GFP-CD39. Altogether, these data suggest that both the NS4B TM1 and TM2 expected to locate between residues 85 and 130, although necessary, are not sufficient to retain the CD39 protein intracellularly and that sequences upstream of these two TMs in the NS4B N-terminal half are required to provide strong ER retention in these chimeras.
Ιmmunofluorescence staining with antibodies directed against the CD39 ecto-domain, the GFP or the NS4B N-terminal 55peptide showed that, with exception of the GFP-130N-CD39 chimera, the CD39 ecto-domain of all the other chimeras was exposed at the cell surface in the majority of the transfected cells () while the GFP or the NS4B N-terminal 55peptide were not (data not shown). This suggests topology models where the NS4B TM1 alone cannot be fully inserted in the membrane (Supplementary Figure S5B) while it can do so when paired with TM2. Indeed the GFP-85-130N-CD39 and the GFP-130N-CD39 chimeras, can have a successful insertion in the ER membrane exposing their TM1 and TM2 connecting loop to the ER lumen (Supplementary Figure S5C) as it has been shown experimentally by others [Citation48]. The topology model suggested in Supplementary Figure S5B (online version only) was further supported by results confirming that GFP translocates correctly across the ER. This was examined by tracking the localization of the NtCD39-GFP polypeptide, consisting of the N-terminal cytoplasmic and transmembrane domain of the hpCD39 (a.a. 1–44) fused to GFP, in transfected cells. In non-permeabilized cells, expressing Nt-CD39-GFP at the plasma membrane, the GFP was detected at the cell surface (data not shown).
Discussion
NS4B is an integral membrane protein essential for HCV RNA replication. Our study addresses for the first time the questions concerning anchoring and retention of this protein in the ER membrane and identifies several regions in the NS4B sequence involved in the NS4B interaction with ER membranes that may be implicated in different steps of the HCV life cycle in the infected cell. Furthermore, it extends previous results on NS4B membrane topology by providing valuable experimental evidence complementing recent in silico analyses [Citation49] that define the boundaries of TM1 and TM2.
The HCV NS4B protein lacks specific ER retention signals identified thus far for membrane proteins, i.e., di-Lys motifs [Citation27] at the C-terminus or di-Arg motifs at the N-terminus [Citation28,Citation29]. In the case of the HCV E1, E2 [Citation50] and NS2 [Citation51] proteins and in a number of other membrane proteins, ER retention or sorting in the Golgi and plasma membrane is often determined by the length and the amino acid composition of the hydrophobic segment in their TM(s) [Citation52–54]. Short TM helices (< 17 residues) promote ER localization. Short TMs are present in the N-terminal half of NS4B () and therefore could account for its ER retention. Here we show that ER retention of NS4B requires the two short TM1 and TM2 helices located at its N-terminal half. Furthermore, our results indicate that the pair of TM1 and TM2 are necessary but not sufficient for NS4B ER retention, in contrast with other multispanning membrane proteins where pairs of TMs were found sufficient for ER retention [Citation55]. Using different NS4B chimeras engineered to express the CD39 ectonucleotidase at the cell surface as a convenient marker of trafficking, we showed that the simultaneous expression of both TM1 and TM2, within the 85-130N domain of NS4B did not interfere with the trafficking of CD39 to the plasma membrane (, ). This suggests that other N-terminal structural features upstream residue 85 are also required for NS4B ER retention. The sequences involved in the NS4B oligomerization [Citation56] could contribute to this, and this aspect merits further investigation. Interestingly, oligomerization has been proposed as ER retention mechanism for other proteins [Citation57]. The possibility that some of the chimeras used in this study were retained non-specifically in the ER due to misfolding cannot be excluded. However, we argue that retention of the 130N-GFP and the GFP-130N-CD39 chimeras in the ER is the result of signaling by specific structural features since both the GFP and the CD39 molecules in the two sets of chimeras used here folded correctly. The latter was verified by the detection of GFP fluorescence and CD39 ATPase activity at the cell surface of the transfected cells.
Figure 9. Suggested boundaries for TM1 and TM2 in the NS4B N-terminal half. The NS4B primary structure between residues 66-135 is shown. The sequences proposed as TM1 and TM2 helices are presented in grey background framed by broken line. The Pro residues, and the putative Nucleotide Binding site (NBD [Citation31]) are underlined and indicated in bold. Under each residue is shown the PROF (http://www.aber.ac.uk/∼phiwww/prof/) prediction for being in a helix (H), in a coil (C) or a b-strand. The helical regions consisting of residues with prediction reliability accuracy for being in a helix > 0.6 (Supplementary Figure S2, online version), are indicated as H1, H2 and H3 and are underlined. Helices H2 and H3 show < 25% solvent accessibility in their entire length and < 5% solvent accessibility in their 10 central residues. Residues 110-113, framed with a solid line, are proposed to form the ER luminal loop connecting TM1 and TM2.
![Figure 9. Suggested boundaries for TM1 and TM2 in the NS4B N-terminal half. The NS4B primary structure between residues 66-135 is shown. The sequences proposed as TM1 and TM2 helices are presented in grey background framed by broken line. The Pro residues, and the putative Nucleotide Binding site (NBD [Citation31]) are underlined and indicated in bold. Under each residue is shown the PROF (http://www.aber.ac.uk/∼phiwww/prof/) prediction for being in a helix (H), in a coil (C) or a b-strand. The helical regions consisting of residues with prediction reliability accuracy for being in a helix > 0.6 (Supplementary Figure S2, online version), are indicated as H1, H2 and H3 and are underlined. Helices H2 and H3 show < 25% solvent accessibility in their entire length and < 5% solvent accessibility in their 10 central residues. Residues 110-113, framed with a solid line, are proposed to form the ER luminal loop connecting TM1 and TM2.](/cms/asset/a0987bca-5c5c-4bdd-8d74-46842835c2bb/imbc_a_442821_f0009_b.gif)
The question of NS4B ER anchoring has not been raised before. For multispanning membrane proteins, the first transmembrane helix often plays this role. In the case of NS4B, the putative TM1 could not be efficiently inserted in the ER membrane in the absence of downstream sequences including TM2 as shown by the topology of the GFP and CD39 ecto-domain of the GFP-113N-CD39 chimera. The 28peptidic linker region between the NS4B N-terminal sequences and the CD39 TM1 contains five positively charged residues which would favor a cytoplasmic localization [Citation60]. This could dictate a type I signal anchor orientation for the NS4B TM1 domain in the GFP-113N-CD39 chimera resulting in translocation of GFP across the ER membrane and its subsequent exposure at the cell surface. However, in our study, GFP was not detected at the surface of intact cells expressing the GFP-113N-CD39 chimera (data not shown) which suggests that the NS4B TM1 is not inserted in the ER membrane. NS4B TM1 probably behaves as a flexible helix that can be inserted in and exit the ER membrane, with no stable integration (Supplementary Figure S5B). The two glycines in the middle of the TM1 helix could confer the required flexibility to the helical structure allowing these orientation changes. A number of proteins exist where a given TM segment will insert properly in the membrane only in the presence of its neighbouring TMs [Citation30,Citation61–65], implying that topogenic information present in more than one TM must be “decoded” simultaneously. For certain proteins the hydrophobic anchors may be too short, as in the case of NS4B TM1 and TM2, and may need to assemble with other TM segments in the channel before they can stably integrate into the lipid phase as one hydrophobic unit [Citation65,Citation66].
NS4B, besides its TMs, contains other sequences in its N-terminal half that mediate membrane binding as indicated by the localization and the membrane binding of the 55N and 70N-GFP chimeras. In accord with previous findings [Citation31], we found that its first 55peptidic N-terminal sequence was sufficient for association with cellular membranes including the ER. However, we demonstrated that the previously identified amphipathic N-terminal helix (a.a. 1–27) [Citation31] is functionally redundant although it could act as an initial ER targeting signal during NS4B synthesis. In our study, the 43-195N-GFP chimera, showed exclusive ER localization. The second amphipathic helix, predicted to fold between residues 45–62, or the highly conserved LRRs-like motif (residue 58–70), could bind first with the ER membrane insertion machinery. The amphipathic helices at the NS4B N-terminus may behave as the N- or C-terminal amphipathic helices of NS5A or NS5B HCV non-structural proteins respectively [Citation67–69] which bind to membranes by adsorption of their hydrophobic phase to the cytoplasmic membrane leaflet. The functional importance of the NS4B N-terminal amphipathic helices in the context of the entire HCV polyprotein probably extends beyond that of ER targeting to interactions with other non-structural HCV proteins essential for formation of the replication complex or with host cell proteins important for virus replication. The second N-terminal amphipathic helix could also participate in the oligomerization of the molecule [Citation56], an aspect that merits further investigation. Lastly, the LRRs like motif, can form amphipathic structures with hydrophobic surfaces capable of interacting with membranes or with other proteins [Citation70,Citation71].
Most importantly in the absence of all four predicted TMs, the 70peptidic C-terminal region of NS4B (a.a. 191–261) mediates association and anchoring of the 191-261-GFP chimera to membranes as shown by detection of the chimera in the membrane fraction after carbonate wash () and localization of the 191-261N-GFP chimera in perinuclear clusters of vesicles positive for ER markers (–). It is possible that the NS4B C-terminal domain is involved in the induction of the membrane alterations described as “membranous web” where the HCV replication complex is localized [Citation23]. Palmitoylation at the NS4B C-terminal Cys 257 and Cys 261 residues [Citation56] may account for membrane binding of the 70peptidic NS4B C-terminal domain.
Finally, the NS4B TM1 and TM2 boundaries assigned by the prediction methods used in this study (Supplementary Table S1) or in previous work [Citation26] were not confirmed by our experimental data. Within the amino acid stretch 66-135 in the NS4B N-terminal half, three helices were predicted with high reliability accuracy to fold between Pro74, Pro89, and Pro112 ( and Supplementary Figure S2). Helix H1 (a.a. 74–84) is too short to be transmembrane, since a minimum of 16 residues are required to form a TM α-helix [Citation58]. However, the helices H2 (93-109) and H3 (a.a. 114–129), with 17 and 16 residues respectively, are sufficiently hydrophobic (, legend), although less hydrophobic than typical TM helices, and fulfil the minimum length requirements to be transmembrane [Citation59]. They could therefore be the first two NS4B TMs. In addition, considering that the putative nucleotide binding site in the NS4B sequence (a.a. 129–135) [Citation31] must be part of a cytoplasmic domain or close to the ER membrane-cytoplasm interface, the putative TM2 should not extend further than residue 129. The 4peptide AAPG, between the proposed TM1 and TM2 helices contains residue 112 shown in the study by Lundin et al. [Citation48] to face the ER lumen and could therefore form the TM1 and TM2 connecting loop. The minimal length of a turn between TM helical hairpins has been found to be two to three residues, with Pro and Gly amongst the most frequently found amino acids in these turns [Citation58].
Taken together, the results of this study indicate multiple levels of interaction of the HCV NS4B protein with ER membranes, suggesting a pleiotropic role for this protein in the implication of HCV with the host cell secretory machinery. Our data contribute significant information to further design more detailed analyses for the structural functional relationships of the NS4B membrane binding elements in the HCV infectious cell culture system. These studies will explore the role(s) of NS4B in robust RNA replication and in the production of infectious particles.
Supplementary Material
Download PDF (463 KB)Acknowledgements
This work was supported by grants from the International Network of the Pasteur Institutes and the Institut Pasteur Hellenique as well by a French INSERM grant (grant number HEPO4O4). The technical assistance of Elina Aslanoglou for the production of the polyclonal antibodies is acknowledged. The authors also thank Dr Ketty Sotiriadou for her invaluable support during the last phase of this work.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Bienz K, Egger D, Pfister T, Troxler M. 1992. Structural and functional characterization of the poliovirus replication complex. J Virol 66:2740–2747.
- Pedersen KW, van der Meer Y, Roos N, Snijder EJ. 1999. Open reading frame 1a-encoded subunits of the arterivirus replicase induce endoplasmic reticulum-derived double-membrane vesicles which carry the viral replication complex. J Virol 73:2016–2026.
- Schaad MC, Jensen PE, Carrington JC. 1997. Formation of plant RNA virus replication complexes on membranes: Role of an endoplasmic reticulum-targeted viral protein. Embo J 16:4049–4059.
- Salonen A, Ahola T, Kaariainen L. 2005. Viral RNA replication in association with cellular membranes. Curr Top Microbiol Immunol 285:139–173.
- Wasley A, Alter MJ. 2000. Epidemiology of hepatitis C: Geographic differences and temporal trends. Semin Liver Dis 20:1–16.
- Seeff LB. 2002. Natural history of chronic hepatitis C. Hepatology 36:S35–46.
- Lindenbach BD, Rice CM. 2001. Flaviviridae, the viruses and their replication. In: Knipe DM, Howley PM, editors. Fields Virology. Lippincott Williams & Wilkins publishers; Philadelphia, USA. pp 991–1042.
- Selby MJ, Choo QL, Berger K, Kuo G, Glazer E, Eckart M, Lee C, Chien D, Kuo C, Houghton M. 1993. Expression, identification and subcellular localisation of the proteins encoded by the hepatitis C viral genome. J Gen Virol 74(Pt 6):1103–1113.
- Lohmann V, Korner F, Koch J, Herian U, Theilmann L, Bartenschlager R. 1999. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 285:110–113.
- Grakoui A, McCourt DW, Wychowski C, Feinstone SM, Rice CM. 1993. Characterization of the hepatitis C virus-encoded serine proteinase: Determination of proteinase-dependent polyprotein cleavage sites. J Virol 67:2832–2843.
- Rosenberg S. 2001. Recent advances in the molecular biology of hepatitis C virus. J Mol Biol 313:451–464.
- Vassilaki N, Mavromara P. 2003. Two alternative translation mechanisms are responsible for the expression of the HCV ARFP/F/core+1 coding open reading frame. J Biol Chem 278:40503–40513.
- Bartenschlager R, Lohmann V. 2001. Novel cell culture systems for the hepatitis C virus. Antiviral Res 52:1–17.
- Blight KJ, McKeating JA, Marcotrigiano J, Rice CM. 2003. Efficient replication of hepatitis C virus genotype 1a RNAs in cell culture. J Virol 77:3181–3190.
- Lindenbach BD, Evans MJ, Syder AJ, Wolk B, Tellinghuisen TL, Liu CC, Maruyama T, Hynes RO, Burton DR, McKeating JA, Rice CM. 2005. Complete replication of hepatitis C virus in cell culture. Science 309:623–626.
- Bartenschlager R. 2006. Hepatitis C virus molecular clones: From cDNA to infectious virus particles in cell culture. Curr Opin Microbiol 9:416–422.
- Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z, Murthy K, Habermann A, Krausslich HG, Mizokami M, Bartenschlager R, Liang TJ. 2005. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med 11:791–796.
- Westaway EG, Mackenzie JM, Kenney MT, Jones MK, Khromykh AA. 1997. Ultrastructure of Kunjin virus-infected cells: Colocalisation of NS1 and NS3 with double-stranded RNA, and of NS2B with NS3, in virus-induced membrane structures. J Virol 71:6650–6661.
- Cho MW, Teterina N, Egger D, Bienz K, Ehrenfeld E. 1994. Membrane rearrangement and vesicle induction by recombinant poliovirus 2C and 2BC in human cells. Virology 202:129–145.
- Irurzun A, Perez L, Carrasco L. 1992. Involvement of membrane traffic in the replication of poliovirus genomes: Effects of brefeldin A. Virology 191:166–175.
- Teterina NL, Bienz K, Egger D, Gorbalenya AE, Ehrenfeld E. 1997. Induction of intracellular membrane rearrangements by HAV proteins 2C and 2BC. Virology 237:66–77.
- van der Meer Y, van Tol H, Locker JK, Snijder EJ. 2005. ORF1a-encoded replicase subunits are involved in the membrane association of the arterivirus replication complex. J Virol 6689–6698.
- Egger D, Wolk B, Gosert R, Bianchi L, Blum HE, Moradpour D, Bienz K. 2002. Expression of hepatitis C virus proteins induces distinct membrane alterations including a candidate viral replication complex. J Virol 76:5974–5984.
- Lindstrom H, Lundin M, Haggstrom S, Persson MA. 2006. Mutations of the Hepatitis C virus protein NS4B on either side of the ER membrane affect the efficiency of subgenomic replicons. Virus Res 121:169–178.
- Hugle T, Fehrmann F, Bieck E, Kohara M, Krausslich HG, Rice CM, Blum HE, Moradpour D. 2001. The hepatitis C virus nonstructural protein 4B is an integral endoplasmic reticulum membrane protein. Virology 284:70–81.
- Lundin M, Monne M, Widell A, Von Heijne G, Persson MA. 2003. Topology of the membrane-associated hepatitis C virus protein NS4B. J Virol 77:5428–5438.
- Teasdale RD, Jackson MR. 1996. Signal-mediated sorting of membrane proteins between the endoplasmic reticulum and the golgi apparatus. Annu Rev Cell Dev Biol 12:27–54.
- Schutze MP, Peterson PA, Jackson MR. 1994. An N-terminal double-arginine motif maintains type II membrane proteins in the endoplasmic reticulum. Embo J 13:1696–1705.
- Zerangue N, Malan MJ, Fried SR, Dazin PF, Jan YN, Jan LY, Schwappach B. 2001. Analysis of endoplasmic reticulum trafficking signals by combinatorial screening in mammalian cells. Proc Natl Acad Sci USA 98:2431–2436.
- Heinrich SU, Rapoport TA. 2003. Cooperation of transmembrane segments during the integration of a double-spanning protein into the ER membrane. Embo J 22:3654–3663.
- Einav S, Elazar M, Danieli T, Glenn JS. 2004. A nucleotide binding motif in hepatitis C virus (HCV) NS4B mediates HCV RNA replication. J Virol 78:11288–11295.
- Kalamvoki M, Miragou V, Hadziyannis A, Georgopoulou U, Varklioti A, Hadzigiannis S, Mavromara P. 2002. Expression of immunoreactive forms of the hepatitis C NS5A protein in E. coli and their use for diagnostic assays. Arch Virol 1733–1745.
- Harlow E, Lane D. 1988. Antibodies. A laboratory manual. Cold Spring Harbor Laboratory.
- Kolykhalov AA, Agapov EV, Blight KJ, Mihalik K, Feinstone SM, Rice CM. 1997. Transmission of hepatitis C by intrahepatic inoculation with transcribed RNA. Science 277:570–574.
- Papanikolaou A, Papafotika A, Murphy C, Papamarcaki T, Tsolas O, Drab M, Kurzchalia TV, Kasper M, Christoforidis S. 2005. Cholesterol-dependent lipid assemblies regulate the activity of the ecto-nucleotidase CD39. J Biol Chem 280:26406–26414.
- Rost B, Fariselli P, Casadio R. 1996. Topology prediction for helical transmembrane proteins at 86% accuracy. Protein Sci 5:1704–1718.
- Tusnady GE, Simon I. 1998. Principles governing amino acid composition of integral membrane proteins: Application to topology prediction. J Mol Biol 283:489–506.
- Tusnady GE, Simon I. 2001. The HMMTOP transmembrane topology prediction server. Bioinformatics 17:849–850.
- Bordier C. 1981. Phase separation of integral membrane proteins in Triton X-114 solution. J Biol Chem 256:1604–1607.
- Graham JM. 1993. Isolation of membranes from tissue culture cells. Methods Mol Biol 19:97–108.
- Fujiki Y, Hubbard AL, Fowler S, Lazarow PB. 1982. Isolation of intracellular membranes by means of sodium carbonate treatment: Application to endoplasmic reticulum. J Cell Biol 93:97–102.
- Lanzetta PA, Alvarez LJ, Reinach PS, Candia OA. 1979. An improved assay for nanomole amounts of inorganic phosphate. Anal Biochem 100:95–97.
- Mezzacasa A, Helenius A. 2002. The transitional ER defines a boundary for quality control in the secretion of tsO45 VSV glycoprotein. Traffic 833–849.
- Mottola G, Cardinali G, Ceccacci A, Trozzi C, Bartholomew L, Torrisi MR, Pedrazzini E, Bonatti S, Migliaccio G. 2002. Hepatitis C virus nonstructural proteins are localized in a modified endoplasmic reticulum of cells expressing viral subgenomic replicons. Virology 293:31–43.
- Melen K, Krogh A, von Heijne G. 2003. Reliability measures for membrane protein topology prediction algorithms. J Mol Biol 327:735–744.
- Kanaji S, Iwahashi J, Kida Y, Sakaguchi M, Mihara K. 2000. Characterization of the signal that directs Tom20 to the mitochondrial outer membrane. J Cell Biol 277–288.
- Matsumoto M, Sakurai Y, Kokubo T, Yagi H, Makita K, Matsui T, Titani K, Fujimura Y, Narita N. 1999. The cDNA cloning of human placental ecto-ATP diphosphohydrolases I and II. FEBS Lett 453:335–340.
- Lundin M, Lindstrom H, Gronwall C, Persson MA. 2006. Dual topology of the processed hepatitis C virus protein NS4B is influenced by the NS5A protein. J Gen Virol 87:3263–3272.
- Welsch C, Albrecht M, Maydt J, Herrmann E, Welker MW, Sarrazin C, Scheidig A, Lengauer T, Zeuzem S. 2007. Structural and functional comparison of the non-structural protein 4B in flaviviridae. J Mol Graph Model 26:546–557.
- Charloteaux B, Lins L, Moereels H, Brasseur R. 2002. Analysis of the C-terminal membrane anchor domains of hepatitis C virus glycoproteins E1 and E2: Toward a topological model. J Virol 76:1944–1958.
- Yamaga AK, Ou JH. 2002. Membrane topology of the hepatitis C virus NS2 protein. J Biol Chem 277:33228–33234.
- Pedrazzini E, Villa A, Borgese N. 1996. A mutant cytochrome b5 with a lengthened membrane anchor escapes from the endoplasmic reticulum and reaches the plasma membrane. Proc Natl Acad Sci USA 93:4207–4212.
- Lundbaek JA, Andersen OS, Werge T, Nielsen C. 2003. Cholesterol-induced protein sorting: An analysis of energetic feasibility. Biophys J 84:2080–2089.
- Barre L, Magdalou J, Netter P, Fournel-Gigleux S, Ouzzine M. 2005. The stop transfer sequence of the human UDP-glucuronosyltransferase 1A determines localisation to the endoplasmic reticulum by both static retention and retrieval mechanisms. Febs J 272:1063–1071.
- Parker AK, Gergely FV, Taylor CW. 2004. Targeting of inositol 1,4,5-trisphosphate receptors to the endoplasmic reticulum by multiple signals within their transmembrane domains. J Biol Chem 279:23797–23805.
- Yu GY, Lee KJ, Gao L, Lai MM. 2006. Palmitoylation and polymerization of hepatitis C virus NS4B protein. J Virol 80:6013–6023.
- Schweizer A, Rohrer J, Hauri HP, Kornfeld S. 1994. Retention of p63 in an ER-Golgi intermediate compartment depends on the presence of all three of its domains and on its ability to form oligomers. J Cell Biol 126:25–39.
- Monné M, Nilsson I, Elofsson A, von Heijne G. 1999. Turns in transmembrane helices: Determination of the minimal length of a “helical hairpin” and derivation of a fine-grained turn propensity scale. J Mol Biol 293:807–814.
- Wallin E, Tsukihara T, Yoshikawa S, von Heijne G, Elofsson A. 1997. Architecture of helix bundle membrane proteins: An analysis of cytochrome c oxidase from bovine mitochondria. Protein Sci 6:808–815.
- Kida Y, Morimoto F, Mihara K, Sakaguchi M. 2006. Function of positive charges following signal-anchor sequences during translocation of the N-terminal domain. J Biol Chem 281:1152–1158.
- Gafvelin G, Sakaguchi M, Andersson H, von Heijne G. 1997. Topological rules for membrane protein assembly in eukaryotic cells. J Biol Chem 272:6119–6127.
- Lu Y, Xiong X, Helm A, Kimani K, Bragin A, Skach WR. 1998. Co- and posttranslational translocation mechanisms direct cystic fibrosis transmembrane conductance regulator N terminus transmembrane assembly. J Biol Chem 273:568–576.
- Zhang JT, Lee CH, Duthie M, Ling V. 1995. Topological determinants of internal transmembrane segments in P-glycoprotein sequences. J Biol Chem 270:1742–1746.
- Chen M, Zhang JT. 1996. Membrane insertion, processing, and topology of cystic fibrosis transmembrane conductance regulator (CFTR) in microsomal membranes. Mol Membr Biol 13:33–40.
- Monné M, Gafvelin G, Nilsson R, von Heijne G. 1999. N-tail translocation in a eukaryotic polytopic membrane protein: Synergy between neighbouring transmembrane segments. Eur J Biochem 263:264–269.
- Lin J, Addison R. 1995. A novel integration signal that is composed of two transmembrane segments is required to integrate the Neurospora plasma membrane H(+)-ATPase into microsomes. J Biol Chem 270:6935–6941.
- Brass V, Bieck E, Montserret R, Wolk B, Hellings JA, Blum HE, Penin F, Moradpour D. 2002. An amino-terminal amphipathic alpha-helix mediates membrane association of the hepatitis C virus nonstructural protein 5A. J Biol Chem 277:8130–8139.
- Dubuisson J, Penin F, Moradpour D. 2002. Interaction of hepatitis C virus proteins with host cell membranes and lipids. Trends Cell Biol 12:517–523.
- Schmidt-Mende J, Bieck E, Hugle T, Penin F, Rice CM, Blum HE, Moradpour D. 2001. Determinants for membrane association of the hepatitis C virus RNA-dependent RNA polymerase. J Biol Chem 276:44052–44063.
- Kobe B, Kajava AV. 2001. The leucine-rich repeat as a protein recognition motif. Curr Opin Struct Biol 11:725–732.
- Kedzierski L, Montgomery J, Curtis J, Handman E. 2004. Lecine-rich repeats in host-pathogen interactions. Arch Immunol Ther Exp (Warsz) 104–112.