Abstract
Since being introduced globally as Aspirin in 1899, acetylsalicylic acid (ASA) has been widely used as an analgesic, immune-regulatory, anti-pyretic and anti-thrombotic drug. ASA and its metabolite, salicylate, were also reported to be able to modulate antigen presenting functions of dendritic cells (DC). However, the intracellular targets of ASA in DC are still poorly understood. Since phagocytosis is the initial step taken by antigen-presenting cells in the uptake of antigens for processing and presentation, ASA might exerts its immune-regulatory effects by regulating phagocytosis. Here we show that ASA inhibits phagocytosis and modulates expression of endosomal SNAREs, such as Vti1a, Vti1b, VAMP-3, VAMP-8 and Syn-8 (but not syn-6 and syn-16) in DC. We further show that the phagocytic inhibitory effect of ASA is dependent on the expression of Vti1a and Vti1b. Consistently, Vti1a and Vti1b localize to the phagosomes and up-regulation of Vti1a and Vti1b inhibits phagocytosis in DC. Our results suggest that ASA modulates phagocytosis in part through the control of endosomal SNARE protein expression and localization in DC. All experiments were performed using either a murine DC line (DC2.4) or primary DC derived from murine bone marrow cells.
Keywords::
Introduction
Since being introduced globally as aspirin in 1899, acetylsalicylic acid (ASA) has been widely used as an analgesic, anti-inflammation, anti-pyretic, and anti-thrombotic drug. Implications of ASA in preventing colon cancer and Alzheimer's disease are of great interest to many researchers (Etminan et al. Citation2003). Initially, most of these effects were thought to closely link to the inhibition of cyclooxygenase (COX-1 and COX-2) activity. New COX-independent anti-inflammation pathways of ASA have been reported, such as the inhibition of the transcription factor NF-κB (Kopp and Ghosh Citation1994, Grilli et al. Citation1996), and its nitric oxide (NO)-dependent effect (Fiorucci et al. Citation2001). Interestingly, Matasic et al. (Citation2000) showed that ASA and its metabolite, salicylate, can affect antigen presentation functions of DC. Hackstein et al. (Citation2001) reported that ASA can inhibit the response of immature DC to inflammatory signals and thus suppresses its maturation. These effects were later shown to be COX-independent.
Since phagocytosis is the initial step taken by antigen-presenting cells for the uptake of antigens for processing and presentation, we envisage that other than down-regulating surface MHC II expression, ASA might exerts its immune-regulatory effects by regulating phagocytosis in the immune cells, DC. Phagocytosis is an active process, where bound pathogen is surrounded by the phagocyte membrane and internalized in a membrane-bound vesicle. Phagocytosis of microorganisms by immature dendritic cells (DC) is an essential component of the host's defence against infection.
The formation of phagosomes requires the targeted delivery of membrane vesicles from the recycling endosome compartment, which contributes to the elongation of membranous pseudopods during the early stages of the phagocytosis process. Thus, it is conceivable that endosomal SNARE proteins, which mediate membrane fusion, have a role to play in this process (Booth et al. Citation2001). Here we show that ASA modulates phagocytosis in DC. Furthermore, our results also suggest that the ASA-mediated modulation of phagocytosis in DC is partly dependent on the expression of endosomal SNARE proteins.
Materials and methods
Cell culture, antibodies and chemicals
A murine dendritic cell line, DC2.4 (Shen et al. Citation1997), was cultured in Dulbecco's Modified Eagle Media (DMEM) (Gibco) supplemented with 10% fetal bovine serum (FBS) (Gibco). DC2.4 cells display dendritic morphology, express dendritic cell-specific markers, MHC molecules, co-stimulatory molecules, and have phagocytic activity as well as antigen-presenting capacity (Shen et al., 1997). Raw 264.7 mouse monocytes/macrophages cell line (TIB-71) were purchased from ATCC. Mouse bone marrow derived DC (murine primary DC) were prepared according to method described previously (Wong et al. Citation2004, Santambrogio et al. Citation2005). Briefly, mice (C57BL/6) were sacrificed and their femorous limbs excised. Bone marrow cells were then flushed out from the femurs, cultured in the presence of murine recombinant GM-CSF for six days to produce immature DC. Mouse anti-c-myc antibodies (Cat #OP10) were purchased from Calbiochem. Rabbit anti-c-myc antibodies (Cat #sc-788) were purchased from Santa Cruz Biotechnology.
cDNA constructs
Vti1a and Vti1b cDNA was amplified by PCR using pfu DNA polymerase and oligomers (AAGCTCGAGCGGTCTTCCGACTTCGAAGGGTACG and GCTCTAGAGCTCAGTGTCCTTTGACG) and digested with XhoI and XbaI. Vti1b cDNA was amplified similarly using the oligomers (GGAATTCCGCCGCCTCCGCCGCC TCCTCCGAGCATTTCGAGAAG and GCTCTAGAGCTCAATGGTGTCGAAAG), and digested with EcoRI and XbaI. Both products were ligated into the vector pDMyc, to be in frame with the C-terminal double myc tag.
Transfection and immunofluorescence microscopy
Growing of cells on cover slips and transfection were performed according to previously established methods (Lowe et al. Citation1997, Wong et al. Citation1998, Xu et al. Citation1998). Briefly, transfected cells (24 h post-transfection) were fixed with 4% paraformaldehyde in PBS, washed, permeabilized with 0.1% saponin for 20 min, prior to incubation in 5 μg/ml of anti-myc antibodies for 1 h. After four washes with PBS, cells were incubated in the dark with goat anti-mouse fluorescein (Chemicon International) for 1 h, washed four times in PBS, mounted and viewed under a fluorescence microscope (Olympus BX60). As for the siRNA transfection study, transfection was performed using Invitrogen LipofectamineTM 2000. SiRNA directed towards Vti1a (catalog number: LQ-040824-01), Vti1b (catalog number: LQ-062978-01), as well as non-targeting siRNA pool (catalog number: D-001810-10), were purchased from Dharmacon. The amount of siRNA used for transfection was optimized at 2000 pmol per 10 cm dish transfection. The cells were assayed for their phagocytic activity 48 h post-transfection.
Phagosome isolation
DC2.4 cells were allowed to internalize fixed GFP-Escherichia coli (0.75 × 109 particles/ml) for 1 h. The cells were then washed three times with ice-cold PBS to remove extracellular bacteria, trypsinized for 10 min at room temperature, and centrifuged at 100 g (720 rpm on Beckman Coulter Allegra X-22R centrifuge) for 5 min at 4°C to further remove extracellular bacteria. DC pellet was resuspended in 3 volumes of H buffer (8.6% sucrose, 25 mM Hepes, 5 mM MgCl2, 1 mM PMSF), and then homogenized with a Dounce Homogenizer. The homogenate was centrifuged at 100 g to remove intact cells, heavier cell debris and nucleus. The supernatant containing phagosomes and lighter membranes were carefully layered on 62% sucrose and centrifuged at 13,000 rpm for 45 min using Beckman Coulter OptimaTM MAX ultracentrifuge (MLS-50 rotor). The lighter membranes remained on top of the 62% sucrose, while the heavier phagosomes were pelleted to the bottom. The phagosomal pellet was washed twice with ice-cold PBS and processed for Western blot analysis.
Phagocytosis assay
Phagocytosis assay was performed using the VybrantTM Phagocytosis Assay Kit (V-6694) purchased from Molecular Probes, with some modifications from the recommended protocol. Briefly, after treatment with various concentrations of ASA, 100 μl of cell suspension containing 2 × 105 viable DC (checked by Trypan blue) were seeded on an ELISA plate and incubated for 4 h to allow the DC to adhere. Fluorescent E. coli (100 μl) was added to each well. After incubation for 2 h at 37°C, the medium was removed and Trypan blue (100 μl) was added to each well to quench un-internalized fluorescent E. coli, and incubated at room temperature for 5 min. The cells were then measured in a fluorescence plate reader (Gemini EM and Tecan, Molecular devices) using 480 nm excitation and 520 nm emission.
In vitro transcription and mRNA electroporation
mRNA was prepared using the Promega RiboMAXTM in vitro transcription system. Primary DC were resuspended in Opti-MEM (Gibco) medium at a concentration of 5 × 107 cells/ml. RNA (40 μg) was added to 200 μl of cell suspension containing 1 × 107 cells/ml and placed in a 2 mm cuvette. The mixture was placed in the electroporator (Electro Square Porator ECM 830, BTX) and a 300 V current was delivered for 500 μs. The cells were allowed to recover at 37°C for 15 min before they were resuspended in complete DMEM containing 10% FBS.
Flow cytometry
Cells were prepared according to the protocols provided in Current Protocols in Immunology (Unit 5.3, basic protocol 1 and 2). Briefly, for detection of surface antigens, cells were washed and resuspended in staining buffer containing 0.1% (w/v) NaN3 and 1% (w/v) bovine serum albumin in PBS. Labelled antibodies were added, mixed and incubated on ice for 20 min. Next, the cells were washed twice with staining buffer, resuspended in fixation solution containing 2% (w/v) paraformaldehyde in PBS and stored at 4°C until they were analyzed by flow cytometry.
Results
ASA inhibits phagocytosis in immature DC
Murine myeloid DC differentiated in the presence of ASA in vitro was shown previously to be less able in presenting antigens to T cells (Matasic et al. Citation2000, Hackstein et al. Citation2001). This was suggested to be one of the ways ASA exerts its immune-regulatory effects. Since phagocytosis is the initial step taken by antigen presenting cells for the up-take of antigens for processing and presentation, we envisage that ASA might exerts its immune-regulatory effects by regulating the phagocytosis process. To determine the effects of ASA on the phagocytic activity of DC in vitro, DC2.4 were incubated with various concentrations of ASA (0, 1, 2, 3, 4, 8, 9, and 10 mM) in DMEM for 18 h prior to incubation with fluorescence-labelled bioparticles (E. coli) in a phagocytosis assay. As shown in , we observed a slight elevation in phagocytic activity at ASA concentrations below 3 mM. However, the phagocytic activity dropped in a dose-dependent manner when DC2.4 were exposed to higher concentrations (above 3 mM) of ASA.
Figure 1. ASA modulates phagocytosis in DC and macrophages. (A) DC2.4 were incubated with various concentrations of ASA (0, 1, 2, 3, 4, 8, 9, and 10 mM) in DMEM for 18 h prior to incubation with fluorescence-labelled bioparticles (E. coli) in a phagocytosis assay. The amount of fluorescent bioparticles (E. coli) ingested by the control and ASA-treated cells was quantified using Gemini EN plate reader. (B) RAW264.7 cells were incubated with various concentrations of ASA (0, 0.1, 0.5, 1, 2, 3, 4, 5, 6, and 7 mM) in DMEM for 18 h prior to incubation with fluorescence-labelled bioparticles (E. coli) in a phagocytosis assay. The amount of fluorescent bioparticles (E. coli) ingested by the control and ASA-treated cells was quantified using Gemini EN plate reader.
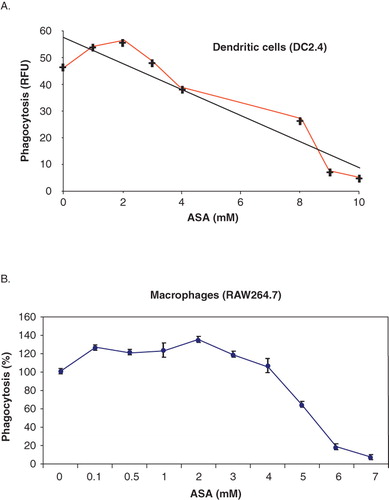
Similar results were also observed in primary DC (data not shown) and the macrophages cell line, RAW264.7 (). The ASA-mediated phagocytosis inhibitory effect observed here at concentrations above 3 mM was suggested to be COX-2/15-epi-lipoxinA4-independent in view of the fact that the ASA-triggered production of 15-epi-lipoxinA4 through modification of COX-2 activity was shown previously to enhance phagocytosis (Godson et al. Citation2000, Mitchell et al. Citation2002). This prompted us to explore the possible mechanism that governs the COX-2/15-epi-lipoxinA4-independent phagocytic inhibitory effect of ASA in DC. We have chosen to use the ASA concentration of 5 mM in the subsequent experiments. Although the ASA concentration of 5 mM is beyond the published in vivo physiological concentration (3 mM) used in the treatment of autoimmune diseases (Hackstein et al., 2001), we observed that at this concentration, the morphology and viability of cells were not altered significantly as compared to those treated with ASA at 3 mM (data not shown). Furthermore, we consistently and repeatedly observed a significant reduction in phagocytosis activity in DC upon treatment with 5 mM ASA.
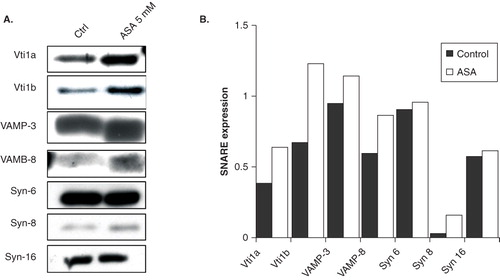
Phagocytosis involves the endosomes, and the endosomal system governs robust membrane vesicles trafficking and protein degradative compartment (lysosomes). The function of SNARE proteins in phagocytosis was highlighted in our study demonstrating the negative regulatory roles of endosomal SNAREs during phagocytosis in DC (Ho et al. Citation2008, Citation2009). Thus, it is possible that endosomal SNAREs (Hong Citation2005) are potential intracellular targets of aspirin. To ascertain this possibility, we determined the effects of 5 mM ASA on the expression of Vti1a (Golgi, trans-Golgi network, and early/late endosomes SNARE), Vti1b (trans-Golgi network and early/late endosomes SNARE), VAMP-3 (recycling endosomes SNARE), VAMP-8 (early/late endosomes SNARE), Syn-6 (trans-Golgi network and early endosomes SNARE), Syn-8 (early/late endosomes SNARE), and Syn-16 (trans-Golgi network and early endosomes SNARE) by Western blotting. Interestingly, ASA up-regulates protein expression of Vti1a, Vti1b, VAMP-3, VAMP-8, and Syn-8, but not Syn-6 and Syn-16 in primary DC (). shows the normalized quantification of protein bands in . Since most of the endosomal SNAREs above share overlapping intracellular localization (in the early/late endosomes) and are potential ASA targets, we have chosen to focus only on Vti1a and Vti1b in the subsequent experiments.
Figure 2. ASA regulates expression of endosomal SNARE proteins (A) Western blot analysis of endogenous Vti1a, Vti1b, VAMP-3, VAMP-8, Syn-6, Syn-8, and Syn-16 expression in control and ASA-treated DC2.4 cells. Lysates were prepared from control and ASA (5 mM)-treated primary DC, separated by 12% SDS-PAGE gel and immunoblotted using Vti1a, Vti1b, VAMP-3, VAMP-8, Syn-6, Syn-8, and Syn-16 specific antibodies. (B) Quantification of protein bands in ‘A’. Representative from three independent experiments is shown.
The ASA-mediated phagocytic inhibitory effect is dependent on Vti1a and Vti1b expression
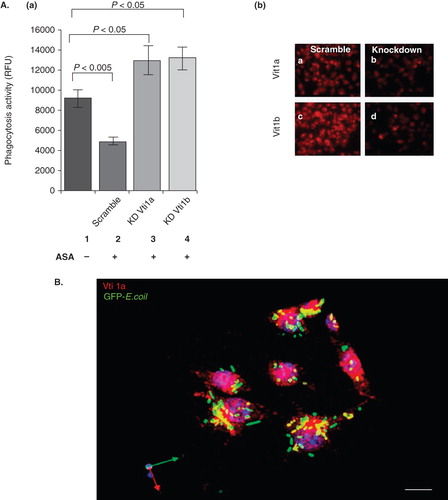
We next asked the question whether the ASA-mediated phagocytosis inhibition was Vti1a and Vti1b-dependent. DC2.4 were treated with siRNAs to knock down the expression of either Vti1a or Vti1b prior to incubation with ASA and phagocytosis analysis. Scrambled siRNA-treated DC2.4 was used as control. DC2.4 was used in this experiment because of its higher susceptibility to siRNA transfection and thus showed a better gene-knockdown result as compared to murine primary DC. As shown in (panel a), down-regulation of either Vti1a or Vti1b significantly reduced the phagocytic inhibitory effects of ASA. Thus, suggesting that Vti1a and Vti1b are key molecules involved in the ASA-mediated inhibition of phagocytosis in DC. Immunofluorescence microscopy analysis using Vti1a- and Vti1b-specific antibodies in (panel b) shows the down-regulation of Vti1a and Vti1b expression in DC2.4 cells upon siRNA treatment.
Figure 3. The ASA-mediated mitigation of phagocytosis in DC is dependent on Vti1a and Vti1b expression. (A) Panel a: Phagocytosis inhibitory effect of ASA on DC2.4 was reduced upon silencing of Vti1a and Vti1b by siRNA transfection. Scramble siRNA was used as control. Panel b: The same set of cells used in the phagocytosis assay in C was stained with Vti1a- and Vti1b-specific monoclonal antibodies to verify successful knock-down of these SNARE proteins. (B) Vti1a recruits to GFP-E. coli-positive membrane compartment. DC2.4 were incubated with GFP-E. coli (green) for 1 h, fixed and viewed under the confocal microscope. Vti1a (red) colocalizes with the GFP-E. coli-positive membrane compartment (in yellow). Experiments were repeated at least two times. Size bar: 10 μm.
Vti1a and Vti1b recruits to the phagosomes and negatively regulates phagocytosis in DC
Since the phagocytic inhibitory effect of ASA is dependent on Vti1a/Vti1b expression and since Vti1a/Vti1b localizes to the phagosomes, it is possible that Vti1a and Vti1b might play important roles in regulating phagocytosis in DC. We first attempted to examine if Vti1a and Vti1b recruites to phagosomes in DC. DC2.4 cells were incubated with GFP-E. coli for 1 h, fixed and viewed under the confocal microscope. As shown in , Vti1a (red) colocalizes with the green GFP-E. coli (in yellow), suggesting that Vti1a recruits to GFP-E. coli-positive membrane compartment. Furthermore, in , stereo 3D convolution images showed the recruitment of Vti1a and Vti1b (in red) to the green GFP-E. coli-positive phagosomes (indicated by white arrows).
Figure 4. Significant amount of Vti1a and Vti1b recruited to the phagosomes upon bacteria ingestion. DC2.4 cells were incubated with GFP-E. coli (green) for 1 h prior to analysis by immunofluorescence microscopy. Cells without GFP-E. coli were used as a control. (A) Recruitment of Vti1a and Vti1b to the phagosomes were determined by immunofluorescense microscopy using antibodies against the respective SNARE proteins (in red). Stereo 3D convolution images showed the recruitment of Vti1a and Vti1b (in red) to the green GFP-E. coli-positive phagosomes (indicated by white arrows). Size bar: 5 μm. (B) Enrichment of Vti1a and Vti1b on phagosomes was quantified using Western blot analysis. DC2.4 cells were allowed to internalize GFP-E. coli for 1 h prior to phagosomal extraction as described in Methods. The phagosomal lysates were prepared and separated by 12% SDS-PAGE gel and immunoblotted using Vti1a, Vti1b and VAMP-3 specific antibodies. The phagosomal extract (PE) showed enrichment of the SNARE protein as compared to the total lysate (TL). Note that four times lower amount of PE (5 μg) was loaded onto the gel as compared to TL (20 μg). The band intensities were quantified using Image J software and normalized to the same amount of protein and presented on the bar chart. Representative from three independent experiments is shown.
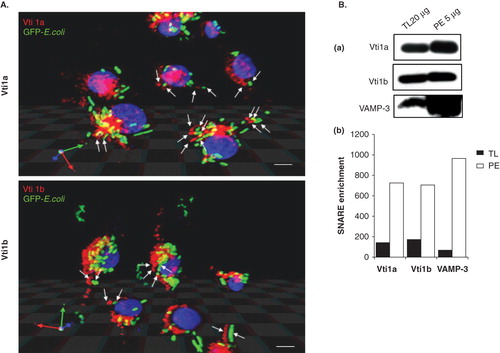
To further confirm the localization of Vti1a and Vti1b to phagosomes, DC2.4 cells were incubated with GFP-E. coli for 1 h and GFP-E. coli-containing phagosomes were purified by centrifugation through a sucrose step gradient. Twenty micrograms of total cell lysate (TL) and 4-fold lower amount (5 μg) of phagosomal fraction (PE) were analyzed by Western blot. As shown in (panel a), both Vti1a and Vti1b were significantly enriched in the phagosomal fraction (PE). VAMP-3 was used as a positive control, as it had previously been shown to localize to phagosomes (Fiorucci et al. Citation2001). The protein bands in panel a were quantified, normalized against the amount of proteins, and presented in panel b.
To determined the role of Vti1a and Vti1b in regulating the phagocytic pathway in DC, c-myc epitope-tagged Vti1a (Myc-Vti1a) and Vti1b (Myc-Vti1b) were expressed in DC2.4 cells. As shown in , like endogenous Vti1a and Vti1b, Myc-Vti1a and Myc-Vti1b were predominantly localized to the perinuclear regions and endosomes in DC. Interestingly, we observed that phagocytic up-take of fluorescence bioparticles was significantly reduced in both pDMyc-Vti1a and pDMyc-Vti1b transfected DC2.4 cells (, panel a). , panel b showed the level of Myc-Vti1a and Myc-Vti1b protein expression by Western blotting.
Figure 5. Up-regulation of Vti1a and Vti1b mitigates phagocytosis in DC. (A) Localization of cells over-expressing Myc-Vti1a and Myc-Vti1b were determined by immunofluorescense microscopy using anti-c-myc monoclonal antibody 24 h post-transfection. Transfection was performed using Effectene reagent. Size bar: 5 μm. (B) Panel a: DC2.4 cells were transfected with the indicated plasmids (pDMyc, pDMyc-Vti1a and pDMyc-Vti1b) and assayed for their phagocytic activity after 24 h post-transfection. Panel b: The corresponding Western blot shows the protein expression of Myc-Vti1a and Myc-Vti1b. Representative from at least three independent experiments was shown. (C) DC2.4 cells transfected with control plasmid and plasmids encoding Myc-Vti1a and Myc-Vti1b were incubated with GFP-E. coli for 15 min at 37°C (0 min time-point). Cells were washed to remove unbound bacteria, and further incubated at 37°C for 1, 1.5 and 2 h prior to quantification using a fluorescence ELISA reader. Panel a: Up-regulation of Vti1a and Vti1b in DC2.4 cells did not increase degradation of GFP-E. coli in the phagosomes. *P < 0.05. Panel b: The corresponding Western blot shows the protein expression of Myc-Vti1a and Myc-Vti1b. Representative from two independent experiments is shown.
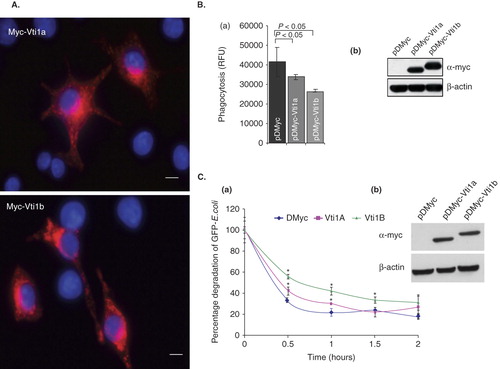
To exclude the possibility that the reduction of intracellular fluorescence label (in our phagocytosis assay) observed in pDMyc-Vti1a and pDMyc-Vti1b transfected DC2.4 cells as compared to control cells was due to an increase in degradation of phagocytosed fluorescence bioparticles (phagosome maturation), DC2.4 cells transfected with control plasmid and plasmids encoding Myc-Vti1a and Myc-Vti1b were incubated with GFP-E. coli for 15 min at 37°C (0 min time-point). Cells were washed to remove unbound bacteria, and further incubated at 37°C for 1, 1.5 and 2 h prior to quantification using a fluorescence ELISA reader (detection of intracellular GFP-proteins). As shown in (panel a), expression of either Myc-Vti1a or Myc-Vti1b did not promote degradation of GFP-proteins (phagosome maturation). In fact, Myc-Vti1a and Myc-Vti1b prevents GFP-protein degradation. , panel b showed the level of Myc-Vti1a and Myc-Vti1b protein expression by Western blotting.
DC2.4 is a DC cell line and to eliminate the possibility of cell lines specificity, the function of Vti1a was examined in murine bone marrow-derived primary DC. Bone marrow-derived primary DC were prepared and cultured as described in the Methods section. Flow-cytometry analysis showed that majority of the day 6 murine primary DC produced were CD11c+ (, panel a). After checking the integrity of the mRNAs (), both the in vitro transcribed control and Myc-Vti1a mRNAs were transfected into murine primary DC by electroporation. (panel b) showed that a significant pool of murine primary DC expressed the Myc-Vti1a proteins (a shift to the right in the histogram). As anticipated, Myc-Vti1a mitigates phagocytosis in murine primary DC as well ().
Figure 6. Vti1a and Vti1b negatively regulates phagocytosis in DC. (A) Mouse bone marrow-derived primary DC (murine primary DC) were transfected with in vitro transcribed Myc-Vti1a mRNA and stained with anti-CD11c and anti-c-myc specific antibodies were analyzed by flow cytometry. Panel a: Shows the CD11c+ murine primary DC population. Panel b: Shows the Myc-Vti1a-expressing murine primary DC populations. (B) Shows the integrity in vitro transcribed Myc-Vti1a mRNA as analyzed by formaldehyde gel. (C) Myc-Vti1a mRNA-transfected DC registered a reduction in phagocytosis activity as compared to control (luciferase mRNA- transfected primary DC). (D) Panel a: Vti1a and Vti1b genes were silenced in DC2.4 cells by siRNA transfection using Lipofectamine 2000. The cells were assayed for their phagocytic activity after 48 h post-transfection. Panel b: The corresponding Western blot analysis indicates successful silencing of Vti1a and Vti1b protein expression. Representative from three independent experiments is shown.
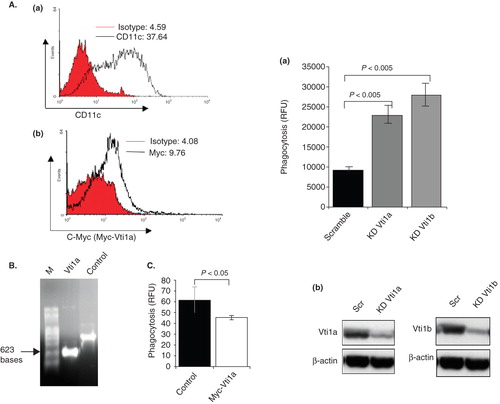
To further confirm the function of Vti1a and Vti1b in regulating phagocytosis in DC, knock-down experiments using siRNA specific for Vti1a and Vti1b were performed. DC2.4 cells were transfected with either scrambled RNA (as control), Vti1a siRNA or Vti1b siRNA and checked for phagocytosis activity. As anticipated, silencing of either Vti1a or Vti1b by siRNA transfection enhances phagocytosis (, panel a) thus establishing their role in negatively regulating phagocytosis. Panel b shows the knock-down of Vti1a and Vti1b expression by siRNA.
ASA induces redistribution of Vti1a, Vti1b, and scavenger receptor CD204 in DC
To determine whether ASA changes intracellular localization of Vti1a and Vti1b in DC, untreated and ASA-treated DC2.4 cells were fixed, stained using anti-Vti1a and -Vti1b monoclonal antibodies, and analyzed by fluorescence microscopy. As shown in (panel a and b), ASA causes accumulation of Vti1a in larger punctate membrane compartments in the cytoplasm as compared to untreated cells. Vti1b proteins were observed to concentrate at the perinuclear region in ASA-treated cells (, panel c and d). Our results show that Vti1a and Vti1b plays important role in regulating phagocytic up-take of bacteria in DC and ASA causes intracellular redistribution of Vti1a and Vti1b. Therefore, the ASA-mediated abrogation of phagocytosis in DC could be due to changes to the endosomal membrane trafficking machinery that govern trafficking of scavenger receptors to and from the cell surface. Scavenger receptor-A or CD204 had been shown recently to be essential for the up-take of E. coli by phagocytosis (Amiel et al. Citation2007). It is interesting to determine if ASA induces redistribution of CD204 protein in DC. Murine primary DC were treated with 5 mM ASA for 24 h in DMEM, fixed, and stained with CD204-specific antibody prior to analysis by immunofluorescense microscopy.
Figure 7. ASA induces redistribution of Vti1a, Vti1b, and scavenger receptor CD204 in DC. (A) Mouse bone marrow-derived primary DC (murine primary DC) were either untreated (Control) or treated with 5 mM ASA for 24 h in DMEM, fixed, and stained with Vti1a- and Vti1b-specific antibodies prior to viewing by immunofluorescense microscopy. ASA promotes redistribution of Vti1a and Vti1b in DC. Size bar: 10 μm. (B) Murine primary DC were treated with 5 mM ASA for 24 h in DMEM, fixed, and stained with CD204-specific antibody prior to viewing by immunofluorescense microscopy. CD204 localized to ASA-induced punctate membrane structures. Size bar: 5 μm. (C) Kinetics of endocytosed CD204 from the cell surface in the absence and presence of ASA. Murine primary DC were either untreated or pre-treated with 5 mM ASA for 4 h in DMEM, incubated with anti-CD204 monoclonal antibody on ice to label the cell surface CD204, transferred to 37°C to allow endocytosis of antibody-tagged cell surface CD204 in the presence of ASA, fixed cells at indicated time-points and stained with VAMP-8-specific rabbit polyclonal antibody prior to analyzing by immunofluorescense microscopy. In the presence of ASA, a significant pool of CD204 accumulated in the early/late endosomes marked by VAMP-8. Size bar: 10 μm.
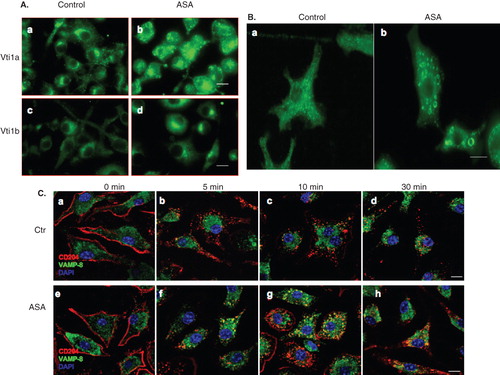
As shown in , ASA induces redistribution of CD204 and formation of CD204-positive punctate membrane structures in murine primary DC. To further determine the effects of ASA on trafficking of CD204 in DC, an experiment was designed to visualize the kinetics of endocytosed CD204 from the cell surface in the absence and presence of ASA. Murine primary DC were either untreated or pre-treated with 5 mM ASA for 4 h in DMEM, incubated with anti-CD204 monoclonal antibody on ice to label the cell surface CD204, transferred to 37°C to allow endocytosis of antibody-tagged cell surface CD204 either in the absence or presence of ASA. Cells were fixed at indicated time-points and stained with VAMP-8-specific rabbit polyclonal antibody prior to analyzing by immunofluorescense microscopy. As shown in (panel a–d), in the absence of ASA, no significant co-localization between endocytosed CD204 (red) and VAMP-8 (green) was observed in all the time-points. However, co-localization (in yellow) could be observed as early as 5 min post-endocytosis of CD204 from the cell surface in cells treated with ASA (, panel f, g and h). Since VAMP-8 localizes to the early/late endosomes, it is possible that endocytosed CD204 accumulate at the early/late endosomes and thus indirectly affects their trafficking to the cell surface upon ASA-treatment. Taken together, our results suggest that the ASA-mediated mitigation of phagocytosis in DC could be associated with the functions of ASA in regulating the expression and intracellular localization of endosomal SNAREs, which indirectly influenced scavenger receptor (CD204) trafficking.
Discussion
In recent years, although much has been known about the importance of phagocytosis in mounting both the innate and cell-mediated immune responses, great effort is still required to determine how the phagocytosis machinery, which is crucial for MHC class I (through cross-priming) and class II restricted antigen presentation are controlled in DC. The formation of phagosomes requires the targeted delivery of membrane vesicles from the recycling endosome compartment, which contributes to the elongation of membranous pseudopods during the early stages of the phagocytosis process. Others (Godson et al. Citation2000, Mitchell et al. Citation2002) have shown that ASA promotes phagocytic up-take of apoptotic neutrophils by macrophages. This enhancement of phagocytosis was shown to be associated with the COX-2-dependent synthesis of ASA-triggered 15-epi-lipoxinA4. Consistently, in this study we also observed a slight increase in phagocytic activity when DC and macrophages were treated with low concentration of ASA (below 3 mM). However, treatment of DC and macrophages with higher concentration of ASA (above 3 mM) significantly abrogates phagocytic up-take of E. coli. Thus, the effect of ASA on the phagocytosis activity in DC and macrophages is biphasic. What is the possible mechanistic of the biphasic effect of ASA on the phagocytic pathway in DC?
Hormesis is a considerably new concept that explains the biphasic dose response to an environmental agent or drug characterized by a low dose stimulation and a high dose inhibitory effect (Kendig et al. Citation2010). In the fields of biology and medicine, hormesis defined as an adaptive response of cells and organisms to a moderate stress. Interestingly, recent findings have elucidated the cellular signaling pathways and molecular mechanisms that mediate hormetic responses which typically involve enzyme such as kinases and deacetylases, and transcription factors such as Nrf-2 and NF-kB (Mattson et al. Citation2007, Marini et al. Citation2008). Thus, it is possible that the biphasic dose response of DC to ASA observed in this study could be explained based on the hormesis concept. It is possible that at a low ASA dosage, DC response to the ASA-mediated ‘moderate stress’ by down-regulating expression of endosomal SNAREs such as Vti1a and Vti1b. This is supported by our observation that serum starvation (a form of moderate stress) down-regulates Vti1b expression in DC2.4 (data not shown). Consistently, we repeatedly observed a downward trend in the protein expression of Vti1b in DC2.4 treated with low dosage of ASA (Supplementary Figure 1, available online). Since lipoxinA4 enhances phagocytosis, it is possible that low dosage of ASA induces synthesis of ASA-triggered 15-epi-lipoxinA4 which in turn down-regulates expression of Vti1a, Vti1b and other endosomal SNAREs. At higher dosages however, ASA activates the lipoxinA4-independent pathway (which enhances expression of Vti1a, Vti1b and other endosomal SNAREs) and mitigates phagocytosis. Quantification of Vti1a and Vti1b expression in untreated and lipoxinA4-treated DC will shed lights on this interesting possibility.
The ability of ASA in inhibiting phagocytosis, an essential pathway for extracellular antigen capture by DC, prompted us to explore the endosomal membrane trafficking pathways in DC. In view of the ability of ASA in up-regulating expression of several endosomal SNARE proteins, we speculated that endosomal SNAREs might modulate phagocytosis by affecting the focal exocytosis of membranes. Since endosomal SNAREs such as Vti1a and Vti1b are known to be involved in the secretory pathway as well, overexpressing these proteins is expected to increase secretion, resulting in a larger amount of membrane receptors delivered to the cell surface, and hence, positively influencing phagocytosis. Interestingly, in contrary, our experimental results show that both Vti1a and Vti1b negatively regulate phagocytosis. Hence, it is likely that Vti1a and Vti1b modulate phagocytosis by other mechanisms, other than replenishing surface membrane via focal exocytosis.
Others (Yang et al. Citation2001, Allen et al. Citation2002) have shown that VAMP-3 does not play a role in regulating phagocytosis. This is consistent with our previous results where up-regulation and silencing (siRNA) of VAMP-3 do not significantly alter bacteria-up-take by DC (Ho et al. Citation2008). Interestingly, like Vti1a and Vti1b, up-regulation of VAMP-8 in DC inhibits phagocytosis (Ho et al. Citation2008). The effect of VAMP-8 up-regulation on phagocytosis correlates well with this study, in view of the fact that VAMP-8 forms SNARE complex with Vti1b/Syn-7/Syn-8 on endosomal membranes (Wade et al. Citation2001). Consistently, Syn-8 is also up-regulated upon ASA treatment. In contrast, Syn-6 and Syn-16 which forms other SNARE complexes was not significantly up-regulated in the presence of ASA ().
Membrane trafficking and fusion not only involves interaction between cognate SNARE proteins on opposing membranes but also tethering factors which interact with SNARE proteins and facilitate SNARE complex assembly. Tethering factors typically form highly extended coiled-coil structures and enhance the efficiency of membrane trafficking by linking the opposing membranes (Pfeffer et al. 1999). In this study, ASA induced intracellular redistribution of Vti1a and Vti1b in DC. Consistently, trafficking of scavenger receptor CD204 (Amiel et al. Citation2007) in DC was also significantly altered upon ASA-treatment. Thus, ASA-treatment and overexpression of endosomal SNAREs might change the expression of scavenger receptors on the cell surface of DC. Since Vti1a containing complex had been demonstrated to mediate homotypic fusion in the early endosomes (Brandhorst et al. Citation2006), there is a possibility that the ASA-dependent increase in Vti1a and Vti1b expression causes homotypic fusion of membranes near to the cell surface. These homotypic fusion events might affect the recycling of membrane proteins to the cell surface and thus possibly perturb the proper formation of membrane projection essential for the phagocytosis process. This is supported by our observation that up-regulation of Vti1a and Vti1b in DC mitigates degradation of phagosomal contents most likely by inhibiting phagosome maturation to become phagolysosome (). Thus, it is possible that inhibition of phagosome maturation abrogates release (from the phagosomes) and recycle (to the cell surface) of membrane proteins (including CD204) which are required for subsequent rounds of phagocytosis.
Conclusion
The major finding of this study is the identification of endosomal SNAREs as key players in the COX-2/15-epi-lipoxinA4-independent regulation of phagocytosis by ASA. ASA up-regulates expression of some endosomal SNAREs and interestingly, our results show that up-regulation of Vti1a and Vti1b expression reduces the rate of bacterial up-take via phagocytosis. The ability of ASA to inhibit phagocytosis (one of the essential route of antigen intake) is consistent with its role as an immune-regulatory drug. Phagocytosis is the initial step taken by antigen-presenting cells for the up-take of antigens. Thus, regulating the efficiency of this process may potentially control antigen processing and presentation.
Figure S1
Download MS Word (127.5 KB)Acknowledgements
We thank Dr Kevin Tan Shyong-wei for the generous gift of GFP-E. coli, and Dr Kenneth Rock for the DC2.4 cell line. We also thank Drs Lim Kah Leong, Lu Jinhua, and Michael Kemeny and members of the Membrane Trafficking and Immunoregulation Research Laboratory for critical reading of the manuscript.
Declaration of interest: This work was supported by research grants from the Academic Research Fund-NUS (R182-000-092-112 to S.H.W) and the Academic Research Council-Ministry of Education, Singapore (R182-000-099-112 to S.H.W). The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Allen LA, Yang C, Pessin JE. 2002. Rate and extent of phagocytosis in macrophages lacking vamp3. J Leukoc Biol 72:217–221.
- Amiel E, Nicholson-Dykstra S, Walters JJ, Higgs H, Berwin B. 2007. Scavenger receptor-A functions in phagocytosis of E. coli by bone marrow dendritic cells. Exp Cell Res 313:1438–1448.
- Booth JW, Trimble WS, Grinstein S. 2001. Membrane dynamics in phagocytosis. Seminars Immunol 13:357–364.
- Brandhorst D, Zwilling D, Rizzoli SO, Lippert U, Lang T, Jahn R. 2006. Homotypic fusion of early endosomes: SNAREs do not determine fusion specificity. Proc Natl Acad Sci USA 103:2701–2706.
- Etminan M, Gill S, Samii A. 2003. Effect of non-steroidal anti-inflammatory drugs on risk of Alzheimer's disease: Systematic review and meta-analysis of observational studies. BMJ 327:128.
- Fiorucci S, Antonelli E, Burgaud JL, Morelli A. 2001. Nitric oxide-releasing NSAIDs: A review of their current status. Drug Saf 24:801–811.
- Godson C, Mitchell S, Harvey K, Petasis NA, Hogg N, Brady HR. 2000. Cutting edge: Lipoxins rapidly stimulate nonphlogistic phagocytosis of apoptotic neutrophils by monocyte-derived macrophages. J Immunol 164:1663–1667.
- Grilli M, Pizzi M, Memo M, Spano P. 1996. Neuroprotection by aspirin and sodium salicylate through blockade of NF-kappaB activation. Science 274:1383–1385.
- Hackstein H, Morelli AE, Larregina AT, Ganster RW, Papworth GD, Logar AJ, Watkins SC, Falo LD, Thomson AW. 2001. Aspirin inhibits in vitro maturation and in vivo immunostimulatory function of murine myeloid dendritic cells. J Immunol 166:7053–7062.
- Hong W. 2005. SNAREs and traffic (2005). Bioch Biophys Acta (BBA) – Molec Cell Res 1744:120–144.
- Ho YHS, Cai DT, Wang CC, Huang D, Wong SH. 2008. Vesicle-associated membrane protein-8/Endobrevin negatively regulates phagocytosis of bacteria in dendritic cells. J Immunol 180:3148–3157.
- Ho YHS, Cai DT, Huang D, Wang CC, Wong SH. 2009. Caspases regulate VAMP-8 expression and phagocytosis in dendritic cells. Biochem Biophys Res Comm 387:371–375.
- Kendig EL, Le HH, Belcher SM. 2010. Defining hormesis: Evaluation of a complex concentration response phenomena. Int J Toxicol 29:235–246.
- Kopp E, Ghosh S. 1994. Inhibition of NF-kappa B by sodium salicylate and aspirin. Science 265:956–959.
- Lowe SL, Peter F, Subramaniam VN, Wong SH, Hong W. 1997. A SNARE involved in protein transport through the Golgi apparatus. Nature 389:881–884.
- Marini AM, Jiang H, Pan H, Wu X, Lipsky RH. 2008. Hormesis: A promising strategy to sustain endogenous neuronal survival pathways against neurodegenerative disorder. Ageing Res Rev 7:21–33.
- Matasic R, Dietz AB, Vuk-Pavlovic S. 2000. Cyclooxygenase-independent inhibition of dendritic cell maturation by aspirin. Immunology 101:53–60.
- Mattson MP, Son TG, Camandola S. 2007. Viewpoint: Mechanisms of action and therapeutic potential of neurohormetic phytochemicals. Dose Resp 5:174–186.
- Mitchell S, Thomas G, Harvey K, Cottell D, Reville K, Berlasconi G, Petasis NA, Erwig L, Rees AJ, Savill J, Brady HR, Godson C. 2002. Lipoxins, aspirin-triggered epi-lipoxins, lipoxin stable analogues, and the resolution of inflammation: Stimulation of macrophage phagocytosis of apoptotic neutrophils in vivo. J Am Soc Nephrol 13:2497–2507.
- Pfeffer SR. 1999. Transport-vesicle targeting: Tethers before SNAREs. Nat Cell Biol 1:E17–22.
- Santambrogio L, Potolicchio I, Fessler SP, Wong SH, Raposo G, Strominger JL. 2005. Involvement of caspase-cleaved and intact adaptor protein 1 complex in endosomal remodeling in maturing dendritic cells. Nat Immunol 6:1020–1028.
- Shen Z, Reznikoff G, Dranoff G, Rock KL. 1997. Cloned dendritic cells can present exogenous antigens on both MHC class I and class II molecules. J Immunol. 158:2723–2730.
- Wade N, Bryant NJ, Connolly LM, Simpson RJ, Luzio JP, Piper RC, James DE. 2001. Syntaxin 7 complexes with mouse Vps10p tail interactor 1b, syntaxin 6, vesicle-associated membrane protein (VAMP)8, and VAMP7 in b16 melanoma cells. J Biol Chem 276:19820–19827.
- Wong SH, Santambrogio L, Strominger JL. 2004. Caspases and nitric oxide broadly regulate dendritic cell maturation and surface expression of class II MHC proteins. Proc Natl Acad Sci USA 101:17783–17788.
- Wong SH, Xu Y, Zhang T, Hong W. 1998. Syntaxin 7, a novel syntaxin member associated with the early endosomal compartment. J Biol Chem 273:375–380.
- Xu Y, Wong SH, Tang BL, Subramaniam VN, Zhang T, Hong W. 1998. A 29-kilodalton Golgi soluble N-ethylmaleimide-sensitive factor attachment protein receptor (Vti1-rp2) implicated in protein trafficking in the secretory pathway. J Biol Chem 273:21783–21789.
- Yang C, Mora S, Ryder JW, Coker KJ, Hansen P, Allen LA, Pessin JE. 2001. VAMP3 null mice display normal constitutive, insulin- and exercise-regulated vesicle trafficking. Mol Cell Biol 21:1573–1580.