Abstract
We have previously shown that a mixture of cerebrosides obtained from dried tubers of herb Typhonium giganteum Engl. plays a neuroprotective role in the ischemic brain through its effect on activation of BKCa channels. It is very curious to know whether a single pure cerebroside compound could activate the BKCa channel as well. This study explored the possible effects of pure cerebroside compounds, termitomycesphins A and B, on the BKCa channel activation. Both termitomycesphins A and B activated the BKCa channels at micromole concentration without significant difference. Termitomycesphin A increased the single channel open probability of the BKCa channels in a dose-dependent manner without modifying the single channel conductance. Termitomycesphin A activated BKCa channel more efficiently when it was applied to the cytoplasmic face of the membrane, suggesting that binding site for termitomycesphin A is located at the cytoplasmic side. Termitomycesphin A shifted the voltage-dependent activation curve to less positive membrane potentials and the Ca2+-dependent activation curve of the channel upwards, suggesting that termitomycesphin A could activate the channels even without intracellular free Ca2+. Furthermore, STREX-deleted BKCa channels were completely insensitive to termitomycesphin A, indicating that STREX domain is required for the activation of the BKCa channel. These data provide evidence that termitomycesphins are potent in stimulating the activity of the BKCa channels. As BKCa channels are associated with pathology of many diseases, termitomycesphins might be used as therapeutic agents for treating these diseases through its regulatory effect on the BKCa channels.
Introduction
The large-conductance Ca2+-activated K+ (BKCa) channels are widely distributed in many tissues such as neurons (Lancaster and Nicoll Citation1987), chromaffin cells (Lingle et al. Citation1996), inner hair cells of cochlea (Issa and Hudspeth Citation1994), chick cardiac myocytes (Ruknudin et al. Citation1993, Kawakubo et al. Citation1999), and skeletal (Pallotta et al. Citation1981) and smooth muscles (Toro et al. Citation1991). Functional BKCa channels are formed by tetramerization of pore-forming α subunits and an auxiliary β subunit (Shen et al. Citation1994, Wang and Sigworth Citation2009). The pore-forming α subunit is encoded by KCNMA1 gene (Butler et al. Citation1993), which is subject to vast tissue-specific (Tseng-Crank et al. Citation1994) and cell-specific alternative pre-mRNA splicing (Zarei et al. Citation2004). Molecular structure of the α subunit consists of seven membrane-spanning segments (S0–S6) at the N-terminus and a large intracellular domain at the C-terminus (Magleby Citation2003). The C-terminus contains multiple regulatory sites, e.g., regulator of conductance for potassium (RCK) domain (Jiang et al. Citation2001), ‘calcium bowl’ region (Schreiber and Salkoff Citation1997), and multiple phosphorylation sites for cAMP- and cGMP-dependent protein kinases (Zhou et al. Citation2001). BKCa channels are activated by membrane depolarization and elevation of intracellular free Ca2+ concentration ([Ca2+]i) (Horrigan and Aldrich Citation2002). These channels have been implicated in many pathological processes. For example, malfunction of BKCa channels can lead to hypertension (Brenner et al. Citation2000, Sausbier et al. Citation2005), epilepsy (Brenner et al. Citation2005, Du et al. Citation2005), cerebellar ataxia (Sausbier et al. Citation2004), incontinence (Meredith et al. Citation2004), and ischemic stroke (Gribkoff et al. Citation2001). They were also involved in diverse physiological processes, including regulation of blood pressure (Brenner et al. Citation2000, Sausbier et al. Citation2005), neurotransmitter release (Raffaelli et al. Citation2004), micturition (Meredith et al. Citation2004), epithelial transport (Sausbier et al. Citation2006), and cerebrovascular circulation (Filosa et al. Citation2006).
We have previously reported that Baifuzi cerebroside (Baifuzi-CB) obtained from dried tubers of herb Typhonium giganteum Engl. (Chinese name: Baifuzi) could potently activate BKCa channels. And this channel activation might be caused by the direct interaction between STREX domain (a 59-amino acid splice insert of BKCa channel) and Baifuzi-CB (Chi et al. Citation2010). However, as we showed in the supplementary figure, Baifuzi-CB is a mixture of cerebrosides, which differ in length of fatty acid, saturation, location and configuration of double bonds on long chain base. Therefore, it is crucial to know whether a single pure cerebroside compound could activate the BKCa channel as well. In the present study, taking termitomycesphins A (Ter A) and B (Ter B) as pure cerebroside compounds, we examined the effect of Ter A and Ter B on BKCa channels. Our results indicated that both Ter A and Ter B activated the BKCa channels at micromole concentration, suggesting that this type of chemicals needs to be studied more extensively to find better BKCa channel openers.
Materials and methods
Cell culture and transfection
Chinese hamster ovary (CHO-K1) cells were cultured in Ham's F-12 nutrient mixture supplemented with 10% FBS at 37°C in a 5% CO2 incubator. Cells were plated onto poly-L-lysine coated coverslips in 35 mm dishes at 60–80% confluence. Then they were transiently cotransfected with the full length chick BKCa channel gene (GenBank accession number AB072618) and GFP (Clontech, Palo Alto, CA, USA), or the STREX-deleted BKCa channel and GFP. The full length chick BKCa channel is used throughout unless where indicated that the STREX-deleted form is used. Transfection was performed with LipofectAMINE Plus reagent (Invitrogen) following the manufacturer's instructions. Cells were used for electrophysiological recordings 1–2 days after the transfection.
Electrophysiology
All the single-channel recordings were sampled at inside-out patch-clamp configuration using an EPC-10 amplifier (HEKA Elektronic, Lambrecht, Germany). Data were acquired at 2–5 kHz and low-pass filtered at 1 kHz. Patch electrodes were pulled from thin walled borosilicate capillary tubes using a Sutter programmable puller (model P-97, Sutter Instrument, Novato, CA, USA) and fire-polished to achieve an electrical resistance in a range of 3–7 MΩ. An Ag/AgCl wire bath electrode was used as ground electrode. The pipette solution was Hanks' balanced salt solution (HBSS, Sigma, in mM): 1.3 CaCl2, 0.5 MgCl2, 0.4 MgSO4, 5.4 KCl, 0.4 KH2PO4, 136.9 NaCl, 0.3 Na2HPO4, 5.6 D-glucose and 4.2 NaHCO3. The bath solution consisted of (in mM): 145 KCl, 10 EGTA, 2 Mg-ATP, 10 HEPES (pH 7.3 with KOH), and CaCl2 adjusted to the desired [Ca2+]i using a free software CALCON3. The [Ca2+]i was 0.1 μM, if not stated otherwise. Continuous recordings of at least 7500 ms were used to determine the single channel open probability (Po). The value of Po in a patch with multiple channels was calculated by using TAC 4.1 (HEKA, Germany), based on the equation: Po = (1 − PC 1/N), where PC is the probability that all of the channel is in closed state, N is the number of channels in the patch, which was estimated from the maximum number of channels observed over a voltage of +60 mV and relatively higher concentration of Ter A when it was consecutively applied. Ter A and Ter B were dissolved in DMSO and stored as a stock solution at −20°C, which was diluted with bath or pipette solution upon use. The final concentration of DMSO in the recording solution did not exceed 0.1% (v/v), which did not affect BKCa channel activity. Unless otherwise stated, all chemicals and reagents were obtained from Sigma. All experiments were done at room temperature (20–25°C).
Extraction and isolation
Termitomyces albuminosus, an edible Chinese mushroom T. albuminosus (Berk.) Heim. (Chinese name: Jizong), were collected in Yanyuan County (Sichuan Province, China). The dried fruiting bodies of the mushroom were powdered and immersed in EtOH with occasional stirring. The EtOH extract of T. albuminosus was washed with hexane, and then partitioned between BuOH and H2O. The active BuOH fraction was chromatographed on octadecylsilyl (ODS), then on silica gel to give a mixture of cerebrosides. Ter A and Ter B () were purified from the mixture by reversed-phase HPLC. The detailed process was described in the previous paper (Qi et al. Citation2000).
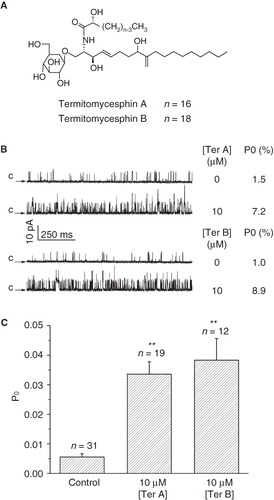
Figure 1. Effect of intracellular Ter A and Ter B on the BKCa channels. (A) Chemical structures of Ter A and Ter B. (B) Representative single channel recordings in the absence and presence of Ter A or Ter B at +20 mV. Arrows indicate the closed state of the channel. The Po values for each trace are indicated at the right. (C) Statistical data for mean Po of the BKCa channels in the presence of Ter A (10 μM) or Ter B (10 μM). **p < 0.005 compared to the control.
Statistics
The results were presented as mean ± SEM. The Origin 7.0 (Originlab, Northampton, MA, USA) was used for statistical analyses and plotting graphs. Statistical comparisons were made by using one-way ANOVA, followed by Bonferroni's post hoc test. The significance was set at p < 0.05.
Results
Effect of intracellular Ter A and B on the BKCa channels
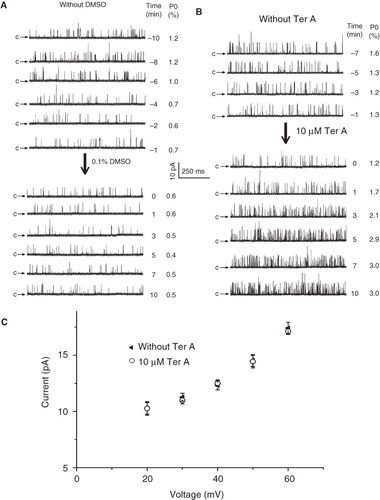
We first compared the effect of Ter A and Ter B on the activation of the BKCa channels. Application of 10 μM Ter A or Ter B to the intracellular face of the membrane significantly increased the BKCa channel activity (). In contrast, there was no significant difference in the single channel current amplitude, indicating that the unitary conductance of the channel was not altered (). Statistical summary in demonstrated that 10 μM Ter A increased Po of the BKCa channels from 0.6 ± 0.1% (n = 31) of control to 3.4 ± 0.4% (n = 19), while the same concentration of Ter B increased the Po to 3.8 ± 0.7% (n = 12), indicating that there were no significant difference between them. As more amount of Ter A than Ter B in hand, we examined the effect of Ter A on the BKCa channels in the following studies. The single channel recordings usually got stable after 5–10 min upon formation of inside-out patches. In the control experiment, 0.1% DMSO was subsequently applied, which did not change the Po of the channel (). To judge the effect of Ter A on the channel activation, Ter A was added only after the recordings getting stable within a 3-min period. Usually, the Po of the channel gradually increased upon intracellular application of Ter A. And the increase reached a plateau in less than 5 min ().
Figure 2. Time course of the BKCa channel activation by intracellular Ter A. Time course of single channel recordings in control condition (A) or upon intracellularly application of Ter A (B). 0 min denotes the time point of intracellularly application of DMSO or Ter A. (C) Current-voltage relationship for single-channel activities before and after application of Ter A. Each point represents mean ± SEM for at least 4 patches.
Dose-dependent effect of Ter A on the BKCa channels
To study the dose-dependent response of the channel, different concentrations of Ter A were applied to the cytosolic side of inside-out patch membrane at +20 mV. As illustrated in , the activity of the channel in the same patch was gradually increased with the increase in [Ter A] from 0–10 μM. showed that the mean Po of the channel was 0.4 ± 0.06% (n = 72) before application of Ter A, and it increased to 1.2 ± 0.2% (n = 15, p < 0.01), 2.0 ± 0.3% (n = 20, p < 0.01), 2.7 ± 0.5% (n = 18, p < 0.005), and 3.4 ± 0.4% (n = 19, p < 0.005) when Ter A was increased to 1.25 μM, 2.5 μM, 5 μM, and 10 μM at the intracellular side of the patch, respectively. The Po described above were plotted against [Ter A] and fitted to the Hill equation: Po = Pmax * [Ter A]N/(EC50 N + [Ter A]N), where Pmax is the maximum Po of the channel at the experimental condition, EC50 is the [Ter A] required to give half-maximal activity of the BKCa channels, and N is the Hill coefficient. The best fit to the data was obtained with values of 4.3% for Pmax, 2.8 μM for EC50, and 1.0 for N, suggesting that there was no cooperation for the stimulation of the BKCa channel by Ter A. This effect was reversible (), the Po of the channel was 0.5 ± 0.06% under control conditions, increased to 3.2 ± 0.3% by 10 μM Ter A, then returned back to 0.9 ± 0.2 % after washout (, n = 5).
Figure 3. Dose-dependent effects of intracellular Ter A on the BKCa channels. (A) Representative single BKCa channel traces at +20 mV in the control and during successive exposure to different concentration of Ter A (1.25–10 μM) in the same patch. (B) Ter A increase the Po of BKCa channels in a dose-dependent manner. The curve is the best fit to Po against the [Ter A] according to the Hill equation. (C) Current traces before, during application and washout of 10 μM Ter A. (D) Statistical summary for the washout experiment. **p < 0.005 compared to the control and washout condition.
![Figure 3. Dose-dependent effects of intracellular Ter A on the BKCa channels. (A) Representative single BKCa channel traces at +20 mV in the control and during successive exposure to different concentration of Ter A (1.25–10 μM) in the same patch. (B) Ter A increase the Po of BKCa channels in a dose-dependent manner. The curve is the best fit to Po against the [Ter A] according to the Hill equation. (C) Current traces before, during application and washout of 10 μM Ter A. (D) Statistical summary for the washout experiment. **p < 0.005 compared to the control and washout condition.](/cms/asset/3bd381ec-859c-4768-96cf-42734fb1d73c/imbc_a_538731_f0003_b.jpg)
Effect of extracellular Ter A on the BKCa channels
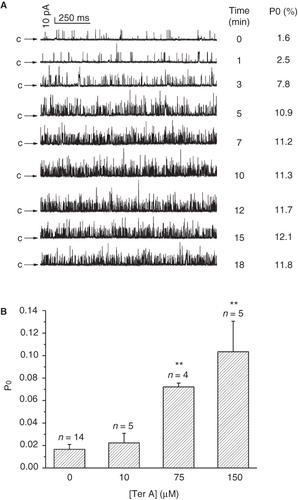
To determine whether the channel activity could be modulated by extracellular Ter A, the tip of the pipette was filled with the pipette solution and then back-filled with the same solution except containing desired [Ter A]. Probably owing to the slow diffusion of Ter A (150 μM) toward the patched membrane, Po of the channel was increased gradually with the increase in time and reached a plateau after about 10 min of the giga-ohm seal formation (). Statistical data in showed that 10, 75 and 150 μM extracellularly applied Ter A increased Po of the channels from 1.7 ± 0.4% (n = 14) of control to 2.2 ± 0.8% (n = 5), 7.2 ± 0.3% (n = 4) and 10.4 ± 2.7% (n = 5), respectively, indicating that extracellular Ter A could activate the BKCa channel as well. Interestingly, the Po of the channel in the presence of 10 μM Ter A in the cytoplasmic side (3.4 ± 0.4%, n = 19) was significantly higher than that obtained with the same concentration of Ter A applied extracellularly (2.2 ± 0.8%, n = 5). Besides, it usually took approximately 10 min to get stable upon extracellularly application of Ter A, much longer than that of intracellular application of Ter A. This result indicated that Ter A activated the BKCa channels more efficiently when it was applied intracellularly, implying that the binding site for Ter A might be located at the cytoplasmic side of the channel.
Figure 4. Effect of extracellularly applied Ter A on the BKCa channels. (A) Effect of 150 μM extracellular Ter A on Po over time at +20 mV. The recordings start (0 min) at the time when the inside-out patch was formed. Arrows indicate the closed state of the channels. (B) Statistical results of extracellular Ter A (10–150 μM) on Po of the channel at 10 min after formation of a giga-ohm seal at +20 mV. Values are mean ± SEM. **p < 0.005 compared to the control.
Effect of Ter A on the Ca2+-dependent activation of the BKCa channels
To understand the effect of Ter A on the Ca2+-dependent activation of the BKCa channel, we examined the BKCa channel activity over a range of [Ca2+]i (1 nM–50 μM) before and after intracellular application of Ter A. Comparing and , it was clear that application of 10 μM Ter A resulted in significant increase in Po of the channel. In , the mean Po of channels was plotted as a function of [Ca2+]i before and after application of Ter A. In the absence of Ter A, the relationship was well fitted with the Hill equation: Po = Pmax * [Ca2+]i N/(KN + [Ca2+]i N), where Pmax is the maximum Po, K is the [Ca2+]i required for the half activation, and N is the Hill coefficient. Values of 8.4% for Pmax, 11.2 μM for K, and 2.3 for N were obtained from the best fit in the absence of Ter A. However, in the presence of Ter A, an additional parameter PTerA needs to be added to the Hill equation as: Po = Pmax * [Ca2+]i N/(KN + [Ca2+]i N) + PTerA to get the best fit. This parameter represents the effect of Ter A on Po of the channel in the condition with no intracellular Ca2+. Values of 20.9% for Pmax, 15.4 μM for K, 2.8 for N, and 3% for PTerA were obtained by the best fitting to the modified Hill equation in the presence of 10 μM Ter A. This result indicated that Ter A shifted the Ca2+-dependent activation curve of the channel upward, suggesting that Ter A could activate the channels even without intracellular Ca2+.
Figure 5. Effect of Ter A on the Ca2+-dependence of BKCa channel activation. Representative single channel current traces of the BKCa channels at [Ca2+]i ranging from 1 nM–50 μM before (A) and after (B) intracellular application of 10 μM Ter A at +20 mV. (C) The relationship between Po and [Ca2+]i in the absence and presence of 10 μM Ter A are fitted to a modified Hill equation (see text for detail). Each point is the mean ± SEM from at least five experiments.
![Figure 5. Effect of Ter A on the Ca2+-dependence of BKCa channel activation. Representative single channel current traces of the BKCa channels at [Ca2+]i ranging from 1 nM–50 μM before (A) and after (B) intracellular application of 10 μM Ter A at +20 mV. (C) The relationship between Po and [Ca2+]i in the absence and presence of 10 μM Ter A are fitted to a modified Hill equation (see text for detail). Each point is the mean ± SEM from at least five experiments.](/cms/asset/2fa1b381-698b-4e54-90a5-3facde629413/imbc_a_538731_f0005_b.jpg)
Effect of Ter A on the voltage-dependent activation of the BKCa channels
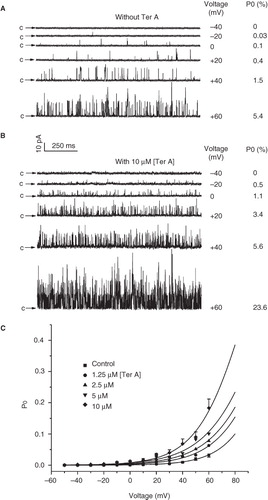
Next, we studied the effect of Ter A on the voltage dependent activation of the BKCa channel. showed current traces at different membrane potentials without Ter A, indicating that the Po of channels increased with the depolarization of the membrane potential. Application of Ter A significantly increased the Po of the channel at every corresponding membrane potential (). The Po of the BKCa channels as a function of the membrane potential with or without Ter A (1.25–10 μM) was fitted by the Boltzmann equation: Po = 1/{1 + e [(V1/2 − V)/k]}, where V1/2 is the voltage required for half-maximal activity of the channel and k is the slope factor of the curve. The best fit to the data was obtained with values of 117.4 mV for V1/2 and 17.0 mV for k in the absence of Ter A and with values of 88.6 mV for V1/2 and 18.4 mV for k in the presence of 10 μM Ter A (). Therefore, Ter A shifted the voltage activation curve toward less positive membrane potentials without affecting the slope factor of the curve, which suggested that Ter A did not alter the voltage-sensitivity of the channel.
Figure 6. Effect of Ter A on the voltage-dependence of the BKCa channel activation. Representative single channel current traces of the BKCa channels at membrane potentials ranging from −50 to +60 mV in the absence (A) and presence (B) of 10 μM Ter A. (C) The relationships between the mean Po of the BKCa channels and the membrane potentials in the absence and presence of Ter A (1.25–10 μM) are fitted with the Boltzmann equation (see text for detail). Only the error bars for the control and 10 μM Ter A are shown for clarity.
STREX domain is required for activation of the BKCa channel by Ter A
We have previously reported that the STREX domain located in the cytoplasmic side of BKCa channel is required for activation of the BKCa channel by a mixture of cerebrosides obtained from Baifuzi (Chi et al. Citation2010). To test whether the STREX domain is also required for the activation of the BKCa channels by a pure cerebroside compound, we compared the effect of Ter A on the wild-type and STREX-deleted BKCa channels (, ). Ter A (10 μM) significantly increased the Po of the wild-type BKCa channel from 3.5 ± 0.4% (n = 7) of control to 7.1 ± 0.4% (n = 7). In contrast, it had no significant effect on the STREX-deleted BKCa channel (). These data indicated that the STREX domain was necessary for Ter A to activate the BKCa channels. This is in accordance with the inference that the binding site for Ter A might be located at the cytoplasmic side of the channel.
Figure 7. Comparison of the effect of Ter A on the activation of wild-type and STREX-deleted BKCa channels. Representative single channel current traces of the wild-type (A) and STREX-deleted (B) BKCa channels in the absence and presence of intracellularly applied Ter A. The membrane potential of the patch is +20 mV and [Ca2+]i is 10 μM. Arrows indicate the closed state of the channels. (C) Summary of the effect of Ter A on the mean Po of the wild-type and STREX-deleted BKCa channel. **p < 0.005 compared to the control.
![Figure 7. Comparison of the effect of Ter A on the activation of wild-type and STREX-deleted BKCa channels. Representative single channel current traces of the wild-type (A) and STREX-deleted (B) BKCa channels in the absence and presence of intracellularly applied Ter A. The membrane potential of the patch is +20 mV and [Ca2+]i is 10 μM. Arrows indicate the closed state of the channels. (C) Summary of the effect of Ter A on the mean Po of the wild-type and STREX-deleted BKCa channel. **p < 0.005 compared to the control.](/cms/asset/f9d0ab56-abe7-4395-b2b5-2aefba1b02e1/imbc_a_538731_f0007_b.jpg)
Discussion
In the present study, we demonstrated that single pure cerebroside compounds, Ter A and Ter B purified from edible mushroom T. albuminosus, could activate the BKCa channel by increasing its Po. Ter A could activate the channel even without intracellular Ca2+ as it shifted the Ca2+-dependent activation curve of the channel upward. Ter A also shifted the voltage-dependent activation curve to the less positive potential. In our previous study, we have shown that a mixture of cerebrosides, isolated from traditional Chinese medicine Baifuzi, could potently activate the BKCa channel (Chi et al. Citation2010). Taken together, these results might suggest that this type of chemicals might be the BKCa channel openers. Therefore, individual cerebroside is merit to be explored to find better BKCa channel openers.
As integral membrane proteins nestled in the lipid environment of the plasma membrane, ion channels are modulated by many lipid-soluble molecules in the membrane (Ordway et al. Citation1989, Barrantes Citation2002, Tillman and Cascio Citation2003, Suh and Hille Citation2005, Maguy et al. Citation2006, Beech et al. Citation2009, Reichow and Gonen Citation2009). BKCa channels have also been shown to be activated by many kinds of lipid-soluble molecules, including cerebrosides (Chi and Qi Citation2006, Chi et al. Citation2010), fatty acid (Denson et al. Citation2000, Clarke et al. Citation2002, Sun et al. Citation2007), steroid (White et al. Citation1995, Nishimura et al. Citation2008), PIP2 (Vaithianathan et al. Citation2008), and sphingomyelin derivate (sphingosine-1-phosphate) (Kim et al. Citation2006). This study demonstrated that single pure cerebroside compounds Ter A and B could activate the BKCa channel as well. Then, what is the molecular mechanism of the BKCa channel activation by termitomycesphins as well as other kinds of cerebrosides? Both our previous and the present study showed that BKCa channels could be activated by cerebrosides even at the inside-out patch configuration. This result strongly suggests that the intracellular signaling milieu is not required for the channel activation. Furthermore, the STREX domain is necessary as its removal abolished the effect of cerebrosides, suggesting that there is interaction between them. How STREX domain that is located in the cytoplasmic side of the membrane could interact with the cerebroside that is highly likely in the lipid membrane? The following two studies indicate that STREX domain could dock on the plasma membrane: (1) STREX-GFP fusion proteins are preferentially located at the plasma membrane (Naruse et al. Citation2009); and (2) Cytoplasmic side of the BKCa channel could interact with the plasma membrane via palmitoylation of its STREX domain (Tian et al. Citation2008). Our recent study has shown that there is a direct interaction between STREX domain and Baifuzi-CB (Chi et al. Citation2010); therefore, it is reasonable to assume that cerebrosides activate the BKCa channel through its interaction with the STREX domain of the channel. A further study to identify the binding sites of cerebrosides on the channel is necessary to confirm this hypothesis.
The blood-brain barrier (BBB) functions to hinder the efficient delivery of many potentially important therapeutic agents to the brain. Overcoming the BBB to deliver therapeutic agents to specific regions of the brain presents a major challenge to treat most brain disorders. Without specific transport systems, only small molecules with high lipid solubility actually cross the BBB (Oldendorf Citation1974, Johansson Citation1990, Pardridge Citation2003). Due to their intrinsic property of crossing the BBB, lipid-soluble small molecules have potential as therapeutic drugs to treat brain disorders. It has been shown that activation of the BKCa channel by its openers could minimize neuronal depolarization, reduced neurotransmitter release and significantly attenuated infarct growth during ischemic stroke in animal models (Gribkoff et al. Citation2001, Hong et al. Citation2006; Chi et al. Citation2010). Moreover, BKCa channels have been suggested to be a specific target for selectively pharmacological modulation of blood-brain tumour barrier to increase delivery of chemotherapeutic drugs to brain metastases (Ningaraj et al. Citation2002, Hu et al. Citation2007). Cerebrosides, such as termitomycesphins, are lipid-soluble small molecules which should cross the BBB effectively. Thus, with its effect on activation of BKCa channels, cerebrosides have potential to become novel therapeutic agents to treat brain diseases that are associated with BKCa channels.
Acknowledgements
We thank Dr Sokabe and Dr Naruse for their generous gift of the BKCa channel and its STREX-deleted form.
Declaration of interest: This work was partly supported by the National Basic Research Program of China (2005CB522804) and the National Science Foundation of China (31070741). The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Barrantes FJ. 2002. Lipid matters: Nicotinic acetylcholine receptor-lipid interactions. Mol Membr Biol 19(4):277–284.
- Beech DJ, Bahnasi YM, Dedman AM, Al-Shawaf E. 2009. TRPC channel lipid specificity and mechanisms of lipid regulation. Cell Calcium 45(6):583–588.
- Brenner R, Chen QH, Vilaythong A, Toney GM, Noebels JL, Aldrich RW. 2005. BK channel β4 subunit reduces dentate gyrus excitability and protects against temporal lobe seizures. Nat Neurosci 8(12):1752–1759.
- Brenner R, Peréz GJ, Bonev AD, Eckman DM, Kosek JC, Wiler SW, Patterson AJ, Nelson MT, Aldrich RW. 2000. Vasoregulation by the β1 subunit of the calcium-activated potassium channel. Nature 407(6806):870–876.
- Butler A, Tsunoda S, McCobb DP, Wei A, Salkoff L. 1993. mSlo, a complex mouse gene encoding “maxi” calcium-activated potassium channels. Science 261(5118):221–224.
- Chi S, Qi Z. 2006. Regulatory effect of sulphatides of BKCa channels. Br J Pharmacol 149(8):1031–1038.
- Chi S, Cai W, Liu P, Zhang Z, Chen X, Gao L, Qi J, Bi L, Chen L, Qi Z. 2010. Baifuzi reduces transient ischemic brain damage through an interaction with the STREX domain of BKCa channels. Cell Death Dis 1:e13–23.
- Clarke AL, Petrou S, Walsh JV Jr, Singer JJ. 2002. Modulation of BKCa channel activity by fatty acids: Structural requirements and mechanism of action. Am J Physiol Cell Physiol 283(5):C1441–1453.
- Denson DD, Wang X, Worrell RT, Eaton DC. 2000. Effects of fatty acids on BK channels in GH3 cells. Am J Physiol Cell Physiol 279(4):C1211–1219.
- Du W, Bautista JF, Yang H, Diez-Sampedro A, You SA, Wang L, Kotagal P, Lüders HO, Shi J, Cui J, Richerson GB, Wang QK. 2005. Calcium-sensitive potassium channelopathy in human epilepsy and paroxysmal movement disorder. Nat Genet 37(7):733–738.
- Filosa JA, Bonev AD, Straub SV, Meredith AL, Wilkerson MK, Aldrich RW, Nelson MT. 2006. Local potassium signaling couples neuronal activity to vasodilation in the brain. Nat Neurosci 9(11):1397–1403.
- Gribkoff VK, Starrett JE, Dworetzky SI, Hewawasam P, Boissard CG, Cook DA, Frantz SW, Heman K, Hibbard JR, Huston K, Johnson G, Krishnan BS, Kinney GG, Lombardo LA, Meanwell NA, Molinoff PB, Myers RA, Moon SL, Ortiz A, Pajor L, Pieschl RL, Post-Munson DJ, Signor LJ, Srinivas N, Taber MT, Thalody G, Trojnacki JT, Wiener H, Yeleswaram K, Yeola SW. 2001. Targeting acute ischemic stroke with a calcium-sensitive opener of maxi-K potassium channels. Nat Med 7(4):471–477.
- Hong KW, Lee JH, Kima KY, Park SY, Lee WS. 2006. Cilostazol: Therapeutic potential against focal cerebral ischemic damage. Curr Pharm Des 12(5)565–573.
- Horrigan FT, Aldrich RW. 2002. Coupling between voltage sensor activation, Ca2+ binding and channel opening in large conductance (BK) potassium channels. J Gen Physiol 120(3):267–305.
- Hu J, Yuan X, Ko MK, Yin D, Sacapano MR, Wang X, Konda BM, Espinoza A, Prosolovich K, Ong JM, Irvin D, Black KL. 2007. Calcium-activated potassium channels mediated blood-brain tumor barrier opening in a rat metastatic brain tumor model. Mol Cancer 6:22–23.
- Issa NP, Hudspeth AJ. 1994. Clustering of Ca2+ channels and Ca2+-activated K+ channels at fluorescently labeled presynaptic active zones of hair cells. Proc Natl Acad Sci USA 91(16):7578–7582.
- Jiang Y, Pico A, Cadene M, Chait BT, MacKinnon R. 2001. Structure of the RCK domain from the E.coli K+ channel and demonstration of its presence in the human BK channel. Neuron 29(3):593–601.
- Johansson BB. 1990. The physiology of the blood-brain barrier. Adv Exp Med Biol 274:25–39.
- Kawakubo T, Naruse K, Matsubara T, Hotta N, Sokabe M. 1999. Characterization of a newly found stretch-activated KCa,ATP channel in cultured chick ventricular myocytes. Am J Physiol 276(6 Pt 2):H1827–1838.
- Kim MY, Liang GH, Kim JA, Kim YJ, Oh S, Suh SH. 2006. Sphingosine-1-phosphate activates BKCa channels independently of G protein-coupled receptor in human endothelial cells. Am J Physiol Cell Physiol 290(4):C1000–1008.
- Lancaster B, Nicoll RA. 1987. Properties of two calcium-activated hyperpolarizations in rat hippocampal neurons. J Physiol 389:187–203.
- Lingle CJ, Solaro CR, Prakriya M, Ding JP. 1996. Calcium-activated potassium channels in adrenal chromaffin cells. Ion Channels 4:261–301.
- Magleby KL. 2003. Gating mechanism of BK (Slo1) channels: So near, yet so far. J Gen Physiol 121(2):81–96.
- Maguy A, Hebert TE, Nattel S. 2006. Involvement of lipid rafts and caveolae in cardiac ion channel function. Cardiovasc Res 69(4):798–807.
- Meredith AL, Thorneloe KS, Werner ME, Nelson MT, Aldrich RW. 2004. Overactive bladder and incontinence in the absence of the BK large conductance Ca2+-activated K+ channel. J Biol Chem 279(35):36746–36752.
- Naruse K, Tang QY, Sokabe M. 2009. Stress-Axis Regulated Exon (STREX) in the C terminus of BKCa channels is responsible for the stretch sensitivity. Biochem Biophys Res Commun 385(4):634–639.
- Ningaraj NS, Rao M, Hashizume K, Asotra K, Black KL. 2002. Regulation of blood-brain tumor barrier permeability by calcium-activated potassium channels. J Pharmacol Exp Ther 301(3):838–851.
- Nishimura I, Ui-Tei K, Saigo K, Ishii H, Sakuma Y, Kato M. 2008. 17β-estradiol at physiological concentrations augments Ca2+-activated K+ currents via estrogen receptor β in the gonadotropin-releasing hormone neuronal cell line GT1-7. Endocrinology 149(2):774–782.
- Oldendorf WH. 1974. Lipid solubility and drug penetration of the blood brain barrier. Proc Soc Exp Biol Med 147(3):813–815.
- Ordway RW, Walsh JV Jr, Singer JJ. 1989. Arachidonic acid and other fatty acids directly activate potassium channels in smooth muscle cells. Science 244(4909):1176–1179.
- Pallotta BS, Magleby KL, Barrett JN. 1981. Single channel recordings of Ca2+-activated K+ currents in rat muscle cell culture. Nature 293(5832):471–474.
- Pardridge WM. 2003. Blood-brain barrier drug targeting: The future of brain drug development. Mol Interv 3(2):90–105.
- Qi J, Ojika M, Sakagami Y. 2000. Termitomycesphins A-D, novel neuritogenic cerebrosides from the edible Chinese mushroom Termitomyces albuminosus. Tetrahedron 56:5835–5841.
- Raffaelli G, Saviane C, Mohajerani MH, Pedarzani P, Cherubini E. 2004. BK potassium channels control transmitter release at CA3-CA3 synapses in the rat hippocampus. J Physiol 557(Pt 1):147–157.
- Reichow SL, Gonen T. 2009. Lipid-protein interactions probed by electron crystallography. Curr Opin Struct Biol 19(5):560–565.
- Ruknudin A, Sachs F, Bustamante JO. 1993. Stretch-activated ion channels in tissue-cultured chick heart. Am J Physiol 264(3 Pt 2):H960–972.
- Sausbier M, Arntz C, Bucurenciu I, Zhao H, Zhou XB, Sausbier U, Feil S, Kamm S, Essin K, Sailer CA, Abdullah U, Krippeit-Drews P, Feil R, Hofmann F, Knaus HG, Kenyon C, Shipston MJ, Storm JF, Neuhuber W, Korth M, Schubert R, Gollasch M, Ruth P. 2005. Elevated blood pressure linked to primary hyperaldosteronism and impaired vasodilation in BK channel-deficient mice. Circulation 112(1):60–68.
- Sausbier M, Matos JE, Sausbier U, Beranek G, Arntz C, Neuhuber W, Ruth P, Leipziger J. 2006. Distal colonic K+ secretion occurs via BK channels. J Am Soc Nephrol 17(5):1275–1282.
- Sausbier M, Hu H, Arntz C, Feil S, Kamm S, Adelsberger H, Sausbier U, Sailer CA, Feil R, Hofmann F, Korth M, Shipston MJ, Knaus HG, Wolfer DP, Pedroarena CM, Storm JF, Ruth P. 2004. Cerebellar ataxia and Purkinje cell dysfunction caused by Ca2+-activated K+ channel deficiency. Proc Natl Acad Sci USA 101(25):9474–9478.
- Schreiber M, Salkoff L. 1997. A novel calcium-sensing domain in the BK channel. Biophys J 73(3):1355–1363.
- Shen KZ, Lagrutta A, Davies NW, Standen NB, Adelman JP, North RA. 1994. Tetraethylammonium block of Slowpoke calcium-activated potassium channels expressed in Xenopus oocytes: Evidence for tetrameric channel formation. Pflugers Arch 426(5):440–445.
- Suh BC, Hille B. 2005. Regulation of ion channels by phosphatidylinositol 4, 5-bisphosphate. Curr Opin Neurobiol 15(3):370–378.
- Sun X, Zhou D, Zhang P, Moczydlowski EG, Haddad GG. 2007. β-subunit-dependent modulation of hSlo BK current by arachidonic acid. J Neurophysiol 97(1):62–69.
- Tian L, Jeffries O, McClafferty H, Molyvdas A, Rowe IC, Saleem F, Chen L, Greaves J, Chamberlain LH, Knaus HG, Ruth P, Shipston MJ. 2008. Palmitoylation gates phosphorylation-dependent regulation of BK potassium channels. Proc Natl Acad Sci USA 105(52):21006–21011.
- Tillman TS, Cascio M. 2003. Effects of membrane lipids on ion channel structure and function. Cell Biochem Biophys 38(2):161–190.
- Toro L, Vaca L, Stefani E. 1991. Calcium-activated potassium channels from coronary smooth muscle reconstituted in lipid bilayers. Am J Physiol 260(6 Pt 2):H1779–1789.
- Tseng-Crank J, Foster CD, Krause JD, Mertz R, Godinot N, DiChiara TJ, Reinhart PH. 1994. Cloning, expression, and distribution of functionally distinct Ca2+-activated K+ channel isoforms from human brain. Neuron 13(6):1315–1330.
- Vaithianathan T, Bukiya A, Liu J, Liu P, Asuncion-Chin M, Fan Z, Dopico A. 2008. Direct regulation of BK channels by phosphatidylinositol 4, 5-bisphosphate as a novel signaling pathway. J Gen Physiol 132(1):13–28.
- Wang L, Sigworth FJ. 2009. Structure of the BK potassium channel in a lipid membrane from electron cryomicroscopy. Nature 461(7261):292–295.
- White RE, Darkow DJ, Lang JL. 1995. Estrogen relaxes coronary arteries by opening BKCa channels through a cGMP-dependent mechanism. Circ Res 77(5):936–942.
- Zarei MM, Eghbali M, Alioua A, Song M, Knaus HG, Stefani E, Toro L. 2004. An endoplasmic reticulum trafficking signal prevents surface expression of a voltage- and Ca2+-activated K+ channel splice variant. Proc Natl Acad Sci USA 101(27):10072–10077.
- Zhou XB, Arntz C, Kamm S, Motejlek K, Sausbier U, Wang GX, Ruth P, Korth M. 2001. A molecular switch for specific stimulation of the BKCa channel by cGMP and cAMP kinase. J Biol Chem 276(46):43239–43245.