Abstract
Lck is a non-receptor tyrosine kinase of the Src family that is essential for T cell activation. Dual N-terminal acylation of Lck with myristate (N-acylation) and palmitate (S-acylation) is essential for its membrane association and function. Reversible S-acylation of Lck is observed in vivo and may function as a control mechanism. Here we identify the DHHC family protein S-acyltransferase DHHC2 as an enzyme capable of palmitoylating of Lck in T cells. Reducing the DHHC2 level in Jurkat T cells using siRNA causes decreased Lck S-acylation and partial dislocation from membranes, and conversely overexpression of DHHC2 increases S-acylation of an Lck surrogate, LckN10-GFP. DHHC2 localizes primarily to the endoplasmic reticulum and Golgi apparatus suggesting that it is involved in S-acylation of newly-synthesized or recycling Lck involved in T cell signalling.
Introduction
S-acylation, also referred to as palmitoylation, is the post-translational addition of a saturated fatty acid, most commonly palmitic acid, to a protein cysteine residue via a thioester bond. Many proteins are S-acylated, including transmembrane proteins like G-protein-coupled receptors (GPCRs), T cell co-receptors CD4 and CD8 and ion channels; cytosolic and membrane-associated proteins like members of the Ras and Src families, the α subunits of heterotrimeric G-proteins, endothelial nitric oxide synthase (eNOS), SNARE vesicle fusion proteins; as well as secreted and viral proteins. Addition of fatty acids to a protein increases the protein's hydrophobicity and facilitates its membrane interaction, frequently a process involved in signalling, trafficking, protein stability, protein-protein interactions and membrane subdomain association (Smotrys and Linder Citation2004, Resh Citation2006a, Citation2006b). When S-acylation takes place on an N-terminal cysteine, the thioester bond is often rearranged to a stable amide bond at the N-terminus, this irreversible type of acylation being referred to as N-acylation (Pepinsky et al. Citation1998, Kleuss and Krause Citation2003). Because palmitate can be added in both S-acyl and N-acyl linkages we prefer the term S-acylation to refer specifically to the thioester-linked mode of acylation.
S-acylation is the only lipid modification that is readily reversible under physiological conditions, and dynamic S-acylation has been demonstrated for many proteins including Ras and the Src kinases Fyn and Lck, PSD−95, eNOS and several GPCRs (Magee et al. Citation1987, Wolven et al. Citation1997, Yeh et al. Citation1999, El-Husseini et al. Citation2002, Zeidman et al. Citation2009). S-acylation is regulated by protein acyl transferases (PATs) that catalyze the attachment of fatty acids to proteins, and by thioesterases (TEs), that remove the acyl chains. The first PATs were identified in Saccharomyces cerevisiae where Akr1p and Erf2p (together with Erf4p) were shown to be PATs for yeast casein kinase 2 (Roth et al. Citation2002) and Ras (Lobo et al. Citation2002), respectively. Akr1p and Erf2p have a conserved zinc finger Asp-His-His-Cys (DHHC) motif which was shown to be essential for the PAT activity (Lobo et al. Citation2002, Roth et al. Citation2002). The 23 DHHC proteins predicted in the mammalian genome have variable substrate specificities (Fukata et al. Citation2006). All substrates identified so far for the DHHC PATs are intracellular proteins (Smotrys and Linder Citation2004, Mitchell et al. Citation2006, Tsutsumi et al. Citation2008) and known PAT/substrate pairs include: DHHC9, together with a Golgi-associated protein GCP16 echoing the yeast Erf2p/Erf4p complex, and H-and N-Ras (Swarthout et al. Citation2005); DHHC21 and eNOS (Fernandez-Hernando et al. Citation2006); HIP14/DHHC17 and huntingtin (Huang et al. Citation2004, Yanai et al. Citation2006); and GODZ/DHHC3 and the γ2 subunit of the GABAA receptor (Keller et al. Citation2004).
Much less is known about the regulation of deacylation. So far, only two TEs that remove acyl chains from proteins have been identified, acyl protein thioesterase 1 (APT1) and protein palmitoyl thioesterase 1 (PPT1) (Zeidman et al. Citation2009). APT1 is a cytosolic protein (Duncan and Gilman Citation1998) with a widespread tissue distribution (Toyoda et al. Citation1999). Ras, various heterotrimeric G-protein α-subunits, eNOS, RGS4 and SNAP-23 have been identified as APT1 substrates in in vitro assays (Duncan and Gilman Citation1998, Yeh et al. Citation1999, Flaumenhaft et al. Citation2007), and APT1 therefore may play a role in the regulation of the function of these proteins. PPT1, on the other hand, does not appear to be involved in regulation of the dynamic acylation seen in many cytosolic proteins, but is a lysosomal protein (Verkruyse and Hofmann Citation1996), removing acyl moieties from proteins as a step in the degradation process. PPT1 deficiency leads to the neurodegenerative disease infantile neuronal ceroid lipofuscinosis (INCL) where there is an accumulation of granular deposits inside the cells (Vesa et al. Citation1995, Lu et al. Citation1996).
T cells have an essential role in acquired immunity as modulators of immune responses through the secretion of cytokines or by killing antigen-bearing target cells. Engagement of the T cell receptor (TCR) by antigen activates a Src-family protein tyrosine kinase pathway, resulting in tyrosine phosphorylation of downstream proteins and activation of signal transduction pathways that lead to cell proliferation, differentiation and survival (Palacios and Weiss Citation2004). Lck (lymphocyte-specific protein tyrosine kinase or p56 lck ) is a non-receptor tyrosine kinase of the Src family. The presence of Lck is essential for TCR signalling; without it downstream signalling is blocked (Straus and Weiss Citation1992, van Oers et al. Citation1996). Once the TCR is engaged, Lck phosphorylates ITAMs of the TCR-associated CD3 chains and ζ chain homodimers. This leads to recruitment of the cytosolic tyrosine kinase ZAP-70 to the TCR, and its phosphorylation and activation by Lck (Palacios and Weiss Citation2004). T cell development has also been shown to be severely impaired in Lck-deficient mice (Molina et al. Citation1992).
Lck is co-translationally modified by N-myristoylation on glycine at position 2, forming an amide bond replacing the initiator methionine residue, and post-translationally by double S-acylation where palmitic acids are added to cysteines at positions 3 and 5 (Koegl et al. Citation1994). The myristoylation is a pre-requisite for S-acylation and required for initial membrane tethering, but myristoylation alone is not sufficient to keep Lck at the membrane. Without the subsequent S-acylation, Lck does not remain attached to the plasma membrane (Koegl et al. Citation1994, Zlatkine et al. Citation1997). Not surprisingly, acylation of Lck is required for its function. Activated but unacylated Lck does not induce interleukin 2 (IL-2) production in T cells, unlike acylated and activated Lck, and the increase of tyrosine phosphorylated proteins is also missing (Yurchak and Sefton Citation1995).
S-acylation of Lck not only targets it to the plasma membrane, but also to specific membrane compartments. The active TCR and its co-receptors CD4 and CD8 are localized to lipid rafts, ordered subdomains in the plasma membrane that are enriched in glycosphingolipids, sphingomyelin and cholesterol, in addition to many signalling proteins, creating an environment favouring signalling (Janes et al. Citation1999, Citation2000, Kabouridis Citation2006). Acylated Lck is also found in these subdomains (Shenoy-Scaria et al. Citation1993, Rodgers et al. Citation1994, Kabouridis et al. Citation1997). In fact, Lck raft localization is necessary, as a reduction in TCR signalling was seen when Lck was acylated by a less hydrophobic analogue of palmitic acid, which allows membrane attachment of Lck but reduces its affinity for lipid rafts (Hawash et al. Citation2002).
Despite the well characterized role of Lck in T cell signalling, and the importance of Lck S-acylation for its function, little is known about the regulation of Lck S-acylation. In this paper we identify DHHC2 as a PAT for Lck in T cells and show that Lck S-acylation and membrane association is decreased when expression of DHHC2 is significantly reduced.
Methods
Cloning and plasmids
An expression vector, pmCFP, enabling C-terminal fusions with monomeric cyan fluorescent protein (mCFP) DNA was created by inserting a Gateway cassette (Invitrogen) and cDNA encoding mCFP into pcDNA3.1. cDNAs encoding full-length DHHC2, 4 and 16 were generated by polymerase chain reaction (PCR) using cDNA from human Jurkat T cells as template and inserted into pmCFP via pENTR/D-TOPO using Gateway reagents (Invitrogen). The pcDNA3- LckN10-GFP expression vector encoding the 10 N-terminal amino acids of Lck is described in (Zlatkine et al. Citation1997), but was used here fused to GFP instead.
Cell culture and transfections
Human Jurkat T cells clone E6.1 (hereby referred to as Jurkat T cells), Lck-negative J.Cam1.6 Jurkat variant and Raji B cells were cultured in RPMI medium (PAA), supplemented with 10% fetal calf serum (FCS; PAA), 100 U/ml penicillin and 100 μg/ml streptomycin (Gibco, Invitrogen). HEK293A and HeLa cells were grown in DMEM (PAA) with the addition of 10% FCS. Human primary T cells and macrophages were freshly isolated and primary B cells were a kind gift from Dr E. Jury, University College London. For transfection, 2 μg of plasmid DNA and 5 μl of Lipofectamine 2000 (Invitrogen) were used for HEK293A cells plated in 35 mm dishes, or 2 μl Fugene 6 (Roche) and 0.5 μg plasmid DNA for HeLa cells plated on glass coverslips in 24-well dishes. For co-transfection of HEK293A cells, LckN10-GFP and DHHC expression vectors were transfected at a 1:2 ratio. Transfected cells were incubated for 24 h before analysis.
RNA extraction and cDNA generation
Total RNA was extracted from 1 × 107 cells with the RNeasy Mini Kit (Qiagen) according to the manufacturer's instructions. Determination of RNA quality and concentration was carried out using 260 nm absorbance analysis in a spectrophotometer and the 18/28S RNA integrity visualized after separation on agarose gels. First strand cDNA from mRNA was generated using oligodT primers and the Ready-To-Go You-Prime First-Strand Beads (GE Healthcare).
mRNA expression analysis
Analysis of the mRNA expression of the human DHHC proteins in various cell lines was carried out by RT-PCR, using cDNA and the primers described in Supplementary Table S1, available online. The forward and reverse primers were derived from different exons. IMAGE expressed sequence tags (EST) vectors (identification numbers in Supplementary Table S2, available online) were used as positive controls for PCR reaction with the human DHHC primer pairs. For DHHC 15, an EST clone was not available. The PCR reactions were carried out using Taq polymerase (Invitrogen) and 10 pmol of each primer and about 500 ng cDNA or 200 ng genomic DNA. Amplification of actin mRNA was used as a positive control for the RT-PCR reaction.
RNAi treatment
Initial RNAi experiments were initially performed using ‘Pool of four’ siRNA oligos for DHHC2, 4, 16 and Lamin A/C (as a positive control for knock-down), and individual siRNA oligos directed against DHHC2 and DHHC4 (purchased from Dharmacon). For introduction of siRNA into Jurkat T cells, the Amaxa V kit was used according to the manufacturer's instructions using cells, passaged no more than 10 times, at a density of 3 × 105 cells/ml and a minimum number of 106 cells. For later experiments, Accell siRNAs Reagents for DHHC2 (cat. # E-018402-00-0005) and DHHC4 (cat. # E-016699-00-0005) and Accell control siRNAs (GAPDH cat # D-001930-01-20 and non-targeting siRNA cat # D-001910-01-20) were purchased from ABgene Limited, Epsom, UK. Gene silencing in Jurkat T cells was performed by addition of 1 μM specific or control Accell siRNA SMART pool in Accell siRNA Delivery Media (cat. # B-005000-100). Cells were analyzed 72 h after addition of siRNA.
Quantitative real-time PCR
mRNA was isolated and reverse transcribed (High Capacity Reverse Transcript Kit, Applied Biosystems). Quantitative real-time PCR was performed using TaqMan® Gene Expression assays Hs00275319_m1 (DHHC2) and Hs01052018_m1 (DHHC4) in combination with TaqMan® Universal PCR master mix (all Applied Biosystems). Relative quantitation was performed using the standard curve method. Standard curves were determined using RNA from untreated cells. Cycle thresholds were normalized to 18S rRNA. All tests were conducted and analyzed in triplicate (7500 Fast Real-time PCR System, Applied Biosystems).
Protein gel electrophoresis and immunoblotting
Proteins were separated on standard 10% SDS-PAGE gels or on 10% Bis-Tris NuPage gels (Invitrogen). For detection of proteins after semi-dry blotting onto PVDF membranes (Millipore), primary antibodies rabbit anti-Lck (Zlatkine et al. Citation1997), mouse anti-tubulin (Abcam, DM1A), mouse anti-actin (Sigma), mouse anti-GFP (Roche), rabbit anti-calnexin (Abcam) or rabbit anti-DHHC2 (see below) were used as indicated, and detected with the appropriate horseradish peroxidase-conjugated secondary antibody (Southern Biotech) and enhanced chemiluminescence.
Subcellular fractionation (S100/P100)
Cells were washed in PBS, incubated on ice for 10 min in swelling buffer (10 mM Tris, pH 7.5, 1 mM EDTA, 1 mM DTT, Complete protease inhibitors [Roche]) and homogenized by repeated passage through a 25-gauge needle. After removal of cell debris and nuclei at 200 g, 4°C for 5 min, the lysate was ultracentrifuged at 100,000 g, 4°C for 60 min in a TLA 45 Beckman rotor. The resulting pellet (P100), containing the membrane fraction, was resuspended in 1 × SDS-PAGE sample buffer, and 5 × SDS-PAGE sample buffer was added directly to the supernatant (S100), corresponding to the cytosolic fraction. The proteins were separated by gel electrophoresis followed by immunoblotting using the indicated primary antibody.
Immunostaining and fluorescence microscopy
HeLa cells, grown on glass coverslips, were washed with ice-cold PBS and fixed for 10 min at room temperature with 4% paraformaldehyde (PFA)/PBS and either mounted with Immunogold mounting medium (Invitrogen) or used for immunostaining, in which case they were blocked and permeabilized in 3% bovine serum albumin (BSA)/0.1% Triton X-100/PBS for 1 h at room temperature (RT). Incubation with primary antibody against Golgin 97 (Invitrogen, diluted 1:50) or calnexin (Santa Cruz, diluted 1:200) was followed by 1 h incubation with Alexa-633-conjugated secondary antibody (Invitrogen, diluted 1:500) in blocking and permeabilization buffer and mounting as above.
HEK293A cells were grown and fixed as for HeLa cells, and permeabilized with 1% Triton X100/PBS for 5 min at RT, blocked for 15 min at 4°C in 3% BSA/PBS, incubated with primary antibody (mouse anti-Golgin 97, diluted 1:50), mouse anti-calreticulin (Abcam, diluted 1:50) or mouse anti-Rab5 (Abcam, diluted 1:50) followed by secondary antibody (FITC-conjugated anti-rabbit antibody or Texas Red-conjugated anti-mouse, 1:50 dilution) in 3% BSA/TBS for 1 h each and mounted using with Citifluor mounting medium (Citifluor Ltd).
For immunofluorescence of endogenous DHHC2 and Lck in Jurkat T cells, 1 × 106 cells were washed and suspended in PBS, added to 13 mm diameter glass coverslips coated with poly-L-lysine (PLL; Sigma) and incubated for 2 min at RT. The PBS was aspirated, followed by fixation, blocking and permeabilization, antibody incubations and mounting as for HeLa cells, described above. Primary antibodies used were rabbit anti-DHHC2 (see below), 1:500 dilution, and mouse anti-Lck (3A5, Santa Cruz, 1:80 dilution) and secondary antibodies were conjugated to Alexa 555 (Invitrogen) for DHHC2 detection and Alexa 647 (Invitrogen) for Lck detection, both used at 1:200 dilutions.
Conjugation of Jurkat T cells and Raji B cells
Raji B cells were incubated with 1 μg/ml of SEE (Toxin Technology) for 1 h at 37°C followed by extensive washing in PBS. Jurkat T cells (5 × 105/slide) were mixed with an equal number of SEE-incubated Raji B cells, briefly centrifuged to enable conjugation and incubated for 15 min at 37°C. Afterwards, conjugates were gently resuspended in PBS and plated onto PLL-coated coverslips and allowed to settle for 2 min. Cells were fixed with 3% PFA/2% sucrose/PBS for 10 min at RT and permeabilized for 8 min with 0.2% Triton X-100/PBS. Conjugates were stained with the indicated antibodies in blocking solution (5% BSA/1% gelatin/PBS) for 1 h at RT. Alexa 488-, Alexa 555- and Alexa 647-labelled secondary antibodies were used accordingly. Conjugates were mounted in ProLong Gold medium with DAPI (Invitrogen).
Confocal microscopy was performed on an inverted laser scanning confocal fluorescence microscope (TCS SP5, Leica Microsystems GmbH, Wetzlar, Germany) using a 63 × 1.2NA oil-immersion objective, except for Jurkat T cell and Raji B cell conjugates, where a Zeiss LSM-510 inverted confocal microscope with 100 × 1.4NA oil objective was used. Images were processed using MetaMorph (Molecular Devices) and Photoshop (Adobe) software. Microscopy was performed in the Facility for Imaging by Light Microscopy (FILM).
Metabolic labelling with 3H-palmitate
1 × 107 Jurkat T or J.Cam1.6 cells or transfected HEK293A cells in six-well dishes were preincubated for 1 h in 1 ml RPMI supplemented with 5% FCS and 5 mM sodium pyruvate for 3H-palmitate labelling. 9,10-3H-palmitic acid (51 Ci/mmol, 200 μCi/ml; GE Healthcare) was added and the cells incubated for a further hour. After cell lysis (in 50 mM Hepes pH 7.5, 150 mM NaCl, 1.5 mM MgCl2, 1 mM EGTA, 1% Triton X100, 10% (v/v) glycerol, 50 mM NaF, 1 mM Na3VO4, 10 mM sodium pyrophosphate and 1% (w/v) octylglucoside), the proteins were separated by SDS-PAGE. Gels containing 3H-palmitate-labelled samples were incubated twice for 30 min in DMSO, then incubated with 22.5% (w/v) 2,5-diphenyloxazole in dimethyl sulphoxide (PPO/DMSO) for 1 h on a shaker, followed by two 30 min washes in water, and then dried and exposed to pre-flashed Kodak BioMax MS film at −80°C (Magee et al. Citation1995). Band densitometry was performed using ImageQuant TL (GE Healthcare) software.
DHHC2 antibody generation
Peptide antibodies against DHHC2 were designed to target the C-terminal variable region and an internal region, between transmembrane domain 3 and 4. Both peptides, C-terminal CKAGMSNPALTMENET and internal CRHGTDKNGFSLGFSK, were synthesized and coupled separately to keyhole limpet haemocyanin and immunized in combination into rabbits (Severn Biotech, UK). The specificity of the peptide antibodies was confirmed by their ability to detect exogenously expressed DHHC2-CFP, in addition to the endogenous proteins, and not having any cross-reactivity to neutrophil proteins, as neutrophils do not express DHHC2 (data not shown).
Results
Restricted expression of DHHC PATs in T cells
In order to investigate which DHHC protein could act as a PAT for Lck (the T lymphocyte-specific Src family kinase, one of the most heavily acylated proteins in T cells) potential candidates were identified. Initially an EST database search suggested that the DHHC genes 2, 4, 15 and 16 had a high probability of expression in Jurkat T cells. In preliminary experiments, these results were confirmed by qualitative RT-PCR using cDNA derived from Jurkat T cells and primers (Supplementary Table S1, available online) specific for each DHHC gene, using actin as a positive control gene (data not shown). EST IMAGE clones (Supplementary Table S2, available online) were used as positive controls for the primers, except for DHHC15, where such a clone did not exist. To exclude the possibility of possible contamination and amplification of genomic DNA, all primers were designed to amplify the smallest part of the DHHC mRNA including an exon-exon boundary. To ensure that the mRNAs encoding DHHC proteins were expressed in their entirety in Jurkat T cells, RT-PCR with primers spanning the whole coding sequence of DHHC2, 4, 15 and 16 was performed. Products corresponding to DHHC2, 4 and 16 mRNA were readily amplified and DNA sequencing confirmed that the PCR products were identical to the reported mRNA sequences of DHHC2, 4 and 16 (accession numbers NM_016353, NM_018106 and NM_032327, respectively). The DHHC15 mRNA, on the other hand, was found to contain a large deletion starting at an exon boundary, which caused a frame shift and would lead to a truncated protein upon expression. Using cDNA from HEK293A cells, the same primers generated a full-length DHHC15 product which was confirmed to be identical to the reported sequence (accession number NM_144969) by DNA sequencing.
The presence of DHHC 2, 4 and 16 mRNA in other cell types of the hematopoietic system was also examined and compared to that of Jurkat T cells. Supporting the validity of Jurkat T cells as a model system for investigation of DHHC proteins and Lck acylation, human primary T cells had the same expression pattern, with production of RT-PCR products corresponding to DHHC2, 4 and 16 mRNA (Supplementary Table S3, available online). Lck plays a crucial role in T cell signalling, and acylation is required for its function (Yurchak and Sefton Citation1995). In primary human B cells, where Lck is not expressed but the related Src family PTK Lyn is involved in the B cell receptor signalling (Scapini et al. Citation2009), DHHC2, 4 and 16 are again present, as judged by RT-PCR (Supplementary Table S3). This expression pattern was not found in macrophages, which are derived from myeloid hematopoietic lineages, where only expression of DHHC2 and 16 was detected by RT-PCR (Table S3). Expression of other members of the DHHC family has not been extensively tested.
Membrane dislocation of Lck by RNAi against DHHC2 and 4
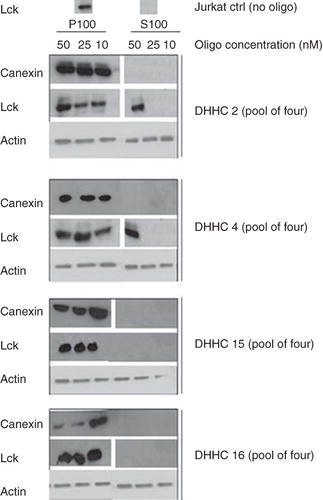
Lck acylation is necessary and sufficient to target it to the plasma membrane (Zlatkine et al. Citation1997). In Jurkat T cells grown under normal cell culture conditions, Lck is almost exclusively found in the particulate fraction after ultracentrifugation (P100 fraction), corresponding mainly to cellular membranes (and cytoskeleton), and not in the soluble fraction (S100) consisting of the cytosol (, top). Introduction of a ‘pool of four’ siRNA oligos designed to target DHHC2 into Jurkat T cells, followed by S100/P100 fractionation, had substantial effects on Lck membrane association. After DHHC2 targeting with the siRNA oligo pool at 50 nM, Lck partially delocalized from the membranes and became cytosolic (), as would be expected if there was a decreased level of acylation of Lck. siRNA against DHHC4 had a similar effect (). Although membrane release was only partial for both DHHC2 and 4 RNAi treatment, this may be because siRNA oligo pools were used – the concentration of individual oligos in the pool may have been suboptimal, and siRNA transfection was probably not efficient in all cells in the population. In contrast, targeting DHHC16 or DHHC15 (which is not expressed in Jurkat cells and thereby acts as a negative control) did not have any effect on the membrane association of Lck (, top). This could either suggest that DHHC16 is not a PAT for Lck, or that the siRNA did not decrease DHHC16 protein levels. As there were no available antibodies against DHHC4, 15 and 16 at the time that this work was done we did not have a direct means of examining the protein levels after siRNA treatment. The distribution between P100 and S100 of calnexin, a non-acylated membrane protein associated with the endoplasmic reticulum (ER), was used both as a control for the efficiency of the fractionation as well as a control that the effects seen after RNAi treatment targeting the DHHCs were specific for acylated proteins like Lck. Treatment with siRNA oligos targeting LaminA/C was used as an additional control for any potential off-target effects caused by siRNA treatment in general, and did not affect the Lck distribution between the S100 and the P100 fractions (data not shown).
Figure 1. DHHC2 and DHHC4 siRNA treatment causes Lck membrane dislocation. Where indicated, ‘pool of four’ siRNA oligos directed towards DHHC2, 4, 15 and 16 were introduced by Amaxa nucleofection into Jurkat T cells at the concentrations indicated. 72 h later, cells were fractionated into a P100 particulate membrane fraction and an S100 soluble cytosolic fraction, the proteins separated by SDS-PAGE and endogenous Lck, actin and calnexin in the fractions were detected by Western blotting. Actin was used as a loading control, and absence of calnexin in the S100 fraction serves as a control for the fractionation.
Characterization of DHHC-fluorescent protein fusions and DHHC2 antibody
The lack of commercial antibodies against DHHCs at the time that this work was done required alternative tools to be created in order to investigate the localization of the potential Lck PATs. Two approaches were taken: the creation of vectors encoding DHHC C-terminal fusion proteins with the cyan fluorescent protein (CFP) and the generation of antibodies (see Materials and methods).
The CFP fusion proteins transiently expressed in HEK293A cells, at varying levels, with DHHC2-CFP expressed at the highest level and DHHC16-CFP at the lowest. The expression of all DHHC-CFP constructs was relatively low compared to CFP alone, suggesting that expression level is determined primarily by protein- and/or mRNA-specific factors rather than by the expression vector. The predicted molecular weight of the fusion proteins was around 70 kDa, although they migrated faster on gels (), consistent with what is seen for many other multispanning transmembrane proteins. Some minor degradation products of the DHHC4-CFP fusion protein can be seen in and . It is possible that DHHC4-CFP is slightly more unstable or its level of expression more tightly controlled compared to the other two DHHCs. However, this may be a feature of the CFP fusion protein which does not reflect the stability of the native DHHC4; we were unable to test this because of the lack of an antibody to DHHC4.
Figure 2. Tools for DHHC protein analysis and membrane association of DHHC proteins. (A) Expression vectors encoding full length DHHC2, 4 and 16 fused to CFP at their C-terminus and CFP alone, were transiently transfected into HEK293A cells. 24 h post transfection, the expression of the DHHC-CFP proteins and CFP were analyzed in total cell lysates by Western blotting using anti-GFP antibody. (B) Specificity of the anti-DHHC2 antibody. DHHC2 (both endogenous and exogenous CFP-fused) was analyzed by Western blotting in cell lysates from HEK293A cells transiently transfected for 24 h with DHHC2-CFP, DHHC4-CFP and CFP. The anti-DHHC2 antibody recognizes endogenous DHHC2 in all lanes and DHHC2-CFP without cross-reactivity to DHHC4. (C) Distribution of DHHC-CFPs and CFP alone between the P100 particulate membrane fraction and the S100 soluble cytosolic fraction of HEK293A cells transiently transfected for 24 h as detected by Western blotting using an anti-GFP antibody.
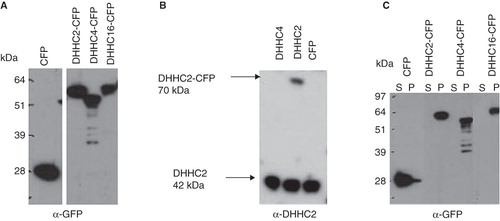
A specific DHHC2 antiserum was produced, detecting both endogenous DHHC2 and exogenous DHHC2-CFP in HEK293A cell lysates without any cross-reactivity to DHHC4 (), and also detecting endogenous DHHC2 from Jurkat T cells and mouse tissues (data not shown; mouse DHHC2 sequence is identical to human in the region corresponding to the peptides used for antibody production).
Overexpression of DHHC2 causes increased S-acylation of LckN10-GFP
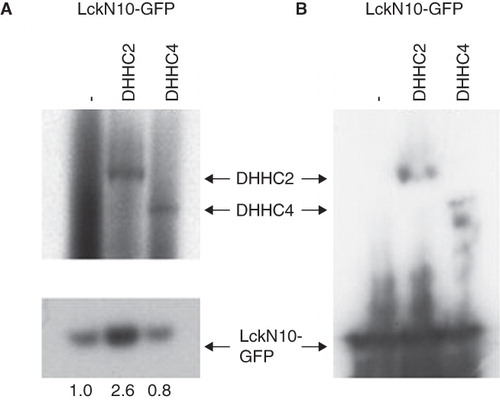
Co-overexpression of DHHC enzymes with potential substrates is frequently used to provide evidence for specific DHHC-substrate acylation (Fukata et al. Citation2006). To test whether DHHC2 and DHHC4 can act as PATs for Lck, the acylation of an overexpressed protein consisting of the 10 N-terminal amino acids of Lck fused to GFP (LckN10-GFP) was assessed upon increasing the levels of these DHHC proteins by transfection with the CFP fusion proteins. LckN10-GFP was more heavily acylated in HEK293A cells when co-expressed with DHHC2-CFP compared to when expressed with empty vector alone (, lower panel); LckN10-GFP expression is similar as seen in the anti-GFP blot (). Densitometric evaluation of the band intensities of the LckN10-GFP acylation suggested that overexpression of DHHC2-CFP caused about 2.6-fold higher acylation of LckN10-GFP in this representative experiment. Co-transfection with DHHC4-CFP, however, did not significantly increase the acylation of LckN10-GFP (, lower panel), perhaps reflecting the lower expression levels of DHHC4-CFP compared to DHHC2-CFP, as shown by Western blotting with anti-GFP (). Interestingly, on overexposed fluorograms, labelling of the DHHC2- and DHHC4-CFP proteins with 3H-palmitic was observed (, upper panel). Autopalmitoylation has been seen for other DHHC proteins (Lobo et al. Citation2002, Fukata et al. Citation2004, Huang et al. Citation2004, Smotrys et al. Citation2005, Swarthout et al. Citation2005, Jennings et al. Citation2009).
Figure 3. Enhanced S-acylation of LckN10-GFP by cotransfected DHHC2-CFP. HEK293A cells were co-transfected with LckN10-GFP and DHHC2-CFP or DHHC4-CFP as indicated. (A) Autoradiography of total cell lysates separated on NuPage gels following metabolic labelling with 3H-palmitate for 2 h. Upper panel – long exposure (21 days) showing 3H-palmitate labelling of transfected DHHC2- and DHHC4-CFP. Lower panel – 2 day exposure showing 3H-palmitate labelling of transfected LckN10-GFP. Numbers represent relative acylation levels normalized to control. (B) Western blotting using anti-GFP antibody detecting LckN10-GFP and DHHC2- and DHHC4-CFP levels in total lysates. The positions of bands corresponding to DHHC2-CFP, DHHC4-CFP and LckN10-GFP are indicated.
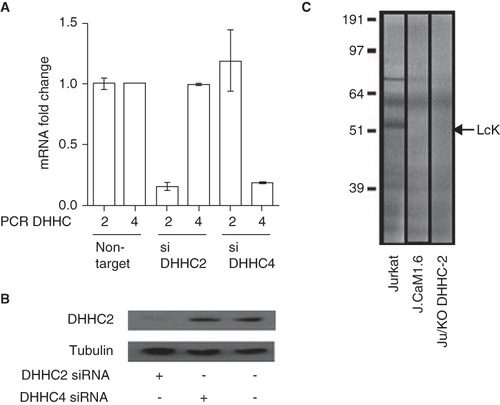
DHHC2 RNAi treatment decreases Lck S-acylation
To provide further evidence for a role of DHHC2 in endogenous Lck S-acylation, we optimized the RNAi effect with Accell siRNAs Reagents (Dharmacon RNAi Technologies) which are modified for stability, enhanced specificity to target and delivery with no transfection reagent required, and high silencing efficiency. Using quantitative real-time PCR we confirmed that the DHHC2 and 4 Accell siRNAs were specific for their respective targets with no effect on the non-cognate DHHC, and reproducibly gave more than 80% knockdown of mRNA after 72 h treatment (). Treatment with DHHC2 siRNA for this length of time causes a substantial knockdown of DHHC2 protein as judged by Western blotting with anti-DHHC2, in contrast to treatment with DHHC4 siRNA where no effects on DHHC2 protein levels were observed (). Following transfection of DHHC2, 4 or control siRNAs into Jurkat T cells and incubation for 70 h, cells were metabolically labelled for the last hour with 3H-palmitate to detect acylation of Lck. siRNA treatment against either DHHC2 or DHHC4, or the control oligos directed against GAPDH, did not have an effect on Lck or global protein levels as judged by 35S-methionine labelling (data not shown). In Jurkat T cells labelled with 3H-palmitic acid the major band around 56 kDa represents Lck as shown by the absence of this band in the Lck-negative J.Cam1.6 variant, in which Fyn is expressed at normal levels (Yamasaki et al. Citation1997). J.Cam1.6 cells have been used by us previously in studies of Lck S-acylation (Kabouridis et al. Citation1997); these cells express low levels of a catalytically inactive deletion mutant of Lck and therefore lack the 56 kDa Lck band. The level of 3H-palmitate incorporation into Lck was consistently reduced in cells treated with siRNA against DHHC2 compared to Lck from cells exposed to control oligos (), consistent with DHHC2 being a major PAT for Lck in Jurkat T cells. There is an additional band in Jurkat cells (∼70–75 kDa) that is lost when cells are treated with DHHC2 siRNA. We do not know the identity of this band but it unlikely to be related to Lck or DHHC2; it could be another DHHC2 substrate. Unfortunately, no consistent effect of DHHC4 siRNA on Lck S-acylation was detected; combined with the lack of enhanced S-acylation seen in co-transfection experiments () we cannot make a firm conclusion that DHHC4 plays a role in Lck S-acylation.
Figure 4. Decreased Lck S-acylation after DHHC2 siRNA treatment. (A) Quantitative real-time PCR was used to assess DHHC2 and DHHC4 mRNA expression in Jurkat cells transfected with Accell siRNA directed against either DHHC2 (siDHHC2) or DHHC4 (siDHHC4). Relative expression compared to Jurkat T cells transfected with a control siRNA (non-targeting) is shown. Error bars are SEM. Results are representative of two independent experiments. (B) Levels of DHHC2 in total protein lysates from Jurkat T cells treated with Accell siRNA against DHHC2 or DHHC4 or without siRNA oligos. DHHC2 was detected by Western blotting using the anti-DHHC2 antibody and detection of tubulin with anti-tubulin antibody was used as a loading control. (C) Total protein lysates from Jurkat T cells Ju or J.Cam1.6 cells treated for 72 h with siRNA oligos directed towards DHHC2 or control siRNA. The cells were labelled with 3H-palmitic acid to show S-acylation. Lck is one of the most abundant S-acylated proteins in Jurkat T cells and its position on the gel is indicated; absence of a corresponding band in Lck-negative J.Cam1.6 cells validates the assignment of the Lck band.
Membrane association and subcellular localization of DHHC proteins
In order for DHHC2 and 4 to be able to acylate Lck, they would have to be present at the location where Lck is acylated as a step in its post-translational modification. Newly synthesized Lck is thought to be acylated at membranes of the early compartments of the exocytic pathway before being transported to the plasma membrane (Bijlmakers and Marsh Citation1999). Fractionation of HEK293A cells transiently expressing DHHC2-, 4- and 16-CFP fusion proteins shows that these DHHC proteins are exclusively associated with the membrane fraction (), as expected for proteins with several membrane spanning regions (Mitchell et al. Citation2006). The P100 fraction contains all membranes and does not provide any information on which particular membrane compartment the DHHC proteins were associated with.
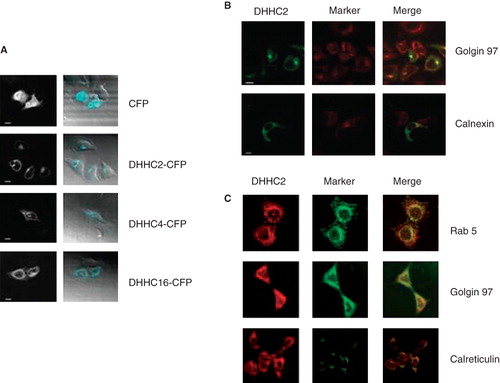
To further examine the localization of DHHC2 and DHHC4, confocal images were taken of HeLa cells transiently expressing DHHC2-, DHHC4- or DHHC16-CFP (). All three proteins could be found in membranous structures in the perinuclear area, unlike CFP alone which had a typical cytosolic and nuclear localization. DHHC2-CFP was found to partially co-localize with both the ER marker calnexin and the Golgi marker Golgin97 (), suggesting that DHHC2 is present in at least some areas of the ER and Golgi. Further strengthening the Golgi and ER localization of DHHC2, we found that the DHHC2 antibody detecting endogenous DHHC2 in HEK293A cells also showed partial co-localization with Golgi and ER markers, especially in the perinuclear area (). Significant overlap with Rab 5, an early endosome marker, was seen again in the perinuclear area where many endomembrane compartments localize, but was absent from the cell periphery where distinct Rab5-positive vesicles could be resolved (); we interpret this to mean that these DHHC proteins are not associated with early endosomes. No significant plasma membrane localization of DHHC2-, 4- or 16-CFP, or plasma membrane staining of endogenous DHHC2 with anti-DHHC2 antibody was seen, consistent with the requirement of Lck S-acylation before trafficking to the plasma membrane.
Figure 5. Subcellular localization of DHHC-CFPs and endogenous DHHC2. (A) Confocal microscopy images of HeLa cells transiently expressing the indicated DHHC-CFPs or CFP alone. Scale bars = 10 μm. (B) Confocal microscopy images of HeLa cells transiently expressing DHHC2-CFP with Golgi apparatus indicated by immunofluorescence using anti-Golgin 97 and ER using anti-calnexin antibodies. Scale bars = 10 μm. (C) Confocal microscopy images of HEK293A cells immunostained for DHHC2, Rab5 as a marker of early endosomes, Golgin 97 as a marker for the Golgi apparatus and calreticulin as a marker for the ER.
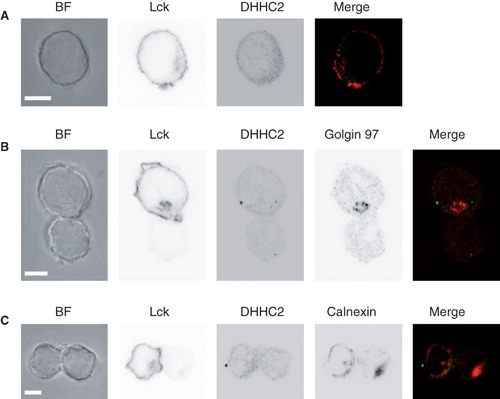
In resting Jurkat T cells, endogenous DHHC2 and Lck are found in close proximity to each other at the cell periphery, but completely non-overlapping (). DHHC2 is localized both to the cell periphery (but not plasma membrane) and in internal perinuclear structures. It also displays a punctate pattern with bright spots in some parts of the cells which do not co-localize with any marker tested. Lck is localized at the plasma membrane, close to DHHC2-positive compartments, but at least in most parts of the cells more peripheral than DHHC2. A portion of the Lck also appears to be found in the same perinuclear structures as DHHC2, but again no complete overlap of the signals was observed under these conditions (). In polarized Jurkat T cells forming immune synapses with Raji B cells loaded with SEE superantigen, endogenous DHHC2 was found in vesicular structures, which were in very close proximity to and frequently overlapping with the ER marker calnexin (). DHHC2-positive vesicles were seen in the Golgi region in 5 out of 23 analyzed cells, identified by anti-Golgin 97, but this was too infrequent to strongly support a localization to the Golgi in these polarized Jurkat T cells. Some polarization of DHHC2 towards the immunological synapse, similar to Lck, was observed. Again, a punctate pattern with bright spots was observed (). Taking all these results together, our interpretation is that these DHHC proteins all reside primarily in the ER with some localization to Golgi.
Figure 6. DHHC2 localization in resting and activated Jurkat T cells. (A) Brightfield (BF) and confocal microscopy images of DHHC2 and Lck detected by immunostaining in Jurkat T cells. Scale bars = 5 μm. (B) and (C) BF and confocal immunofluorescence images of Jurkat T cells (top in B, left in C; marked with anti-Lck) conjugated with SEE-loaded Raji B cells. Greyscale images are shown for clarity. In (B) the Golgi apparatus is visualized by staining with anti-Golgin 97 and in (C) the ER is visualized by staining with anti-calnexin. Merged images are also shown; green – DHHC2; red – Golgin 97 (B) or calnexin (C). Accumulation of Lck at the T-B cell interface confirms productive formation of the immunological synapse. Images are representative of n = 23 (B) and n = 24 (C) cell pairs. Scale bars = 5 μm.
Discussion and conclusion
A wide range of intracellular signalling proteins are S-acylated, affecting their localization and function. In humans there are 23 potential PATs of the DHHC type, thought to be responsible for the S-acylation of these signalling proteins. Substrates have been identified for some of DHHC PATs, but many are still not yet confirmed as active PATs. Several of the DHHC PATs that have been shown to mediate S-acylation have multiple and overlapping substrates. The S-acylation of Lck is a requirement for its proper membrane attachment and consequently for its function. Despite the fact that Lck depends on S-acylation for its function has been known for almost 15 years, the actual Lck PAT has not been identified. In this paper we show that DHHC2 is a PAT for Lck in vivo. The most common approach to identify DHHC/substrate pairs has been co-transfection (e.g. Fukata et al. Citation2006). However, this suffers from the problem that inappropriate enzyme/substrate combinations that do not occur in vivo may be observed due to promiscuity introduced by overexpression and the loss of the correct intracellular localization. This may partly account for the apparently low specificity of DHHC proteins. We have therefore combined co-expression with knockdown of DHHC proteins to provide a more physiological view.
The first step in finding the PATs for Lck was to identify the DHHC proteins expressed in cells where Lck S-acylation is present and relevant. Fortunately, hematopoietic cells have a limited expression of DHHC proteins, restricting the number of possible Lck PAT candidates. EST database searches suggested that DHHC2, 4, 15 and 16 are expressed in Jurkat T cells and RT-PCR experiments confirmed that DHHC2, 4 and 16 are expressed in both Jurkat and primary human T cells, validating Jurkat T cells as a model cell line for identification of an Lck PAT of the DHHC type. The expression of DHHC2, 4 and 16 in B cells suggests that there is a possibility that one or more of them could also be PATs for another Src family protein tyrosine kinase, Lyn, which is involved in B cell signalling. The sequence of the N-terminus of Lyn contains, like Lck, the cysteine that is the site for S-acylation in close proximity to a myristoylated glycine residue (Milligan et al. Citation1995).
In addition to being expressed in the relevant cell type to be potential Lck PATs, DHHC2 and 4 were also found to co-localize with the known compartments through which Lck passes during its transport to the cell surface, i.e. ER and Golgi complex (Bijlmakers et al. Citation1997, Bijlmakers and Marsh Citation1999). Not surprisingly, considering that DHHC proteins have at least four transmembrane domains (Mitchell et al. Citation2006), the expressed CFP-tagged DHHCs were associated with the particulate fraction after subcellular fractionation. Both endogenous and exogenous DHHC2 also partially co-purify with flotillin-1, a lipid raft marker found to associate with detergent-resistant membranes (DRMs; data not shown). This indicates that a small amount of DHHC2 is concentrated either in, or in close proximity to, areas enriched with cholesterol and other S-acylated proteins where active Lck is found (Shenoy-Scaria et al. Citation1993, Rodgers et al. Citation1994, Kabouridis et al. Citation1997, Bijlmakers Citation2009). DRMs are believed to represent a part of the plasma membrane derived from lipid rafts and thought to be areas of high signalling activity. As Lck undergoes cycles of acylation and deacylation (Paige et al. Citation1993, Zeidman et al. Citation2009), the presence of an Lck PAT in these domains could mean that even a small amount of DHHC2 is sufficient for acylation of Lck in lipid rafts, or that deacylated Lck quickly dissociates from rafts and is transported elsewhere in the cell for reacylation, or both. However, the majority of DHHC2 was not associated with DRMs; this would be consistent with DHHC2 being involved in the S-acylation of newly synthesized Lck rather than reacylation of Lck at the plasma membrane. Other studies in simple cultured cells such as fibroblasts support our localization of DHHC2 to the ER/Golgi complex (Fernandez-Hernando et al. Citation2006, Ohno et al. Citation2006). Several other recent reports have suggested that DHHC2 may also target to post-Golgi membranes and the plasma membrane in more highly differentiated and polarized cells such as neurons, and cycle dynamically between them (Noritake et al. Citation2009, Greaves et al. Citation2011, Jia et al. Citation2011, Levy et al. Citation2011). These differences in localization may be due to the highly divergent cell types employed. It is possible that DHHC2 contains localization signals that are silent in simple unpolarized cells as recently identified by Greaves et al. (Citation2011), resulting in ER/Golgi targeting, but are recognized by cell-specific factors in polarized differentiated cells allowing transport to specialized domains in the plasma membrane.
siRNA treatment against DHHC2 in Jurkat T cells clearly abrogated the S-acylation of Lck, evidenced by membrane dissociation and a reduction in incorporated 3H-palmitate into endogenous Lck. However, siRNA for DHHC16 had no effect on Lck membrane association, indicating specificity in the effects of DHHC2 siRNA. We were able to confirm effective siRNA knockdown of DHHC2 using antibodies that we prepared to this protein, but no similar reagents were available for DHHC 4 and 16 so we cannot at present confirm knockdown of these proteins. Conversely, increasing levels of DHHC2 by co-transfection of a CFP fusion lead to an increase in 3H-palmitate incorporation into an LckN10-GFP fusion protein, suggesting that DHHC2 can be an Lck PAT in vivo. Published in vitro assays using purified Lck fragments and DHHC2 support our findings that DHHC2 is an Lck PAT (Jennings et al. Citation2009). The observed effects on Lck delocalization of siRNA against DHHC4 initially suggested that DHHC4 may also be an Lck PAT. However, cotransfection of DHHC4-CFP with LckN10-GFP did not result in increased incorporation of 3H-palmitate into the latter, and also siRNA-mediated knockdown of DHHC4 (confirmed at the mRNA but not protein level) did not have a reproducible effect on Lck S-acylation. Thus, we cannot confirm a role for DHHC4 in Lck S-acylation at this time. Nevertheless, the membrane delocalization of Lck after DHHC4 siRNA treatment was reproducible and this could mean that DHHC4 is indeed a PAT for Lck or that the delocalization was an off-target effect unrelated to DHHC4 and possibly to Lck S-acylation. This will require further investigation. Whether any of these DHHC gene products are PATs for the other T cell Src family kinase Fyn is not yet clear.
The predominant localization of DHHC2 and 4 to intracellular membrane compartments suggests that they may be involved in S-acylation of newly synthesized Lck during its transport to the plasma membrane. However, they may also play a role in reacylation of mature Lck during an acylation-deacylation cycle, which is relatively slow (Zeidman et al. Citation2009) and could occur by internalization of deacylated Lck and reacylation on intracellular compartments, such as occurs with Ras proteins (Rocks et al. Citation2005). Acylation-deacylation cycles are known to occur for many proteins (Wolven et al. Citation1997, Yeh et al. Citation1999, El-Husseini et al. Citation2002), with impact on their function. This is an attractive prospect for Lck; DHHC 2 could play an important role in such an Lck acylation cycle, as it does for the RGS7 family-binding protein (Jia et al. Citation2011). The identity of the thioesterase(s) that catalyse(s) Lck deacylation remains to be determined.
Several other substrates of DHHC2 have been identified previously. These range from cytoskeleton-associated protein 4 (CKAP4) (Zhang et al. Citation2008), to the transmembrane proteins CD9 and CD151 of the tetraspanin family (Sharma et al. Citation2008), the neuronal adaptor/scaffold protein PSD95 (Fukata et al. Citation2004), the SNARE proteins SNAP-23/25 (Greaves et al. Citation2010) and the intracellular signalling proteins GAP43 (Fukata et al. Citation2004), Gαi2 (Tsutsumi et al. Citation2009), R7BP (Jia et al. Citation2011) and eNOS (Fernandez-Hernando et al. Citation2006). Interestingly, there is no apparent structural similarity between the reported substrates of DHHC2, or even any sequence similarities surrounding the residues being S-acylated. DHHC2 can thus apparently mediate S-acylation of cysteines located in the N-terminal region (PSD-95, GAP-43 and Gα), internally in the protein sequence (SNAP-23/25), in the juxtamembrane region of transmembrane proteins (CD9 and CD151) and close to an N-terminal myristoylated glycine (Lck and eNOS). This argues against the simple assumption that different DHHC family members might S-acylate subsets of target proteins on related motifs. This is consistent with recent work from Bastiaens’ group (Rocks et al. Citation2010) suggesting that substrate specificity is not essential for reversible S-acylation and that the cycle can be controlled by the spatial and temporal segregation of acylation (PATs on intracellular membranes, e.g. of the ER and Golgi) and deacylation (TEs in the cytoplasm). If this is the case, why there are 23 mammalian DHHC proteins remains to be explained. Firstly, specificity could be partially regulated by differential expression of DHHC proteins, as found in this study. It could also be that DHHC proteins, many of which are localized to the ER or Golgi at the organelle level, have subtly different sub-organellar organization that could contribute to their functional specificity; this would need to be addressed by much higher resolution microscopy techniques recently developed (Cebecauer et al. Citation2010). Alternatively, DHHC enzyme activities may be differentially regulated in cells.
Other DHHC PATs than DHHC2 have been suggested to S-acylate Lck. Co-expression of DHHC17 and DHHC18 with Lck in HEK293 cells increased 3H-palmitate incorporation into Lck, in contrast to co-expression of Lck with DHHC1, DHHC7 and DHHC15 (Fukata et al. Citation2004). Furthermore, there is a report, supported by unpublished data, that another DHHC PAT, DHHC21, can mediate S-acylation of both Lck and Fyn (Iwanaga et al. Citation2009), which could indicate that single DHHC PATs could S-acylate more than one Src kinase. However, our own analysis and EST data do not support the expression of DHHC21 in T cells so we feel that it is unlikely that DHHC21 can be a physiologically relevant PAT for Lck. These results may be due to the overexpression of exogenous proteins which can cause mis-localization or loss of specificity, as mentioned above.
In conclusion, reducing DHHC2 level in Jurkat T cells using siRNA causes decreased Lck S-acylation levels and its dislocation from membranes, and conversely overexpression of DHHC2 increases S-acylation of an Lck surrogate, LckN10-GFP. This suggests that DHHC2 is a bona fide PAT for Lck. However, so far we have been unable to unequivocally demonstrate that DHHC4 knockdown has been successful, due to the lack of antibodies to this protein, and also overexpression of DHHC4-CFP does not enhance LckN10-GFP S-acylation. We therefore have to be cautious in assigning DHHC4 as an Lck PAT.
Acknowledgements
We thank Dr E. Jury, University College London, for the kind gift of primary human B cells. Work in the Magee laboratory was supported by grants G0100471 and G0400710 from the UK Medical Research Council. D.M. Davis wishes to acknowledge an EMBO Young Investigator award. We thank the Facility for Imaging by Light Microscopy (FILM) for access to fluorescence microscopes and advice.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Bijlmakers M-J, Isobe-Nakamura M, Ruddock LJ, Marsh M. 1997. Intrinsic signals in the unique domain target p56lck to the plasma membrane independently of CD4. J Cell Biol 137:1029–1040.
- Bijlmakers M-J, Marsh M. 1999. Trafficking of an acylated cytosolic protein: Newly synthesized p56lck travels to the plasma membrane via the exocytic pathway. J Cell Biol 145:457–468.
- Bijlmakers MJ. 2009. Protein acylation and localization in T cell signaling. Mol Membr Biol 26:93–103.
- Cebecauer MC, Spitaler M, Sergé A, Magee AI. 2010. Signalling complexes and clusters: Functional advantages and methodological hurdles. J Cell Sci 123:309–320.
- Duncan JA, Gilman AG. 1998. A cytoplasmic acyl-protein thioesterase that removes palmitate from G protein α subunits and p21RAS. J Biol Chem 273:15830–15837.
- El-Husseini AE-D, Schnell E, Dakoji S, Sweeney N, Zhou Q, Prange O, 2002. Synaptic strength regulated by palmitate cycling on PSD-95. Cell 108:849–863.
- Fernandez-Hernando C, Fukata M, Bernatchez PN, Fukata Y, Lin MI, Bredt DS, 2006. Identification of Golgi-localized acyl transferases that palmitoylate and regulate endothelial nitric oxide synthase. J Cell Biol 174:369–377.
- Flaumenhaft R, Rozenvayn N, Feng D, Dvorak AM. 2007. SNAP-23 and syntaxin-2 localize to the extracellular surface of the platelet plasma membrane. Blood 110:1492–1501.
- Fukata M, Fukata Y, Adesnik H, Nicoll RA, Bredt DS. 2004. Identification of PSD-95 palmitoylating enzymes. Neuron 44:987–996.
- Fukata Y, Iwanaga T, Fukata M. 2006. Systematic screening for palmitoyl transferase activity of the DHHC protein family in mammalian cells. Methods 40:177–182.
- Greaves J, Gorleku OA, Salaun C, Chamberlain LH. 2010. Palmitoylation of the SNAP25 protein family: Specificity and regulation by DHHC palmitoyl transferases. J Biol Chem 285:24629–24638.
- Greaves J, Carmichael JA, Chamberlain LH. 2011. The palmitoyl transferase DHHC2 targets a dynamic membrane cycling pathway: Regulation by a C-terminal domain. Mol Biol Cell 22:1887–1895.
- Hawash IY, Hu XE, Adal A, Cassady JM, Geahlen RL, Harrison ML. 2002. The oxygen-substituted palmitic acid analogue, 13-oxypalmitic acid, inhibits Lck localization to lipid rafts and T cell signaling. Biochim Biophys Acta 1589:140–150.
- Huang K, Yanai A, Kang R, Arstikaitis P, Singaraja RR, Metzler M, 2004. Huntingtin-interacting protein HIP14 is a palmitoyl transferase involved in palmitoylation and trafficking of multiple neuronal proteins. Neuron 44:977–986.
- Iwanaga T, Tsutsumi R, Noritake J, Fukata Y, Fukata M. 2009. Dynamic protein palmitoylation in cellular signaling. Prog Lipid Res 48:117–127.
- Janes PW, Ley SC, Magee AI. 1999. Aggregation of lipid rafts accompanies signaling via the T cell antigen receptor. J Cell Biol 147:447–461.
- Janes PW, Ley SC, Magee AI, Kabouridis PS. 2000. The role of lipid rafts in T cell antigen receptor (TCR) signalling. Semin Immunol 12:23–34.
- Jennings BC, Nadolski MJ, Ling Y, Baker MB, Harrison ML, Deschenes RJ, 2009. 2-bromopalmitate and 2-(2-Hydroxy-5-nitro-benzylidene)-benzo[b]thiophen-3-one inhibit DHHC-mediated palmitoylation in vitro. J Lipid Res 50:233–242.
- Jia L, Linder ME, Blumer KJ. 2011. Gi/o signaling and the palmitoyl transferase DHHC2 regulate palmitate cycling and shuttling of RGS7 family-binding protein. J Biol Chem 286:13695–13703.
- Kabouridis PS. 2006. Lipid rafts in T cell receptor signalling (Review. Mol Membr Biol 23:49–57.
- Kabouridis PS, Magee AI, Ley SC. 1997. S-acylation of LCK protein tyrosine kinase is essential for its signalling function in T lymphocytes. EMBO J 16:4983–4998.
- Keller CA, Yuan X, Panzanelli P, Martin ML, Alldred M, Sassoe-Pognetto M, 2004. The γ2 subunit of GABA(A) receptors is a substrate for palmitoylation by GODZ. J Neurosci 24:5881–5891.
- Kleuss C, Krause E. 2003. Galpha(s) is palmitoylated at the N-terminal glycine. EMBO J 22:826–832.
- Koegl M, Zlatkine P, Ley SC, Courtneidge SA, Magee AI. 1994. Palmitoylation of multiple Src-family kinases at a homologous N-terminal motif. Biochem J 303:749–753.
- Levy AD, Devignot V, Fukata Y, Fukata M, Sobel A, Chauvin S. 2011. Subcellular Golgi localization of stathmin family proteins is promoted by a specific set of DHHC palmitoyl transferases. Mol Biol Cell 22:1930–1942.
- Lobo S, Greentree WK, Linder ME, Deschenes RJ. 2002. Identification of a Ras palmitoyltransferase in Saccharomyces cerevisiae. J Biol Chem 277:41268–41273.
- Lu JY, Verkruyse LA, Hofmann SL. 1996. Lipid thioesters derived from acylated proteins accumulate in infantile neuronal ceroid lipofuscinosis: correction of the defect in lymphoblasts by recombinant palmitoyl-protein thioesterase. Proc Natl Acad Sci USA 93:10046–10050.
- Magee AI, Gutierrez L, McKay IA, Marshall CJ, Hall A. 1987. Dynamic fatty acylation of p21N-ras. EMBO J 6:3353–3357.
- Magee AI, Wootton J, de Bony J. 1995. Optimised methods for detecting radiolabelled lipid-modified proteins in polyacrylamide gels. Methods Enzymol 250:330–336.
- Milligan G, Parenti M, Magee AI. 1995. The dynamic role of palmitoylation in signal transduction. Trends Biochem Sci 20:181–187.
- Mitchell DA, Vasudevan A, Linder ME, Deschenes RJ. 2006. Protein palmitoylation by a family of DHHC protein S-acyltransferases. J Lipid Res 47:1118–1127.
- Molina TJ, Kishihara K, Siderovskid DP, van Ewijk W, Narendran A, Timms E, 1992. Profound block in thymocyte development in mice lacking p56lck. Nature 357:161–164.
- Noritake J, Fukata Y, Iwanaga T, Hosomi N, Tsutsumi R, Matsuda N, 2009. Mobile DHHC palmitoylating enzyme mediates activity-sensitive synaptic targeting of PSD-95. J Cell Biol 186:147–160.
- Ohno Y, Kihara A, Sano T, Igarashi Y. 2006. Intracellular localization and tissue-specific distribution of human and yeast DHHC cysteine-rich domain-containing proteins. Biochim Biophys Acta 1761:474–483.
- Paige LA, Nadler MJ, Harrison ML, Cassady JM, Geahlen RL. 1993. Reversible palmitoylation of the protein-tyrosine kinase p56lck. J Biol Chem 268:8669–8674.
- Palacios EH, Weiss A. 2004. Function of the Src-family kinases, Lck and Fyn, in T-cell development and activation. Oncogene 23:7990–8000.
- Pepinsky RB, Zeng C, Wen D, Rayhorn P, Baker DP, Williams KP, 1998. Identification of a palmitic acid-modified form of human Sonic hedgehog. J Biol Chem 273:14037–14045.
- Resh MD. 2006a. Palmitoylation of ligands, receptors, and intracellular signaling molecules. Sci STKE 2006:1–12.
- Resh MD. 2006b. Trafficking and signaling by fatty-acylated and prenylated proteins. Nat Chem Biol 2:584–590.
- Rocks O, Peyker A, Kahms M, Verveer PJ, Koerner C, Lumbierres M, 2005. An acylation cycle regulates localization and activity of palmitoylated Ras isoforms. Science 307:1746–1752.
- Rocks O, Gerauer M, Vartak N, Koch S, Huang Z-P, Pechlivanis M, 2010. Cell. 141:458–471.
- Rodgers W, Crise B, Rose JK. 1994. Signals determining protein tyrosine kinase and glycosyl-phosphatidylinositol-anchored protein targeting to a glycolipid-enriched membrane fraction. Mol Cell Biol 14:5384–5391.
- Roth AF, Feng Y, Chen L, Davis NG. 2002. The yeast DHHC cysteine-rich domain protein Akr1p is a palmitoyl transferase. J Cell Biol 159:23–28.
- Scapini P, Pereira S, Zhang H, Lowell CA. 2009. Multiple roles of Lyn kinase in myeloid cell signaling and function. Immunol Rev 228:23–40.
- Sharma C, Yang XH, Hemler ME. 2008. DHHC2 affects palmitoylation, stability, and functions of tetraspanins CD9 and CD151. Mol Biol Cell 19:3415–3425.
- Shenoy-Scaria AM, Gauen LK, Kwong J, Shaw AS, Lublin DM. 1993. Palmitylation of an amino-terminal cysteine motif of protein tyrosine kinases p56lck and p59fyn mediates interaction with glycosyl-phosphatidylinositol-anchored proteins. Mol Cell Biol 13:6385–6392.
- Smotrys JE, Linder ME. 2004. Palmitoylation of intracellular signaling proteins: Regulation and function. Annu Rev Biochem 73:559–587.
- Smotrys JE, Schoenfish MJ, Stutz MA, Linder ME. 2005. The vacuolar DHHC-CRD protein Pfa3p is a protein acyltransferase for Vac8p. J Cell Biol 170:1091–1099.
- Straus DB, Weiss A. 1992. Genetic evidence for the involvement of the lck tyrosine kinase in signal transduction through the T cell antigen receptor. Cell 70:585–593.
- Swarthout JT, Lobo S, Farh L, Croke MR, Greentree WK, Deschenes RJ, 2005. DHHC9 and GCP16 constitute a human protein fatty acyltransferase with specificity for H- and N-Ras. J Biol Chem 280:31141–31148.
- Toyoda T, Sugimoto H, Yamashita S. 1999. Sequence, expression in Escherichia coli, and characterization of lysophospholipase II. Biochim Biophys Acta 1437:182–193.
- Tsutsumi R, Fukata Y, Fukata M. 2008. Discovery of protein-palmitoylating enzymes. Pflugers Arch 456:1199–206.
- Tsutsumi R, Fukata Y, Noritake J, Iwanaga T, Perez F, Fukata M. 2009. Identification of G protein {alpha} subunit-palmitoylating enzyme. Mol Cell Biol 29:435–447.
- van Oers NS, Killeen N, Weiss A. 1996. Lck regulates the tyrosine phosphorylation of the T cell receptor subunits and ZAP-70 in murine thymocytes. J Exp Med 183:1053–1062.
- Verkruyse LA, Hofmann SL. 1996. Lysosomal targeting of palmitoyl-protein thioesterase. J Biol Chem 271:15831–15836.
- Vesa J, Hellsten E, Verkruyse LA, Camp LA, Rapola J, Santavuori P, 1995. Mutations in the palmitoyl protein thioesterase gene causing infantile neuronal ceroid lipofuscinosis. Nature 376:584–587.
- Wolven A, Okamura H, Rosenblatt Y, Resh MD. 1997. Palmitoylation of p59fyn is reversible and sufficient for plasma membrane association. Mol Biol Cell 8:1159–1173.
- Yamasaki S, Tachibana M, Nobukata S, Iwashima M. 1997. Lck-independent triggering of T-cell antigen receptor signal transduction by staphylococcal enterotoxins. J Biol Chem 272:14787–14791.
- Yanai A, Huang K, Kang R, Singaraja RR, Arstikaitis P, Gan L, 2006. Palmitoylation of huntingtin by HIP14 is essential for its trafficking and function. Nat Neurosci 9:824–831.
- Yeh DC, Duncan JA, Yamashita S, Michel T. 1999. Depalmitoylation of endothelial nitric-oxide synthase by acyl-protein thioesterase 1 is potentiated by Ca(2+)-calmodulin. J Biol Chem 274:33148–33154.
- Yurchak L, Sefton B. 1995. Palmitoylation of either Cys-3 or Cys-5 is required for the biological activity of the Lck tyrosine protein kinase. Mol Cell Biol 15:6914–6922.
- Zeidman R, Jackson CS, Magee AI. 2009. Protein acyl thioesterases. Mol Membr Biol 26:32–41.
- Zhang J, Planey SL, Ceballos C, Stevens SM Jr, Keay SK, Zacharias DA. 2008. Identification of CKAP4/p63 as a major substrate of the palmitoyl acyltransferase DHHC2, a putative tumor suppressor, using a novel proteomics method. Mol Cell Proteomics 7:1378–1388.
- Zlatkine P, Mehul B, Magee A. 1997. Retargeting of cytosolic proteins to the plasma membrane by the Lck protein tyrosine kinase dual acylation motif. J Cell Sci 110:673–679.