Abstract
Orthologous proteins do not necessarily share the same function in all species and those sharing the same function might employ a modified catalytic mechanism. Thus, comparative analysis of homologous or orthologous proteins from different organisms can provide detailed information on the function and the mechanism of an entire protein family. The sensor kinase ETR1 from Arabidopsis thaliana has been well characterized by genetic, physiological and biochemical studies. However, as further model plants are coming into focus for plant hormone research, a general protocol for isolation and purification of orthologous ETR1 proteins seems instrumental for a detailed molecular analysis of this protein family. In this study, we describe the native purification of recombinant ETR1 from Arabidopsis thaliana by mild solubilization with the zwitter-ionic detergent Fos-Choline-14 and single-step purification by immobilized metal ion affinity chromatography. The same protocol was successfully applied for the purification of the orthologous proteins from the moss Physcomitrella patens subsp. patens and the tomato Lycopersicon esculentum. The successful transfer of the purification protocol to proteins of the same family which share sequence identity of 63–80% only suggests that this protocol presents a general purification strategy which is likely to apply also to the purification of other members of the sensor histidine kinase family.
Introduction
Membrane proteins play critical roles in metabolic, regulatory and intercellular processes such as transport, cell adhesion or signal transduction (Calderwood et al. Citation2000, White Citation2009). Protein kinases are a large and functionally diverse family in the group of membrane proteins and are currently one of the major drug targets. The sensor kinase ETR1 of Arabidopsis thaliana belongs to a family of membrane bound hybrid-type histidine kinases which also occur in pathogenic organisms like Yersinia, Salmonella, or Candida. In plants, ETR1 and the four isoforms ERS1, ETR2, ERS2 and EIN4 control perception and signaling of the plant hormone ethylene which mediates numerous physiological and developmental processes in plants including senescence and abscission of petals, leafs and fruits (Bleecker and Kende Citation2000, Chen et al. Citation2005). Furthermore, ethylene signaling is involved in the response to various biotic and abiotic stresses like pathogen infection or draught (Johnson and Ecker Citation1998).
ETR1 is considered the prototype of the ethylene receptor family in Arabidopsis and has been characterized in most detail in the past. Sequence analysis suggests that the receptor is composed of an amino-terminal transmembrane sensor domain, a catalytic transmitter domain and a carboxyl-terminal response regulator domain (Johnson and Ecker Citation1998, Bleecker and Kende Citation2000). According to signaling in typical two-component systems (Hwang et al. Citation2002) signal perception by the amino-terminal domain of the ethylene receptor is supposed to control autophosphorylation of a conserved histidine residue in the catalytic transmitter domain of the receptor. Then the phosphoryl group is thought to be transferred to a conserved aspartate in the carboxyl-terminal receiver domain of the receptor. In order to trigger cellular signal response the phosphoryl group has to be further transferred from the receptor onto nuclear response regulator proteins via histidine phosphotransfer proteins forming a multi-step his-asp phosphotransfer cascade (phosphorelay). Still, the molecular mechanism by which binding of the plant hormone triggers the downstream events is not recognized yet, even though it seems evident that conformational changes in the receptor protein promote these processes (Bleecker et al. Citation1998).
Plants are sessile organisms that have to adapt to different environmental conditions such as water, light and nutrient supply. They inhabit an enormous range of ecological habitats. As a consequence of this adaptation plants that originate from different ecological niches show differences in gene expression and also in protein regulation. Hence, analysis of proteins from different species can expose regulatory mechanism or even the basic molecular mechanism of a certain protein family. In this context analysis of ethylene receptor proteins from different species is expected to reveal the common molecular processes and conformational changes that are connected to ethylene binding and signal transduction. Receptors from different plant species might also show different sensitivity towards the plant hormone reflecting their adaptation to different environments. Comparison of ethylene sensitivity and binding capacity of different receptors to their sequence will also help to pinpoint the binding site of the plant hormone in the transmembrane amino-terminal sensor domain.
To address these issues production of recombinant receptor proteins will be necessary for detailed in vitro studies. In order to avoid a time-consuming development of individual purification protocols for ETR1 receptor orthologs, we have set up a general protocol that was first used to purify ETR1 from A. thaliana. Later on the same protocol was successfully applied to obtain ETR1 orthologs from the moss Physcomitrella patens subsp. patens (Cove et al. Citation2009) and the tomato Lycopersicon esculentum (Kimura and Sinha Citation2008) at high purity and homogeneity.
Materials and methods
Reagents, detergents and materials
All chemicals were obtained at analytical grade. Pwo DNA polymerase peqGOLD from peqlab (Erlangen, Germany) and dNPTs from New England Biolabs (NEB, Frankfurt, Germany) were used in the PCR reactions. T4-DNA polymerase and restriction enzymes were purchased from NEB and used in their appropriate buffers. Primers were synthesized at Sigma-Aldrich (Munich, Germany). DNA fragments were extracted from agarose gels using the illustra GFX PCR DNA and gel band purification kit from GE Healthcare (Munich, Germany). Plasmid isolation was performed with the QIAprep spin miniprep kit from Qiagen (Hilden, Germany). Detergents used in solubilization studies were purchased from Anatrace (Maumee, OH, USA), except for n-dodecyl-β-d-maltoside which was obtained from Glycon (Luckenwalde, Germany). Radiolabelled γ32P-ATP with a specific activity of 3000 Ci/mmol was ordered from Hartman Analytic (Braunschweig, Germany). Horseradish peroxidase conjugated anti his antibodies were purchased from Miltenyi Biotec (Bergisch Gladbach, Germany). A primary rabbit antibody against ETR1 from A. thaliana was kindly provided by A.B. Bleecker (University of Wisconsin, USA). Rabbit IgG HRP linked secondary antibodies were obtained from GE Healthcare. Immobilon western chemiluminescent HRP substrate and centrifugal filter units (Amicon Ultra, MWCO 30 kDa) were purchased from Millipore (Schwalbach, Germany). Sequi-Blot polyvinylidene fluoride (PVDF) membranes from Bio-Rad Laboratories (Munich, Germany) were used for immobilisation of proteins. Ni-NTA agarose was obtained from Qiagen, PD-10 desalting columns were from GE Healthcare. Protein quantification was carried out with Pierce's BCA protein assay kit according to the manufacturer's instruction.
Bacterial strains
For cloning and plasmid amplification Escherichia coli strain XL1-Blue from Stratagene was used (La Jolla, CA, USA). Protein expression studies were performed with BL21 (DE3) from Stratagene and derivate strains C41 (DE3) and C43 (DE3) (Miroux and Walker Citation1996).
Cloning of expression vectors
DNA fragments encoding ethylene receptor ETR1 from Physcomitrella patens (subsp. patens) and ETR1 from Lycopersicon esculentum were synthesized at GenScript (Piscataway, NJ, USA). Synthetic DNA was codon-optimized for expression in E. coli and supplied in vector pUC57. For expression studies synthetic DNA fragments were cloned into expression vector pTEV-16b, a vector based on vector pET-16b from Novagen (Darmstadt, Germany). In the modified vector, a TEV protease cleavage site was substituted for the endogenous factor Xa site in order to allow removal of the N-terminal deca His-tag after affinity purification. The modified vector was constructed from pET-16b by digesting the vector with restriction enzymes NcoI and BamHI resulting in the excision of the coding sequences for His-tag, factor Xa recognition site and multiple cloning site. A double-stranded fragment containing a deca His-tag followed by a TEV recognition sequence (ENLYFQG) and sites for NdeI and XhoI was inserted at this position by sequence and ligation independent cloning (SLIC) (Li and Elledge Citation2007). The double-stranded insert was generated from the complementary nucleotides TEV-for (5′-ATTTTGTTTAACTTTAAGAAGGAGATATACCATGGGCCATCATCATCATCATCATCATCATCATCACAGCAGCGGCCATG-3′) and TEV-rev (5′-TTCCTTTCGGGCTTTGTTAGCAGCCGGATCCTCGAGCATATGTCCCTGAAAATACAGGTTTTCATGGCCGCTGCTGTGAT-3′). For cloning of the synthetic etr1 genes the following SLIC primer sets were used to amplify the coding sequence for ETR1 receptor from P. patens (Pp-for 5′-CATGAAAACCTGTATTTTCAGGGACATATGGATTCTTGCAATTGTGTGGA-3′ and Pp-rev 5′-TTTGTTAGCAGCCGGATCCTCGAGCATATGTTAGGTTTTGCGACGGGTTT-3′) and the receptor from L. esculentum (Le-for 5′-CATGAAAACCTGTATTTTCAGGGACATATGGGCAGTCTGCTGCGTATGAA-3′ and Le-rev 5′-TTTGTTAGCAGCCGGATCCTCGAGCATATGTTAACTTTCCAGAACCACGC-3′). The etr1 synthetic genes were amplified from pUC57 by standard PCR (Sambrook and Russell Citation2006). Vector pTEV-16b was linearized with restriction enzyme NdeI. The vector and the amplified gene sequences of ETR1 were gel purified and assembled by homologous recombination (Li and Elledge Citation2007).
By standard, XL1-Blue competent cells were transformed with 100–150 ng DNA and grown on selective 2YT agar plates supplied with 100 μg/ml ampicillin (Sambrook and Russell Citation2006). Colonies were selected for successful integration of the insert by colony PCR. DNA of positive clones was amplified and isolated with Qiagen's QIAprep spin miniprep kit. The correct nucleotide sequence was confirmed by sequencing at StarSEQ (Mainz, Germany).
Expression of ETR1 in E. coli
For liquid cultures 2YT medium supplied with 100 μg/ml ampicillin (2YT/ampicillin) was used. Agar plates were prepared with the same medium containing 1.5% (w/v) agar. Overnight liquid cultures were grown in non baffled flasks, main expression cultures were grown in baffled flasks. Receptor protein ETR1 from Arabidopsis thaliana was expressed in C43 (DE3) cells as described previously (Voet-van-Vormizeele and Groth Citation2008).
Expression vectors containing gene sequences for ETR1 from P. patens and L. esculentum were transformed into E. coli strains BL21 (DE3), C41 (DE3) and C43 (DE3). Colonies were selected on 2YT/ampicillin agar plates. For screening experiments, a single colony was used to inoculate 5 ml of 2YT/ampicilin liquid medium. The culture was grown by shaking at 37°C and 180 rpm over night. At the next day, 100 ml 2YT/ampicilin medium in baffled 250 ml flasks was inoculated with the overnight grown culture to an OD600 of 0.05. The cells were incubated at 30°C. At OD600 0.5–0.8 protein expression was induced by addition of isopropyl-β-d-1-thiogalactopyranoside (IPTG) to a final concentration of 1 mM. For test expressions at 16°C, the incubation temperature was reduced 30 min prior to induction. Deduced from the optical density of the cell culture, expression samples with similar amounts of cells were analyzed by SDS-PAGE and immunoblotting. At preparative scale, the volume of over night and main cultures was increased to 500 ml in 1 l flasks. Main cultures were grown by shaking at 30°C up to OD600 0.4. The incubation temperature was then reduced to 16°C and protein expression was induced after 30 min by addition of 0.1–1 mM IPTG. Cells were harvested 24 h after induction by centrifugation at 10,000 g and 4°C for 6 min. Collected cells were stored at −80°C.
Solubilization of ETR1 from bacterial membranes
Cells were thawed on ice, resuspended at 0.4 g/ml in 50 mM Tris/HCl pH 8.0, 200 mM sodium chloride and 0.002% PMSF (w/v). DTT at 10 mM and 10% glycerol (w/v) were added for cells of strain BL21, while 2 mM DTT and 15% glycerol (w/v) were used for strains C41 (DE3) and C43 (DE3). Resuspended cells were passed through a pre-cooled French pressure cell at 1,500 psi. The resulting homogenate of C41 and C43 cells was centrifuged for 20 min at 40,000 g to obtain the membrane fraction. Membranes from BL21 cells were pelleted at a higher centrifugation force of 230,000 g. For testing the solubilization efficiency of 23 different detergents, membrane pellets were resuspended in 10 ml resuspension buffer. Aliquots of 400 μl were supplied with detergents at concentrations of 1% (w/v) for Anapoe-20, Anapoe-35, Anapoe-58, Anapoe-C10E6, Anapoe-X-114, Anapoe-X-405, Anzergent 3-12, Anzergent 3-14, Cyclofos-5, Cyclofos-6, Cyclofos-7, Fos-Choline-11, Fos-Choline-12, Fos-Choline-13, Fos-Choline-14 or n-dodecyl-β-d-maltopyranoside and with 2% (w/v) for detergents Anzergent-3-10, C-Dodecafos, Cyclofos-4, Fos-Choline-9, Fos-Choline-10, n-octyl-β-d-glucopyranoside or n-octyl-β-d-thiomaltopyranoside, respectively (Ren et al. Citation2009). The samples were incubated for 2 h at 20°C. Non-solubilized material was removed by centrifugation at 150,000 g and 4°C for 30 min. The supernatant was collected and the pellet was resuspended in 400 μl resuspension buffer. Aliquots (10 μl) of each fraction were analyzed by dot blot and immunodetection. Large-scale solubilization of ETR1 was obtained in a maximum volume of 10 ml of a buffer containing 50 mM sodium phosphate pH 8.0, 100 mM sodium chloride, 0.002% PMSF and 1% (w/v) Fos-Choline-14. Solubilization of the receptor proteins was supported by gentle stirring for 2 h at 20°C or by sonication on ice for 20 min with 50% duty cycle. After centrifugation at 230,000 g and 4°C for 30 min solubilized proteins were collected from the supernatant.
Purification of ETR1 by immobilized metal affinity chromatography
Gravity flow purification of His-tagged ETR1 proteins was obtained at room temperature using 4 ml Ni-NTA that was equilibrated with 10 column volumes (CV) of 50 mM sodium phosphate pH 8.0, 2 M sodium chloride, 0.002% (w/v) PMSF and 0.015% (w/v) Fos-Choline-14. The supernatant from solubilization was supplied with 2 M sodium chloride and mixed with the resin. The suspension was slightly agitated for 15 min and then filled into an empty column (inner diameter: 1.5 cm). The resin was washed with 20 CV of the sodium phosphate buffer followed by 20 CV of the same buffer supplied with 50 mM imidazole. Bound ETR1 receptor was eluted with 5 CV of 50 mM sodium phosphate, 150 mM sodium chloride, 250 mM imidazole, 0.002% (w/v) PMSF and 0.015% (w/v) Fos-Choline-14.
SDS-PAGE and immunoblotting
Samples from expression studies, solubilization and purification were analyzed by discontinuous SDS-PAGE under reducing conditions (Laemmli Citation1970). Samples from expression cultures (500 μl) were centrifuged for 5 min at 14,000 g. The resulting pellet was resuspended in 150 μl water and 50 μl loading buffer (30 mM Tris, 30 mM boric acid, 0.7 mM EDTA, 50 mM DTT, 5 mM magnesium chloride, 6.7% (w/v) SDS, 16.7% (w/v) sucrose and 0.16% (w/v) bromophenol blue. Samples were incubated at 95°C for 10 min for cell disruption.
Proteins separated by electrophoresis on 10% (w/v) acrylamide gels were either silver stained (Heukeshoven and Dernick Citation1988) or transferred to a PVDF membrane by semi-dry Western blotting (Towbin et al. Citation1979). Samples from solubilization studies were mixed with loading buffer and pipetted onto the membrane without previous separation by SDS-PAGE (dot blot). Transfer membranes were treated with either hexa histidine antibodies conjugated to horseradish peroxidase or with a rabbit antibody directed against an epitope of ETR1 from A. thaliana (amino acids 401-738) (Schaller et al. Citation1995). This primary antibody was used together with a peroxidase linked secondary antibody to visualize the receptor proteins by incubation with Immobilon western chemiluminescent HRP substrate according to the manufacturer's protocol.
Autophosphorylation of ETR1
Autokinase activity of ETR1 was tested by a radioactive assay in a buffer containing 50 mM Tris/HCl pH 7.5, 100 mM potassium chloride, 0.1% (w/v) n-dodecyl-β-d-maltopyranoside (DDM) (Voet-van-Vormizeele and Groth Citation2008). Detergent exchange to DDM was achieved during affinity purification on Ni-NTA by replacing Fos-Choline-14 in washing and elution buffers with 0.03% (w/v) DDM. Purified protein was concentrated to 2.5 ml by ultra filtration and buffer was changed to the assay buffer. The solution containing the purified receptor was centrifuged for 30 min at 150,000 g and 4°C. Protein was adjusted to 0.5 mg/ml in a reaction volume of 50 μl and incubated with 1 mM manganese chloride for 10 min at room temperature. The phosphorylation reaction was initiated by adding 0.2 mM ATP containing γ32P-ATP to result in a concentration of the radioactive label of 0.4 μCi/μg protein. The reaction was terminated after 30 min by adding DTT-free loading buffer and samples were stored at −20°C for 24 h. DTT was added to a final concentration of 60 mM prior to SDS-PAGE. After electrophoresis proteins were transferred to a PVDF membrane by Western blotting and dried membranes were exposed to a phosphor imaging plate for 4–7 days to visualize incorporation of 32P.
CD spectroscopy
Far-UV CD spectra were recorded at protein concentrations of up to 0.3 mg/ml using Suprasil cuvettes from Hellma (Müllheim, Germany) and a Jasco-715 spectropolarimeter (Groß-Umstadt, Germany). Measurements with natively purified proteins were recorded at room temperature in 50 mM potassium phosphate pH 7.5 and 0.05% (w/v) DDM. Measurements were also performed with AtETR1 incubated in 50 mM potassium phosphate pH 7.5 supplied with 4 M guanidinium hydrochloride over night at 4°C. For each sample 10 spectra were recorded in the range from 260 – 200 nm. Bandwidth of 2 nm, step resolution of 1 nm and a scan speed of 50 nm/min were used in these measurements. Secondary structure of the purified ETR1 protein was deduced from the spectra by the program CDPro (Sreerama and Woody Citation2000).
Results and discussion
A purification protocol was established for the sensor kinase ETR1 from the model plant Arabidopsis thaliana and was used as a blueprint for the purification of two other plant hybrid type histidine kinases. Based on their different degree of sequence homology ethylene receptors from the moss Physcomitrella patens subsp. patens and from tomato Lycopersicon esculentum were selected to prove the general applicability of this protocol. BLAST analysis showed sequence identity of 80% and similarity of 90% for AtETR1 and LeETR1 (Altschul et al. Citation1997). For PpETR1 substantial lower similarity to AtETR1 was found (63% identity and 77% similarity). Sequence alignment of the three orthologous receptors () reveals high conservation in the amino-terminal domain which contains the transmembrane helices that are involved in ethylene binding via the copper cofactor bound to C65 and H69 (Rodríguez et al. Citation1999). Putative phosphorylation sites suggested for AtETR1 (amino acids H353 and D659 (Gamble et al. Citation1998)) are also present in the orthologous receptor LeETR1, while the conserved histidine in the kinase domain of PpETR1 is replaced by tyrosine. Hence, different molecular signaling mechanisms might be realized in early divergent lineages. Analysis of the different receptors might provide important information on the evolution of ethylene sensor kinases.
Figure 1. Alignment of primary sequences of orthologous ETR1 proteins from A. thaliana, P. patens subsp. patens and L. esculentum. Conserved amino acids in all three sequences are shaded in black, while a grey background indicates similar residues. The bold arrows indicate catalytic residues H353 and D659 in AtETR1, the dashed arrows show amino acids C65 and H69 that are involved in binding of a copper cofactor necessary for ethylene binding.

Expression of AtETR1, PpETR1 and LeETR1
To verify whether receptors PpETR1 and LeETR1 can be expressed at similar conditions as described for AtETR1 (Voet-van-Vormizeele and Groth Citation2008), coding sequences for both orthologs were cloned into expression vector pTEV-16b by sequence and ligation independent cloning (Li and Elledge Citation2007). In contrast to AtETR1 which was isolated from an Arabidopsis cDNA library (Schaller et al. Citation1995), synthetic gene sequences were obtained for the orthologs that were codon-optimized for E. coli expression. Expression vectors encoding PpETR1 or LeETR1 were transformed into cells of E. coli strains BL21 (DE3), C41 (DE3) and C43 (DE3). Strain C43 (DE3) has been successfully applied for expression of AtETR1 (Voet-van-Vormizeele and Groth Citation2008). Together with C41 (DE3) this strain has been used for the expression of several membrane proteins in the past (Miroux and Walker Citation1996). Analytical scale cell cultures were grown at 30°C and 16°C and protein expression was induced by the addition of IPTG to a final concentration of 1 mM. Samples from the cultures were analyzed for expression of PpETR1 and LeETR1 by SDS-PAGE and immunoblotting using an antibody directed against the deca histidine affinity tag of the receptor proteins.
In cell cultures of strain BL21 (DE3) grown at 16°C increasing expression of proteins with molecular weights of about 90 kDa (PpETR1 culture) and 75 kDa (LeETR1 culture) was observed over a period of 24 h (). Both receptors were identified by immunoblotting and expression bands were assigned to PpETR1 (calculated molecular weight of 89.1 kDa including the affinity tag) and LeETR1 (87.1 kDa including the tag) (). For LeETR1 the shift in calculated and apparent molecular weight observed in SDS-PAGE might be related to the fact that the receptor is embedded into SDS micelles rather than completely unfolded. Such a difference in analytical and apparent molecular weight is frequently observed when membrane proteins are analyzed by SDS-PAGE (Rath et al. Citation2009).
Figure 2. Effect of temperature on the expression of PpETR1 and LeETR1 in BL21 (DE3) at 30°C and 16°C. All cell cultures contained 1 mM IPTG. Equal amounts of cells from liquid cultures were analyzed by SDS-PAGE. Proteins were silver-stained (A) and receptor proteins were identified by Western blot analysis using antibodies directed against the deca His-tag (B). Protein bands representing the receptor proteins are framed by black boxes. Note that at 16°C expression temperature the protein band of receptor PpETR1 is overlayed by an endogenous protein of the bacterial host.
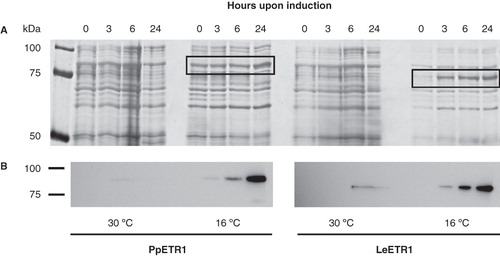
Faint expression bands for both receptors were observed when cells were grown at 30°C. However, the expression level was very low and protein was only detectable by immunostaining. No expression of both orthologs was found in strains C41 (DE3) and C43 (DE3) (data not shown) even though strain C43 (DE3) was successfully applied to produce recombinant AtETR1 (Voet-van-Vormizeele and Groth Citation2008). These results emphasize that expression protocols have to be adapted for orthologous proteins. However, this was achieved with minor efforts by a set of three different strains and by testing only two growth temperatures and a single inductor concentration (1 mM IPTG).
The inductor IPTG can act as competitive inhibitor of β-galactosidase and can interfere with the meta-bolism of the bacterial host (Kosinski et al. Citation1992). To facilitate protein synthesis and bacterial growth, IPTG was applied at a reduced concentration of 0.1 mM in preparative scale expressions. For LeETR1 the amount of cells per litre of medium was only slightly raised from 3 – 4 g after reducing the inductor concentration. In contrast, PpETR1 was not expressed at 0.1 mM IPTG (data not shown). In consequence the initial concentration of 1 mM was chosen for PpETR1 cultures and a cell mass of 6 g per litre medium was obtained.
Solubilization of recombinant ETR1 receptors from E. coli membranes
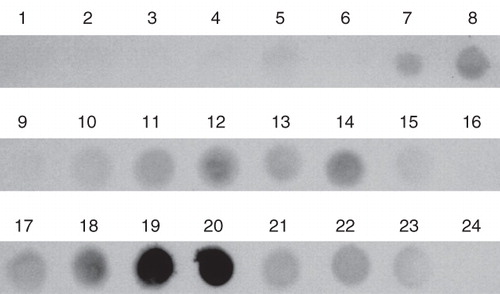
In analytical studies to identify a suitable detergent for mild but efficient solubilization of the ETR1 receptor, membrane pellets containing AtETR1 were treated with a set of 23 non-ionic and zwitter-ionic agents that have been successfully applied for solubilization of membrane proteins in the past (Ren et al. Citation2009). Depending on their critical micelle concentration (cmc) detergents were used in the solubilization process either at 2% w/v (for detergents with cmc >0.3%) or at 1% (w/v) for detergents whose cmc is smaller than 0.3% (w/v). Non-solubilized material was removed by centrifugation and solubilized membrane proteins were collected from the supernatant. The supernatants were analyzed by dot blot () using an antibody directed against the affinity tag of the receptors in order to identify the detergent which shows the highest efficiency in solubilization. Two zwitter-ionic detergents, Fos-Choline-13 and Fos-Choline-14 (No. 19 and 20 in ) turned out to be most efficient in solubilization of AtETR1 from the E. coli membranes in these experiments.
Figure 3. Solubilization efficiency of various ionic and non-ionic detergents used for extraction of AtETR1 from E. coli membranes. Supernatant fractions after centrifugation were analyzed by dot blot. Solubilized receptor proteins were monitored by antibodies directed against the hexa histidine tag. The following detergents were applied in the solubilization study: (1) Anapoe-20, (2) Anapoe-35, (3) Anapoe-58, (4) Anapoe-C10E6, (5) Anapoe-X-114, (6) Anapoe-X-405, (7) Anzergent 3-10, (8) Anzergent 3-12, (9) Anzergent 3-14, (10) C-Dodecafos, (11) Cyclofos-4, (12) Cyclofos-5, (13) Cyclofos-6, (14) Cyclofos-7, (15) Fos-Choline-9, (16) Fos-Choline-10, (17) Fos-Choline-11, (18) Fos-Choline-12, (19) Fos-Choline-13, (20) Fos-Choline-14, (21) n-octyl-β-d-glucopyranoside, (22) n-dodecyl-β-d-maltopyranoside, (23) n-octyl-β-d-thiomaltopyranoside, (24) no detergent.
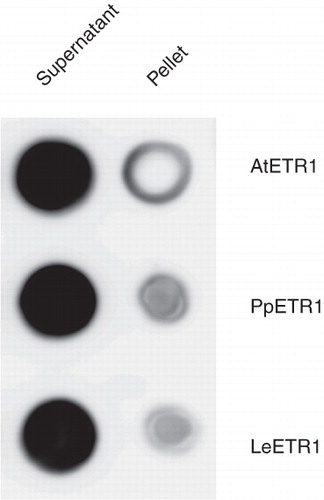
Fos-Choline-14 was chosen for preparative scale solubilization because of its lower cmc (0.0046% compared to 0.027% for Fos-Choline-13, all w/v). The solubilization protocol established for AtETR1 was subsequently tested for the orthologous receptors PpETR1 and LeETR1. shows that both orthologs were solubilized with a similar efficiency as AtETR1. This result underlines the general applicability of the solubilization protocol for other hybrid type sensor kinases.
Figure 4. Solubilization efficiency of Fos-Choline-14 on recombinant AtETR1, PpETR1 and LeETR1. Supernatant and pellet fractions after solubilization and centrifugation were analyzed by dot blot. The receptor proteins were identified by immunostaining with an antibody directed against the deca His-tag.
Purification of solubilized ETR1 receptor proteins
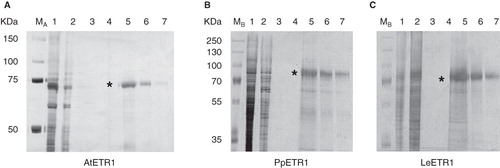
A purification protocol was developed for AtETR1 and then tested on the two orthologs PpETR1 and LeETR1. Solubilized AtETR1 was subjected to immobilized metal affinity chromatography using Ni-NTA resin. Binding of the recombinant protein to the affinity matrix was achieved only when 2 M sodium chloride was added to the washing buffers which increases the ion strength of the medium and promotes hydrophobic interactions (Li et al. Citation2001). Highly pure AtETR1 was obtained after two washing steps with buffers containing 0 mM and 50 mM imidazole, respectively. The receptor protein was eluted from the affinity column at 250 mM imidazole (). A single band on SDS-PAGE confirmed purity and homogeneity of the recombinant ETR1. Again, a gel shift of the receptor protein was observed in SDS-PAGE. The protein band was detected at about 70 kDa although the theoretical molecular weight of the receptor is 85 kDa. Solubilized PpETR1 and LeETR1 were purified at the same conditions to test the general applicability of the purification protocol. SDS-PAGE analysis of samples from both preparations revealed that highly pure protein was also obtained for the orthologous receptors (). However, protein yield differed substantially among the receptors. For AtETR1 about 0.5 mg pure protein was obtained per gram cells. On average 0.25 mg PpETR1, but only about 0.1 mg LeETR1 were isolated per gram cells. This difference might be related to the different E. coli strains used for expression. BL21 (DE3) used for PpETR1 and LeETR1 contains fewer membranes than derivative strains C41 (DE3) and C43 (DE3) which both form a complex internal membrane system (Miroux and Walker Citation1996).
Figure 5. Purification of solubilized, His-tagged ethylene receptors AtETR1 (A), PpETR1 (B) and LeETR1 (C) by immobilized metal affinity chromatography. Samples from all purification steps were separated on 10% SDS-PAGE gels and proteins were visualized by silver-staining. Lane 1, supernatants after solubilization and centrifugation; lane 2, fractions of unbound protein after application to the affinity column; lane 3, washing fractions (no imidazole); lane 4, washing fractions with buffer supplied with 50 mM imidazole; lanes 5–7, elution fractions (lanes 5 and 6: 2 CV each, lane 7: 1 CV) containing ETR1 proteins (indicated by asterisk). Molecular weight markers: lane M A, Precision Plus Dual Color Protein Standards, Bio-Rad Laboratories (Munich, Germany); lane M B, PageRuler Plus Prestained Protein Ladder, Fermentas (St. Leon-Rot, Germany).
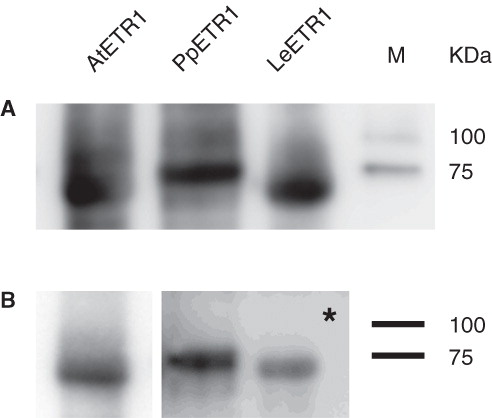
Samples collected from the affinity chromatography were also analyzed by Western blot and immunostaining. ETR1 was identified in all samples by its N-terminal deca His-tag () and by an ETR1 specific antibody (). Signals arising from detection of the affinity tag of the receptors are similar in their intensity indicating equal levels of recombinant protein. As expected, the signals for PpETR1 and LeETR1 were less pronounced when using the antibody that was raised against the carboxy-terminus of AtETR1. shows the related epitope in the carboxy-terminus of the receptor orthologs and confirms the lower degree of conservation in this domain.
Figure 6. Western blot analysis of elution fractions from IMAC purification of AtETR1 (85 kDa), PpETR1 (89 kDa) and LeETR1 (87 kDa). The receptor proteins were identified by immunostaining with an antibody directed against the deca His-tag (A) and by an antibody directed against an epitope in the C-terminal domain of receptor AtETR1 (B) (Schaller et al. Citation1995). Signals were detected after 30 sec and 10 min (indicated by asterisk), respectively. Molecular weight marker: lane M, Precision Plus Dual Color Protein Standards.
Molecular characterization of the purified receptor proteins
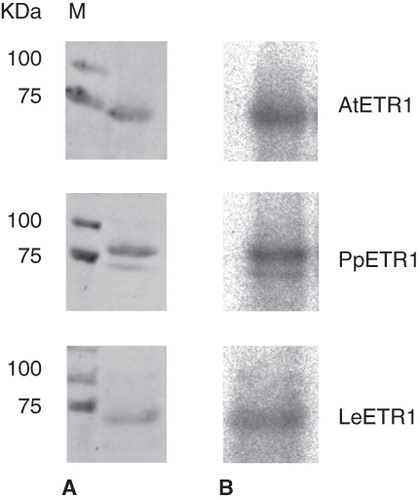
The three orthologous ETR1 proteins were solubilized from their bacterial host membranes with Fos-Choline-14 and purified by IMAC. When incubated with γ32P-ATP in a radioactive phosphorylation assay, incorporation of radiolabelled 32P was observed for all orthologs () indicating that all recombinant receptors were obtained in a functional and active state.
Figure 7. Autoradiography showing kinase activity of purified receptors AtETR1, PpETR1 and LeETR1. The receptor proteins were detected by Ponceau S staining (A) and incorporation of 32P (B). Molecular weight marker: lane M, Precision Plus Dual Color Protein Standards.
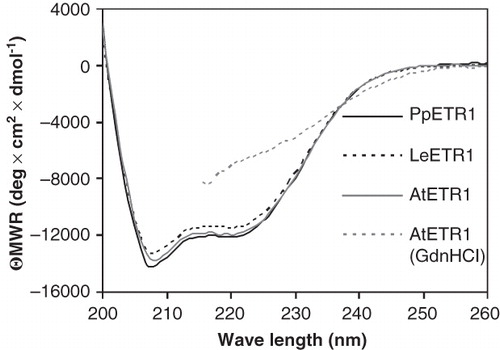
Purified receptors were also analyzed by CD spectroscopy to obtain information on protein secondary structure and folding. With a minimum at 208 nm and zero crossing at 201 nm, the three ETR1 orthologs had almost identical CD spectra that are characteristic of typical α-helical proteins (). Secondary structure calculations by CDPro (Sreerama and Woody Citation2000) suggest an α-helical content of 41–52% and a β-sheet content of 9–14%. Both figures agree well with the numbers predicted from the primary structure of the receptors calculated with SOPMA (Geourjon and Deléage Citation1995) (α-helices: 46–48%, β-sheets: 15–16%) and are indicator for a native protein structure. In accordance, receptor AtETR1 treated with 4 M guanidinium hydrochloride shows a much less pronounced CD spectrum between 215 and 260 nm when compared to the natively prepared proteins. Data collection beyond this point was not possible due to high absorption of the denaturant.
Figure 8. Far-UV CD spectra of purified AtETR1, PpETR1 and LeETR1. Mean residue weight ellipticity was calculated from collected data taking into account the molecular weight and the protein concentration to allow for comparison of individual spectra. Data of AtETR1 treated with 4 M guanidinium hydrochloride indicate that the receptor is unfolded in the presence of the chemical denaturant. Due to high absorption of the denaturant no data were collected below 215 nm.
Conclusion
The objective of this study was to establish a native purification protocol for the sensor kinase AtETR1, a membrane protein involved in ethylene signaling in plants, and to demonstrate the general applicability of this protocol for the purification of orthologous hybrid type sensor kinases from P. patens subsp. patens and L. esculentum. Our study demonstrates that even though slight variations in the expression protocols were necessary for the individual receptors, all orthologs could be solubilized and purified to homogeneity and high purity from the E. coli membranes by the same protocol. Up to 0.5 mg of purified recombinant membrane protein were obtained per gram of bacterial cells – quantities that allow further biochemical and biophysical studies on the receptor kinases. CD spectroscopy and autokinase activity of the purified recombinant proteins underline the native fold and functionality of the receptors.
Our study emphasizes that a purification strategy and established protocol for a complex membrane protein can be transferred and adopted for the purification of orthologous proteins. Due to the sequence similarity and similar modular structure of the ETR1 sensor kinase with typical bacterial two-component signaling kinases, our protocol might be also applicable without or with only minor adjustments to those proteins when produced recombinant in E. coli as well.
Acknowledgements
We thank Nicole Voet van Vormizeele and Patricia Robel for technical assistance. We also thank Benjamin Scharein for assistance with the CD measurements.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, 1997. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res 25(17):3389–3402.
- Bleecker AB, Esch JJ, Hall AE, Rodríguez FI, Binder BM. 1998. The ethylene-receptor family from Arabidopsis: Structure and function. Phil Trans R Soc B 353(1374):1405–1412.
- Bleecker AB, Kende H. 2000. Ethylene: A gaseous signal molecule in plants. Annu Rev Cell Dev Biol 16(1):1–18.
- Calderwood DA, Shattil SJ, Ginsberg MH. 2000. Integrins and actin filaments: Reciprocal regulation of cell adhesion and signaling. J Biol Chem 275(30):22607–22610.
- Chen YF, Etheridge N, Schaller GE. 2005. Ethylene signal transduction. Ann Bot 95(6):901–915.
- Cove DJ, Perroud PF, Charron AJ, McDaniel SF, Khandelwal A, Quatrano RS. 2009. The moss Physcomitrella patens: A novel model system for plant development and genomic studies. Cold Spring Harb Protoc 2009; doi:10.1101/pdb.emo115.
- Gamble RL, Coonfield ML, Schaller GE. 1998. Histidine kinase activity of the ETR1 ethylene receptor from Arabidopsis. Proc Nat Acad Sci USA 95(13):7825–7829.
- Geourjon C, Deléage G. 1995. SOPMA: Significant improvements in protein secondary structure prediction by consensus prediction from multiple alignments. Comput Appl Biosci 11(6):681–684.
- Heukeshoven J, Dernick R. 1988. Improved silver staining procedure for fast staining in PhastSystem Development Unit. I. Staining of sodium dodecyl sulfate gels. Electrophoresis 9(1):28–32.
- Hwang I, Chen HC, Sheen J. 2002. Two-component signal transduction pathways in Arabidopsis. Plant Physiol 129(2):500–515.
- Johnson PR, Ecker JR. 1998. The ethylene gas signal transduction pathway: A molecular perspective. Annu Rev Genet 32:227–254.
- Kimura S, Sinha N. 2008. Tomato (Solanum lycopersicum): A model fruit-bearing crop. Cold Spring Harb Protoc 2008; doi: 10.1101/pdb.emo105.
- Kosinski M, Rinas U, Bailey J. 1992. Isopropyl-β-d-thiogalactopyranoside influences the metabolism of Escherichia coli. Appl Microbiol Biotechnol 36(6):782–784.
- Laemmli UK. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227(5259):680–685.
- Li MZ, Elledge SJ. 2007. Harnessing homologous recombination in vitro to generate recombinant DNA via SLIC. Nat Methods 4(3):251–256.
- Li Y, Agrawal A, Sakon J, Beitle RR. 2001. Characterization of metal affinity of green fluorescent protein and its purification through salt promoted, immobilized metal affinity chromatography. J Chromatogr A 909(2):183–190.
- Miroux B, Walker JE. 1996. Over-production of proteins in Escherichia coli: Mutant hosts that allow synthesis of some membrane proteins and globular proteins at high levels. J Mol Biol 260(3):289–298.
- Rath A, Glibowicka M, Nadeau VG, Chen G, Deber CM. 2009. Detergent binding explains anomalous SDS-PAGE migration of membrane proteins. Proc Nat Acad Sci USA 106(6):1760–1765.
- Ren H, Yu D, Ge B, Cook B, Xu Z, Zhang S. 2009. High-level production, solubilization and purification of synthetic human GPCR chemokine receptors CCR5, CCR3, CXCR4 and CX3CR1. PLoS One 4(2):e4509.
- Rodríguez FI, Esch JJ, Hall AE, Binder BM, Schaller GE, Bleecker AB. 1999. A copper cofactor for the ethylene receptor ETR1 from Arabidopsis. Science 283(5404):996–998.
- Sambrook J, Russell DW. 2006. The condensed protocols from Molecular cloning: A laboratory manual. New York: Cold Spring Harbor Laboratory Press.
- Schaller GE, Ladd AN, Lanahan MB, Spanbauer JM, Bleecker AB. 1995. The ethylene response mediator ETR1 from Arabidopsis forms a disulfide-linked dimer. J Biol Chem 270(21):12526–12530.
- Sreerama N, Woody RW. 2000. Estimation of protein secondary structure from circular dichroism spectra: Comparison of CONTIN, SELCON, and CDSSTR methods with an expanded reference set. Anal Biochem 287(2):252–260.
- Towbin H, Staehelin T, Gordon J. 1979. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: Procedure and some applications. Proc Nat Acad Sci USA 76(9):4350–4354.
- Voet-van-Vormizeele J, Groth G. 2008. Ethylene controls autophosphorylation of the histidine kinase domain in ethylene receptor ETR1. Mol Plant 1(2):380–387.
- White SH. 2009. Biophysical dissection of membrane proteins. Nature 459(7245):344–346.