Abstract
Membrane proteins are intrinsically involved in both human and pathogen physiology, and are the target of 60% of all marketed drugs. During the past decade, advances in the studies of membrane proteins using X-ray crystallography, electron microscopy and NMR-based techniques led to the elucidation of over 250 unique membrane protein crystal structures. The aim of the European Drug Initiative for Channels and Transporter (EDICT) project is to use the structures of clinically significant membrane proteins for the development of lead molecules. One of the approaches used to achieve this is a virtual high-throughput screening (vHTS) technique initially developed for soluble proteins. This paper describes application of this technique to the discovery of inhibitors of the leucine transporter (LeuT), a member of the neurotransmitter:sodium symporter (NSS) family.
Introduction
Membrane proteins are intrinsically involved in both human and pathogen physiology, and are the target of 60% of all marketed drugs. This makes them hugely attractive for the development of new therapies for a range of diseases including cancer and heart disease, as well as bacterial infections and malaria. Historically, most new drugs were found by identifying the active ingredient from traditional remedies or by serendipitous discovery.
In the modern era, the understanding of how disease and infection are controlled at the molecular and physiological level has led to target specific entities based on this knowledge. This process is lengthy and incredibly expensive, with the average new molecular entity having taken 10–12 years to develop, typically costing in excess of US $1.5 billion to develop. The use of High Throughput Screening (HTS) of large compound libraries against a particular protein target began in the 1990s, when large pharmaceutical companies compiled libraries of several million compounds and screened them against a wide range of soluble proteins using robots. This technology was heralded as a new dawn for drug discovery, allowing the rapid identification of hit molecules which could be tuned to produce a lead molecule with excellent in vivo activity and desirable physicochemical properties. Whilst this technique has seen some success (Fox et al. Citation2006), it has not produced the wealth of new drugs that was initially anticipated. This has been, in part, due to the somewhat variable quality of these original libraries and also the targets which are suitable to screen via this approach. Membrane proteins have not really been investigated using HTS as the biochemical assays using purified soluble membrane proteins needed to screen for inhibitors cannot often be automated and performed on the scale required for these screens (Cuatrecasas Citation2006, Payne et al. Citation2007). Also, the costs involved in these screens are considerable, a single HTS campaign using a 500,000 compound library costs around $1 million. This makes such screens challenging to achieve in an academic setting, although there are a few notable exceptions (Verkman Citation2012). However, with the advent of high performance computing, new computational algorithms have been developed to allow high throughput screening to be done ‘virtually'.
The technique of molecular docking has been well established for some time. However, recent advances in both hardware and software algorithms have made possible the rapid docking of very large collections of small molecules into the chosen molecular target, a technique known as virtual high-throughput screening (vHTS). The speed of some of these programs is such that up to 100,000 molecular structures can be docked per day when using a cluster of parallel processors. However, as with all docking algorithms, the scoring function that is used to assess the validity of specific docking poses is paramount, and each program has its own unique scoring function. These scoring functions will necessarily place different weightings on the various factors involved in ligand binding. As none of these functions are considered faultless, a consensus scoring approach is the best way to identify potential lead molecules. Consensus scoring uses several scoring functions to predict binding affinity. If a compound is predicted to bind tightly to a chosen protein using several docking algorithms, this provides higher confidence in the prediction. Among the most prominent docking programs available for virtual screening are:
AutoDock (http://autodock.scripps.edu/);
GOLD (http://www.ccdc.cam.ac.uk/products/life_sciences/gold/);
DOCK (http://dock.compbio.ucsf.edu/); and
The European Drug Initiative for Channels and Transporters (EDICT) sought to use these vHTS techniques to identify inhibitors of various membrane proteins including several members of the secondary active transporter family.
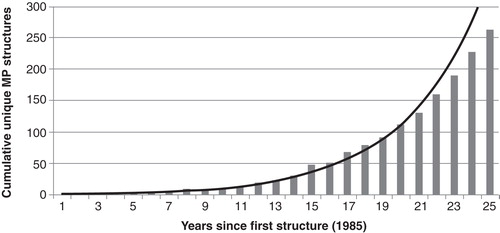
We have recently demonstrated the potential of the application of in silico approaches to the identification of inhibitors of soluble proteins such as the antimalarial target DHODH (Davies et al. Citation2009), the aspartic acid protease BACE-1 (Yi Mok et al. Citation2009) and penicillin-binding proteins (Woon et al. Citation2011). Usually, for these proteins high-resolution crystal structures were available as well as robust and well-documented biochemical assays allowing the activity of proposed inhibitors to be readily probed. However, during the past decade, advances in the study of membrane proteins using X-ray crystallography, electron microscopy and NMR-based techniques have led to the elucidation of over 250 unique membrane protein crystal structures by 2010 ().
Figure 1. Exponential growth of the number of unique membrane protein X-ray structures available since 1985. Produced using data from: http://blanco.biomol.uci.edu/mpstruc/listAll/list.
Our initial goal was to identify structurally characterized and clinically relevant membrane proteins in order to apply vHTS techniques. Due to the inherent difficulties involved in crystallizing membrane proteins (Torres et al. Citation2003), many of the available X-ray crystal structures are at a poorer resolution than is desired for vHTS. We would ideally only wish to use structures with resolution below 2 Å as these have the most accurate representation of the protein's arrangement; however, for membrane proteins, we decided to use structures with resolution <4 Å to increase the number of suitable structures available for us to work with. In order to carry out a successful vHTS screen on a selected target protein, we also required existence of an assay allowing for robust and at least medium throughput screening of chosen compounds. Hence, after careful consideration of the targets list supplied for EDICT, we decided to consider a selection of proteins, including the bacterial member of the neurotransmitter: sodium symporter (NSS) family, the leucine transporter (LeuT).
Our case study: LeuT
A suitable protein for conducting a vHTS is LeuT, a bacterial homologue of the NSS family characterized by their ability to use the sodium gradient across the plasma membrane to drive the transport of its solute against its concentration gradient. Other members of the NSS family include transporters for the neurotransmitters dopamine, norepinephrine and serotonin. The first high-resolution crystal structure of LeuT (Yamashita et al. Citation2005) (PDB code 2A65) revealed that leucine and two sodium ions are located in close proximity within a dehydrated protein core (). LeuT fulfils our criteria as a suitable test protein model in the following ways: (i) A high-resolution crystal structure (1.65 Å) is available for LeuT as well as different conformational states have been resolved; (ii) LeuT can be purified in large quantities in an active form; and (iii) we are able to measure the binding of [3H]-leucine to LeuT and thus the possible inhibition by synthesized compounds that will be generated based on positive hits from our vHTS.
Figure 2. LeuT structure. (a) Overall structure of the LeuT protein with the S1-binding site containing L-leucine (green) and two sodium ions (magenta) and the S2-binding site containing the tricyclic antidepressant imipramine (cyan). (b) Close-up of the S1-binding site; the leucine molecule sits in a very tight binding site and makes several hydrogen bonding interactions with Ala22, Gly26, Thr254 and Ser256. There is also a large hydrophobic interaction between the ligand and Tyr108, Phe259 and Ile359. This Figure is reproduced in colour in Molecular Membrane Biology online.
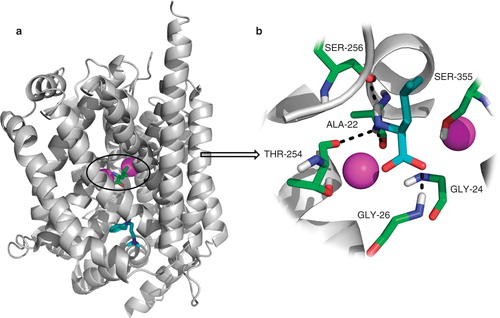
Partial unwinding of the transmembrane helices in LeuT is apparent in the vicinity of the ligand-binding site. This feature is also seen in the more recent crystal structure by Singh et al. (Citation2007) (PDB code 2Q72) which has been used in the present investigation.
The alternating access mechanism (Jardetzk Citation1966) is applicable to the LeuT protein and this crystal structure is seen as an intermediate occluded state where both the extracellular and intracellular gates are closed, preventing access to the bound substrate. Shi et al. (Citation2008) identified a second substrate binding site (S2) located in the extracellular vestibule around 11 Å from the primary binding site (S1). This site is comprised of residues shown recently to participate in low affinity binding of tricyclic antidepressants. Binding and flux experiments showed that the two binding sites can be occupied simultaneously. The substrate in the secondary site allosterically triggers intracellular release of sodium and substrate from the primary site, thereby functioning as a ‘symport effector'. Because tricyclic antidepressants (e.g., clomipramine [CMI]) bind differently to this secondary site, they do not allow substrate release from the primary site and thus act as symport uncouplers, inhibiting transport. Moreover, Quick et al. (Citation2009) showed that n-octyl-β-D-glucopyranoside (OG), the detergent used to crystallize LeuT, impairs the sodium-coupled symport mechanism. They also demonstrated that OG detergent overlaps with residues predicted to constitute the S2 site inhibiting leucine binding. Thus, all LeuT crystal structures, which have been solved in OG, represent functionally blocked forms of the transporter, whereas having substrate bound in the S2 binding site promotes a different state that is essential for sodium-coupled symport, as it was shown in functional experiments performed using dodecyl-β-D-maltopyranoside (DDM)-solubilized LeuT Quick et al. (Citation2009).
LeuT also binds other hydrophobic amino acids such as alanine or valine. The fundamental requirement for a molecule to be a LeuT substrate has been linked to fit in the occluded substrate-binding cavity which is not easy given the nature of the binding site (Ryan and Mindell Citation2007, Singh et al. Citation2007). Amino acids such as tryptophan, which are too big to fit the occluded binding site, can still bind, but trap the transporter in an open-out conformation (Singh et al. Citation2008).
Methods
Virtual high-throughput screening using eHiTS
The electronic high-throughput screening program eHiTS (Zsoldos et al. Citation2006, Citation2007) utilizes an exhaustive systematic search algorithm that considers all docking poses that lack severe steric clashes with the protein. The system employs a unique graph matching algorithms and dock tables which are stored in SQL databases. Unlike DOCK or FlexX (http://www.biosolveit.de/FlexX-C/), eHiTS does not use an incremental construction method but instead attempts to find the global optimum by enumerating combinations of independent fragment dockings.
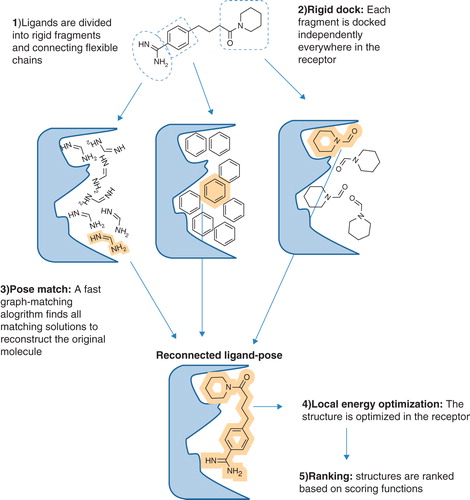
The eHiTS software has a novel flexible docking ligand method that exhaustively produces all ligand poses that avoid severe steric clashes between the protein receptor and the ligand. In the first instance, the binding pocket is defined by a steric grid which divides regions into separate pockets and identifying all possible interaction sites. The ligand is divided into rigid fragments and connecting flexible chains. Each fragment is wrapped into a polyhedron shape with chemical properties assigned to the vertices of the polyhedron. All rigid fragments are docked to every possible place in the S1 site of LeuT and scored for predicted binding affinity ().
Figure 3. Schematic overview of the eHiTS docking strategy. In eHiTS, ligands are divided into rigid fragments and connecting flexible chains (Step 1). These fragments are docked individually into the binding site of the target protein (Step 2) and a fast graph-matching algorithm finds all matching solutions to reconstruct the original molecule (Step 3), which can then be optimized (Step 4), scored and ranked (Step 5). Reproduced with permission from Nature Reviews Microbiology (Simmons et al. Citation2010). This Figure is reproduced in colour in Molecular Membrane Biology online.
Initially, a simple and fast chemical flag based statistical scoring function is used during the rigid fragment-docking and pose-matching phases. This function is not too sensitive to small variations in the interaction geometry, interaction distance and hydrogen-bonding angle.
All ring systems are considered rigid and their conformation is preserved as given in the input. Therefore, it is desirable to use multiple ring conformers (e.g., chair, boat and twist boat for a cyclohexane) for complete conformational sampling. These can be generated using a conformer generation program such as Corina (Schwab Citation2010). An exhaustive matching of compatible rigid fragment pose sets is performed to yield complete structures. Typically, the program evaluates several million mappings of the rigid fragment polyhedra to cavity polyhedra. The reconstructed solutions define a rough binding pose which is then refined using a local energy minimization in the targeted site of the protein receptor. A more sensitive, empirical-scoring function is used during the final local energy minimization phase. This scoring function has smooth curves representing the distance and angle dependency of the interactions while supporting efficient gradient-based optimization.
The final resulting poses are evaluated using a third, more time-consuming scoring function that combines both statistical and empirical components, plus additional grid based geometrical terms as well as entropy loss estimation and another novel scoring element based on the coverage of receptor surface area. This final scoring function attempts to estimate the binding free energy more accurately. The result of the final accurate scoring is used to rank the generated solutions (Zsoldos et al. Citation2007).
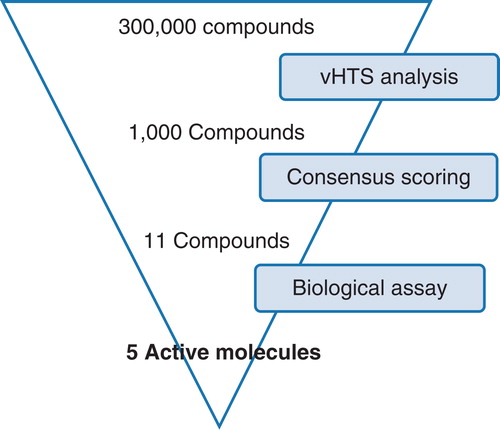
Using the high-throughput screening mode within the program (version 9.0), three compound libraries containing a total of 300,000 commercially available compounds from Maybridge (http://www.maybridge.com), Chembridge (http://www.chembridge.com) and KeyOrganics (http://www.keyorganics.co.uk) were screened against the X-ray crystal structure of LeuT co-crystallized with sodium ions and L-leucine (PDB code 2Q72). This crystal structure is at 1.70 Å resolution which makes it a good quality model of the protein. The water molecules present in this crystal structure were removed, as was the L-leucine ligand. The OG detergent molecule and the tricyclic antidepressant CMI were not removed from the model as they are structurally distinct from the S1 leucine binding site which we focussed our investigation on. We chose to focus our investigations initially on the S1-binding site as it is the main site involved in amino acid transport. Further investigations could focus on identifying new ligands for the S2 binding site; however, this is outside of the scope for the current publication.
From the library of commercially available molecules, 1000 compounds were selected for further evaluation and consensus scoring using the de novo design program SPROUT (Gillet et al. Citation1994, Citation1995, Law et al. Citation2003, Zsoldos et al. Citation2003). These 1000 compounds were selected based on their predicted binding to the protein as predicted using the eHiTS scoring algorithm. From this set, 11 molecules were selected for purchase. These 11 compounds were chosen based upon their predicted binding affinity to the protein as determined using the eHiTS and SPROUT scoring functions and also their predicted binding pose, as visualized using the SPROUT protein boundary surface representation. Another key factor for consideration is the synthetic accessibility of the molecules; this is key for developing further structure-activity relationships ().
Figure 4. vHTS screening cascade. Starting from a commercially available compound library of 300,000 compounds, initial vHTS identified 1000 compounds which were evaluated further using consensus scoring and also a knowledge of compound solubility. A total of 11 compounds were purchased from Maybridge and evaluated for activity against the LeuT. This Figure is reproduced in colour in Molecular Membrane Biology online.
High-resolution mass spectroscopy was used to confirm the integrity of the purchased compounds. Of the 11 compounds assayed, five showed activity against LeuT with four showing enough activity to allow determination of dose-response curves ().
Figure 5. Dose-response inhibition of [3H]-leucine ([3H]-Leu) binding to LeuT by compounds 2, 4, 8 and 9. Normalized results are given as mean ± SEM on the basis of four independent experiments.
![Figure 5. Dose-response inhibition of [3H]-leucine ([3H]-Leu) binding to LeuT by compounds 2, 4, 8 and 9. Normalized results are given as mean ± SEM on the basis of four independent experiments.](/cms/asset/23a078be-2f98-407b-922c-3589b6b7665b/imbc_a_710341_f0005_b.jpg)
LeuT expression and purification
Wild-type LeuT from Aquifex aeolicus was produced in Escherichia coli C41(DE3) harbouring pET16b encoding LeuT-His8, essentially as described by Chae et al. (Citation2010). Plasmid was kindly provided by E. Gouaux (Vollum Institute, Portland, Oregon, USA). Briefly, after isolation of bacterial membranes followed by solubilization in 1% DDM, LeuT was bound to Ni2+-affinity Chelating Sepharose Fast Flow resin (GE Healthcare, USA), following elution in buffer composed of 20 mM Tris-HCl (pH 8.0), 1 mM NaCl, 199 mM KCl, 0.05% DDM, 300 mM imidazole and 20% glycerol. Eluted LeuT was subsequently aliquoted, frozen at −80°C and stored until use (no activity losses of frozen batches were detected as compared to unfrozen protein).
Effects of inhibitors on binding
Inhibitory effects of tested compounds on [3H]-leucine binding were measured using scintillation proximity assay (SPA) (Quick and Javitch Citation2007). Briefly, SPA reaction mixture consisted of 0.0002 mg/ml LeuT, 10 nM [3H]-leucine and copper chelate (His-Tag) YSi beads (PerkinElmer, USA). Binding was assessed in the buffer composed of 20 mM Tris-HCl (pH 8.0), 190 mM NaCl, 10 mM KCl and 0.05% DDM in the absence or presence of tested compounds or CMI used as the control pharmacological inhibitor with the final solvent (DMSO) concentration of 10%, and monitored using MicroBeta liquid scintillation counter (PerkinElmer). Normalized results are expressed as mean ± SEM from at least two independent experiments.
Results
The LeuT protein from A. aeolicus provided us with an excellent vehicle to test our vHTS methods on membrane proteins. From an initial purchase set of 11 compounds, five compounds were found to exhibit activity against the protein when tested at 1 mM, a hit rate of 45%. This hit rate is very pleasing; a hit rate of greater than 20% from vHTS work is classed as very successful and this sort of study would not expect to achieve a 100% success rate. The four most active compounds were tested for their dose-response inhibition of the [3H]-leucine binding ().
Three of the compounds, 4, 8 and 9 inhibited approximately 100% of leucine binding, suggesting they either block both the S1- and S2-binding sites, or alter the protein conformation in such a way that leucine can no longer bind. This is an interesting result especially as compared to control pharmacological inhibitor employed in this study (CMI), known to inhibit only the S2-binding site (i.e., only approximately 50% of the total binding). An inhibitor at the S2-binding site blocks substrate binding at the site, but unlike the substrate molecule, does not couple inward release of S1 to the sodium gradient, thereby acting as a symport uncoupler (Quick et al. Citation2009). Moreover, for all three compounds, leucine binding was inhibited with comparable half-maximal inhibitory concentrations (IC50; ); whereas the estimated IC50 value for CMI was 5-to 6-fold higher.
Table I. Identification of inhibitors of LeuT. % of total [3H]-leucine binding in the presence of 1 mM tested compounds and IC50 values for the inhibition of [3H]-leucine binding by compounds 2, 4, 8 and 9 or clomipramine (CMI) are given as mean ± SEM. Numbers in parentheses (N) indicate number of independent experiments.
The inhibitory properties of compound 2 were weaker compared to the other active molecules in the test set; the compound did not block 100% of leucine binding within the tested concentration range and the estimated IC50 value was much higher ().
Compound 2 is very closely related to tryptophan, which was shown to bind LeuT with low affinity and trap it in an open-to-out conformation. The mode of action of compound 2 could be similar.
Based our successes in screening a small number of compounds, we purchased a second generation of compounds exploring the structure-activity relationship of compound 1 as we were keen to identify ‘non-amino acid like' inhibitors of LeuT; however, none of these compounds showed any appreciable activity (data not shown). The active compounds were selected based on their predicted binding affinity to the S1-binding site and are able to make several key interactions with the protein, including H-bonding interactions with Gly26, Tyr108 and Ser355 and hydrophobic interaction with Ile359, Phe253 and Phe259 ().
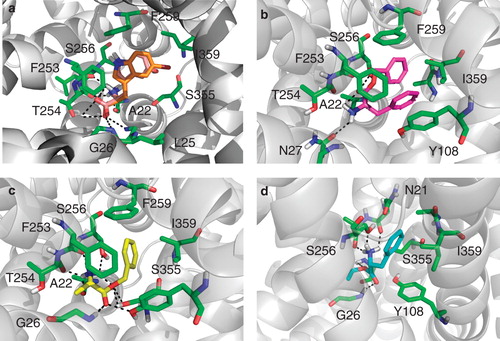
Figure 6. Binding pose of compound 2, 4, 8 and 9. (a) Compound 2 (orange) overlaid with its inactive variant compound 10 (salmon) docked into the crystal structure of the LeuT bound to L-tryptophan and sodium ions (PDB code 3F3A). The compounds makes H-bonding interactions with Leu25, Gly26 and Thr254 as well as filling a hydrophobic pocket created by Phe259 and Ile359, this pocket is violated by compound 10. (b) Compound 4 (magenta) docked into the crystal structure of the LeuT used for all vHTS (2Q72). The compound makes H-bonding interactions with Ala22, Asn27 and Ser256. (c) Compound 8 (yellow) docked into the crystal structure of the LeuT used for all vHTS (2Q72). The compound makes H-bonding interactions with many active site residues including Ala22, Tyr108 and Ser256. (d) Compound 9 (cyan) docked into the crystal structure of the LeuT used for all vHTS (2Q72). The compound makes H-bonding interactions with Asn21, Ala22, Gly24 and 26 and Ser256. This Figure is reproduced in colour in Molecular Membrane Biology online.
Discussion
The application of computational modelling to gain insight into membrane proteins has recently seen increasing success due to the availability of high-quality data sets and atomic resolution structures of several major facilitator proteins. Until now, models for these systems could not robustly predict putative inhibitors or substrates based on the protein structure, more often ligand-based approaches and pharmacophore models were used to screen large databases and identify novel transporter ligands (Giacomini et al. Citation2010).
As part of the EDICT project, we have also identified selective inhibitors of the potassium channel Kv1.1–1.2(3) using a consensus docking virtual screening approach (Wacker et al. Citation2012). From 89 electrophysiologically evaluated compounds, 14 novel compounds were found to inhibit the current carried by Kv1.1–1.2(3) channels by more than 80% at 10 μM. This represents a hit rate of around 15% using much more stringent cut-off values for the hit molecules, however this target had previously been validated and we were able to ‘train' our scoring functions based on known Kv1.1–1.2(3) inhibitors.
On this completely blind docking study, the hit rate we achieved from our initial vHTS was very pleasing at around 45%, validating the use of our approach against membrane proteins and giving us the confidence to move forward and screen against other suitable targets. This hit rate was higher than that seen for other soluble proteins which we have targeted using the same approach (Davies et al. Citation2009, Yi Mok et al. Citation2009, Woon et al. Citation2011) although the definition of a hit molecule is potentially more relaxed in the current study. An interesting observation from this study was that only compounds that contained an amino acid functionality showed any significant binding activity – we purchased a second generation of compounds exploring the structure-activity relationship of compound 1; however, none of these compounds showed any appreciable activity.
It is hoped that future efforts involving co-crystallization of these compounds could further validate these docking results and perhaps suggest some further insight into the mechanism of action of this class of transporters.
It was particularly interesting, given the spectrum of amino acids that have been shown to bind to LeuT that compound 2; 5-bromotryptophan, is active whereas the hydroxyl derivative 10 shows no appreciable binding affinity. This can be rationalized by examining the docking pose (http://autodock.scripps.edu/, http://www.simbiosys.ca/ehits/index.html). The bromine moiety is located in a hydrophobic pocket in LeuT in the open-to-out conformation within the crystal structure of LeuT bound to L-tryptophan and sodium ions (PDB code 3F3A), makes a favourable interaction with the protein (). The hydroxyl containing compound however, is more hydrophilic and therefore makes an unfavourable interaction within the hydrophobic region of the protein.
The functional state of LeuT in reported structures solved in OG detergent and the number of existing substrate binding sites in detergent-solubilized transporter are still highly controversial issues. Although in the present study we compared inhibitory properties of the compounds with only one pharmacological inhibitor (CMI), it appears that several of these molecules may inhibit both the S1 and S2 binding sites. However, further biochemical characterization of the compounds using other pharmacological inhibitors or detergents known to disrupt binding (e.g., OG) is required to prove this. Finally, it is unclear as to whether these compounds are true inhibitors of LeuT or if they are rather novel substrates for the protein. Further testing would be required to determine this. The boundary between a substrate and a competitive inhibitor depends on the size, shape and rigidity of the ligand relative to the constraints imposed by the binding pocket (Singh et al. Citation2008).
Conclusions
We have demonstrated that eHiTS is a powerful screening tool to identify inhibitors of membrane proteins. This is very exciting as around 60% of all known drug targets are membrane proteins and the molecules identified here are not only relevant in aiding our understanding of the bacterial leucine transporter, but are also relevant to other NSS family members or closely related glucose and nucleobase transporters (Weyand et al. Citation2008).
The remit of the EDICT project was to focus on applying high throughput approaches for structure-function analysis of membrane transporters and channels. This large multidisciplinary and integrated effort has used many different technologies for structure determination in combination with computational and experimental analyses to identify potential new target sites for drugs to alleviate the burden of the diseases involving membrane-transporters and channels.
Whilst we are still some way from having a lead molecule for further clinical development, the EDICT project has been very successful in identifying inhibitors of a range of different membrane proteins. It has also confirmed that the vHTS technologies used in the past to develop inhibitors of soluble proteins can successfully be applied to more challenging targets. This is a very exciting finding and it is hoped that future projects can build on these initial findings to identify clinical candidates.
Acknowledgements
The research leading to these results has received funding from the European Community's Seventh Framework Programme FP7/2007-2013 under grant agreement no HEALTH-F4-2007-201924, EDICT Consortium. This work was partially supported by the Danish National Research Council (C.J. L., U.G.) and the Lundbeck Foundation (C.J. L., U.G.).
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Chae PS, Rasmussen SGF, Rana RR, Gotfryd K, Chandra R, Goren MA, 2010. Maltose-neopentyl glycol (MNG) amphiphiles for solubilization, stabilization and crystallization of membrane proteins. Nat Methods 7:1003–1008.
- Cuatrecasas P. 2006. Drug discovery in jeopardy. J Clin Investig 116:2837–2842.
- Davies M, Heikkila T, Mcconkey GA, Fishwick CWG, Parsons MR, Johnson AP. 2009. Structure-based design, synthesis, and characterization of inhibitors of human and plasmodium falciparum dihydroorotate dehydrogenases. J Med Chem 52:2683–2693.
- Fox S, Farr-Jones S, Sopchak L, Boggs A, Nicely HW, Khoury R, 2006. High-throughput screening: Update on practices and success. J Biomol Screen 11:864–869.
- Giacomini KM, Huang S-M, Tweedie DJ, Benet LZ, Brouwer KLR, Chu X, & 2010. Membrane transporters in drug development. Nat Rev Drug Discov 9:215–236.
- Gillet VJ, Myatt G, Zsoldos Z, Johnson AP. 1995. SPROUT, HIPPO and CAESA: Tools for de novo structure generation and estimation of synthetic accessibility. Perspect Drug Discov Des 3:34–50.
- Gillet VJ, Newell W, Mata P, Myatt G, Sike S, Zsoldos Z, 1994. SPROUT – recent developments in the de-novo design of molecules. J Chem Info Computer Sci 34:207–217.
- Jardetzk O. 1966. Simple allosteric model for membrane pumps. Nature 211:969–970.
- Law JMS, Fung DYK, Zsoldos Z, Simon A, Szabo Z, Csizmadia IG, 2003. Validation of the SPROUT de novo design program. J Mol Struct-Theochem 666:651–657.
- Payne DJ, Gwynn MN, Holmes DJ, Pompliano DL. 2007. Drugs for bad bugs: Confronting the challenges of antibacterial discovery. Nat Rev Drug Discov 6:29–40.
- Quick M, Javitch JA. 2007. Monitoring the function of membrane transport proteins in detergent-solubilized form. Proc Natl Acad Sci USA 104:3603–3608.
- Quick M, Winther AML, Shi L, Nissen P, Weinstein H, Javitch JA. 2009. Binding of an octylglucoside detergent molecule in the second substrate (S2) site of LeuT establishes an inhibitor-bound conformation. Proc Natl Acad Sci USA 106:5563–5568.
- Ryan RM, Mindell JA. 2007. The uncoupled chloride conductance of a bacterial glutamate transporter homolog. Nat Struct Mol Biol 14:365–371.
- Schwab CH. 2010. Conformations and 3D pharmacophore searching. Drug Discov Today Technol 7:e245–e253.
- Shi L, Quick M, Zhao YF, Weinstein H, Javitch JA. 2008. The mechanism of a neurotransmitter: sodium symporter – inward release of Na+ and substrate is triggered by substrate in a second binding site. Mol Cell 30:667–677.
- Simmons KJ, Chopra I, Fishwick CWG. 2010. Structure-based discovery of antibacterial drugs. Nat Rev Microbiol 8:501–510.
- Singh SK, Piscitelli CL, Yamashita A, Gouaux E. 2008. A competitive inhibitor traps LeuT in an open-to-out conformation. Science 322:1655–1661.
- Singh SK, Yamashita A, Gouaux E. 2007. Antidepressant binding site in a bacterial homologue of neurotransmitter transporters. Nature 448:952–956.
- Torres J, Stevens TJ, Samso M. 2003. Membrane proteins: The ‘Wild West' of structural biology. Trends Biochem Sci 28:174.
- Verkman AS. 2012. Aquaporins in clinical medicine. In: Caskey CT, Austin CP, Hoxie JA, editors. Annual Review of Medicine. Vol. 63. Palo Alto, CA: Annual Reviews.
- Wacker SJ, Jurkowski W, Rolland J-F, Simmons KJ, Fishwick C, Johnson P, 2012. Identification of novel selective inhibitors of the potassium channel Kv1.1–1.2(3) by high-throughput virtual screening and electrophysiology. ChemMedChem.
- Weyand S, Shimamura T, Yajima S, Suzuki S, Mirza O, Krusong K, 2008. Structure and molecular mechanism of a nucleobase-cation-symport-1 family transporter. Science 322:709–713.
- Woon ECY, Zervosen A, Sauvage E, Simmons KJ, Zivec M, Inglis SR, 2011. Structure guided development of potent reversibly binding penicillin binding protein inhibitors. ACS Med Chem Lett 2:219–223.
- Yamashita A, Singh SK, Kawate T, Jin Y, Gouaux E. 2005. Crystal structure of a bacterial homologue of Na+/Cl−dependent neurotransmitter transporters. Nature 437:215–223.
- Yi Mok N, Chadwick J, Kellett KAB, Hooper NM, Johnson AP, Fishwick CWG. 2009. Discovery of novel non-peptide inhibitors of BACE-1 using virtual high-throughput screening. Bioorg Med Chem Lett 19:6770–6774.
- Zsoldos Z, Reid D, Simon A, Sadjad BS, Johnson AP. 2006. eHITS: An innovative approach to the docking and scoring function problems. Curr Protein Pept Sci 7:421–435.
- Zsoldos Z, Reid D, Simon A, Sadjad SB, Johnson AP. 2007. eHiTS: A new fast, exhaustive flexible ligand docking system. J Mol Graph Model 26:198–212.
- Zsoldos Z, Szabo I, Szabo Z, Johnson AP. 2003. Software tools for structure based rational drug design. J Mol Struct Theochem 666:659–665.