Abstract
The Na,K-ATPase is essential to all animals, since it maintains the electrochemical gradients that energize the plasma membrane. Naturally occurring inhibitors of the pump from plants have been used pharmaceutically in cardiac treatment for centuries. The inhibitors block the pump by binding on its extracellular side and thereby locking it. To explore the possibilities for designing an alternative way of targeting the pump function, we have examined the structural requirements for binding to a pocket that accommodates the two C-terminal residues, YY, in the crystal structures of the pump. To cover the sample space of two residues, we first performed docking studies with the 400 possible dipeptides. For validation of the in silico predictions, pumps with 13 dipeptide sequences replacing the C-terminal YY were expressed in Xenopus laevis oocytes and examined with electrophysiology. Our data show a significant correlation between the docking scores from two different methods and the experimentally determined sodium affinities, which strengthens the previous hypothesis that sodium binding is coupled to docking of the C-terminus. From the dipeptides that dock the best and better than wild-type YY, it may therefore be possible to develop specific drugs targeting a previously unexplored binding pocket in the sodium pump.
Introduction
The Na,K-ATPase maintains the sodium and potassium gradients across the plasmamembrane in virtually all animal cell types (Jorgensen et al. Citation2003). The gradients are of fundamental importance for signaling and secondary transport, control of cell volume, osmoloarity, and energization of the membrane. The Na,K-ATPase was one of the earliest membrane protein enzymes discovered (Libet Citation1948, Skou Citation1957), and it founded the P-type ATPase family that also encompasses, e.g., proton and calcium pumps, as well as heavy-metal pumps and lipid flippases (Axelsen and Palmgren Citation2001).
Crystal structures of potassium-bound forms of the Na,K-ATPase have revealed an intimate docking of the α-subunit carboxy terminus at the transmembrane domain (Morth et al. Citation2007, Shinoda et al. Citation2009). The C-terminus was proposed to be a key regulator of a previously unrecognized ion pathway between the ion binding sites and the cytoplasm (Poulsen et al. Citation2010).
Specific inhibitors of the Na,K-ATPase termed cardiotonic steroids are produced by plants like Digitalis and Strophanthus and by Bufo toads (Schatzmann Citation1953). They have been used for centuries, and digoxin is the oldest cardiac medication still in use for treatment of heart failure with atrial fibrillation (Mijatovic et al. Citation2007). The mechanism of action is believed to involve a slowing of the heart rate by increased vagal efferent activity and an anti-adrenergic influence. A detailed molecular understanding of the mechanism is, however, still lacking, and the extent of digoxin use has fallen due to its complicated pharmacokinetic properties and narrow therapeutic index (Gheorhgiade et al. Citation2006, Ehle et al. Citation2011). Furthermore, the effect is affected by the extracellular potassium concentration, with patients suffering from hypokalemia experiencing an increased effect (Shapiro et al. Citation2006).
Retrospective epidemiological studies have revealed that surprisingly few of the patients with heart problems maintained on cardiotonic steroid treatment died from cancer (Stenkvist Citation2001). One reason for the beneficial effect of prolonged cardiotonic steroid treatment may be that very low amounts of the drugs can regulate cell junction contacts (Larre et al. Citation2010) and proliferation (Tian et al. Citation2009). Such reports have spurred an interest in Na,K-ATPase inhibitors as potential chemotherapeutic agents and suggest that the Na,K-ATPase might be an attractive drug target, not only for its role in ionic balances but also for its role as a hormone receptor (Mijatovic et al. Citation2007). So far, their narrow therapeutic window has prevented cardiotonic steroids from being developed into anti-cancer drugs, but improved subunit specificity might be a means of reducing cardiotoxicity and improving anti-tumor activity. This strategy has in part been demonstrated with the hemi-synthetic analogue UNBS1450 derived from the naturally occurring cardiotonic steroid 2-oxovoruscharin (Van Quaquebeke et al. Citation2005).
Both the cardiotonic steroids ouabain and digoxin induce vasoconstriction when administered in vivo, but only ouabain raises the blood pressure after prolonged administration in rats. Digoxin and the related cardiotonic steroid digitoxin are actually very effective in lowering blood pressure in rats with ouabain-induced hypertension (Levinsky et al. Citation1975, Hulthen et al. Citation1984, Kimura et al. Citation2000, Manunta et al. Citation2000). This suggests a more complex relationship between the various effects observed for cardiotonic steroids than inhibition of the pump alone can account for. Secondary signaling pathways must also be affected, e.g., by cardiotonic steroids activating the Src kinase in a signal-transduction pathway where the Na,K-ATPase acts as a receptor (Haas et al. Citation2000, Li et al. Citation2009a).
A compound that acts on the Na,K-ATPase, but with a different mechanism of action, different pharmacokinetics and a broader therapeutic index would therefore be of interest for the indications known for cardiotonic steroids, but possibly also for novel indications. We have explored the structural requirements for a ligand in the potential regulatory C-terminal pocket by docking of dipeptides and by electrophysiological studies of pumps with the two C-terminal residues mutated. This has allowed us to test the initial assumption that the functional cycle is affected by how well the ligand binds in the C-terminal pocket and to explore the basis for designing a compound with higher affinity than the intramolecular ligand. For two of the docking programs, we find a significant correlation between the docking scores and the extracellular sodium affinity, and we have identified dipeptide sequences with higher affinity for the binding pocket than the wild-type YY.
Materials and methods
In silico docking of dipeptide ligands
The full length Na,K-ATPase has large domains on either side of the lipid bilayer. For docking, a smaller system focused on the transmembrane part was used. The N-terminal 749 residues of the α-subunit, the 64 C-terminal residues of the β-subunit and all of the γ subunit were removed. The truncation is expected to preserve the immediate environment surrounding the C-terminal pocket and thus be applicable to shorter time-scale simulations of the region.
The protein was embedded in a pre-equilibrated patch of solvated bilayer of 102 POPC lipids using the g_membed tool in Gromacs (Hess et al. Citation2008). The system was equilibrated for 5 ns using position restraints on all heavy atoms. The final structure of the equilibration was used to construct a protein mutant by removing the eight C-terminal residues. Since we have used strong position restraints on the protein, its structure after the equilibration was within 0.2 Å of the original crystal structure. This was the structure used for docking.
The dipeptide library was generated in pymol. For Glide docking, the library was additionally pre-processed by the software LIGPREP from the MAESTRO suite to assign atomic partial charges, define possible tautomerisaton states, stereoisomers and protonation in 5.5–8 pH range.
Glide (Friesner et al. Citation2004) performs a guided solution space search by an initial rough guess of the ligand conformation and a subsequent energy optimization on a non-bonded potential grid based in the OPLS-AA force field. The best candidates, which are defined by the scoring function, are further refined by Monte Carlo sampling of the ligand pose. Intermolecular interactions were pre-calculated on a grid representing the extracellular half of receptor and centred on the cavity where the C-terminal residues are present in the wild-type structure to ensure correct placement of docking poses and to include long range interactions (up to 20 Å). Flexibility was derived by in place temporary alanine mutations and van der Waals radii scaling. All dipeptide ligands were docked with full flexibility on the grid. For each ligand, ten poses were generated and subsequently clustered (RMSD < 0.5 A). Glide Score and E-model scoring functions were applied to rank all poses. Glide Score is an empirically derived scoring function, which combines lipophilicity, hydrogen bonding, ionic interactions, van der Waal's interactions and solvation terms, each term representing the weighted sum based on nominal optimal distances typical for the interaction type (Friesner et al. Citation2004). E-model is the sum of the Glide Score, ligand-receptor molecular mechanics interaction energy and the ligand strain energy (Friesner et al. Citation2004).
AutoDock (Morris et al. Citation1998, Huey et al. Citation2007) and AutoDock Vina (Trott and Olson Citation2010) have similar approaches to representing protein and ligand structures as well as the docking algorithm. AutoDock Vina implementation of global optimization is modified and is two orders of magnitude faster according to authors. Due to an optimized scoring function, it has proven more accurate performance in benchmarking (Trott and Olson Citation2010). The scoring function of AutoDock is a semi-emiprical force field including evaluations of van der Waal's dispersion/repulsion, hydrogen bonding, electrostatics and desolvation weighted by training on a set of experimentally derived complexes, whereas AutoDock Vina does not formulate steric, hydrophobic and H-bonding terms as potentials, but derives them from best fit experimental affinity measurements and conformational preferences of receptor-ligand complexes.
For protein preparation, intermolecular interactions were pre-calculated on a grid centred on the C-terminal cavity and spanning up to 20 Å to include long range interactions without searching for suboptimal solutions outside the target cavity.
The fully flexible ligands were docked and the search for the optimal combination of position and geometry of a given ligand was driven by a genetic algorithm that uses iterative enrichment of best scoring solutions. Docked poses were optimized, and significant minima were combined for structure refinement and clustering.
Electrophysiology
Plasmids encoding human α1 and β1 subunits of the Na,K-ATPase were purchased from Origene (http://www.origene.com) and subcloned into the pXOON vector (Jespersen et al. Citation2002) using EcoRI/NotI. Mutations Q116R and N127D were introduced into α1 by PCR to reduce ouabain sensitivity (Price and Lingrel 1988), yielding the construct referred to as wild type. The chosen mutations at the C-terminal were introduced by PCR using QuikChange™ Site-Directed Mutagenesis Kit. The constructs were sequenced by using a reverse primer in the pXOON vector close to the C-terminal insertion site. From NheI digested plasmids, RNAs were transcribed with the mMESSAGE ULTRA kit (Ambion). Oocytes from Xenopus laevis were isolated and defoliated. β1 (1 ng) and α1 (10 ng) RNAs were co-injected into Stage V–VI oocytes. After 1–3 days at 17°C, oocytes were loaded with sodium in 95 mM Na, 90 mM sulphamic acid, 5 mM HEPES, 10 mM TEACl, 0.1 mM EGTA, pH 7.6. Electrophysiological measurements were performed using the two-electrode voltage-clamp technique with an OC-725C voltage-clamp apparatus (Warner Instrument Corp.) The measurements was performed in 115 mM Na, 110 mM sulphamic acid, 1 mM MgCl2, 0.5 mM CaCl2, 5 mM BaCl2, 10 mM HEPES, pH 7.4 as well as 10 μM ouabain to inhibit the endogenous oocyte Na+/K+-ATPase. NMDG replaced Na in sodium-free solutions. To determine steady-state currents, 15 mM K replaced 15 mM Na, a series of 200-ms voltage steps was run and the 10 mM ouabain background was subtracted. Charge movement was determined from a single exponential fitting of the difference trace between recordings with and without 10 mM ouabain.
Data were recorded and analyzed using pClamp 10.3 (Axon Instruments).
Results
The 400 possible dipeptides were analyzed by docking. For the starting structure, molecular dynamics (MD) was used to equilibrate a model of the pump in the [K2]E2.Pi state (PDB code 2ZXE) with the transmembrane domains intact, but with much of the intracellular and extracellular parts removed. A snapshot at the end of the equilibration showed no large changes compared to the crystal structure, suggesting that the deletions do not disturb the structure on the time-scale used. The C-terminal eight residues of the α subunit were removed, and the dipeptide library was screened with four docking programs, giving four separate rankings. To generate an average ranking, the score distributions were normalized for each of the procedures, and the consensus function was calculated as an average of the z-scores of the individual procedures (Supplementary , available online).
Table I. Characteristics of the dipeptide sequences selected.
The four docking procedures differ in their methodologies of the stochastic searches for a ligand's geometry and position in the complex as well as in the scoring functions applied on intermediate selections, and they give very different rankings. To evaluate the predictive powers of the procedures, 13 dipeptides were selected for electrophysiological studies. The selection contained both high and low scoring sequences with polar, non-polar, positively and negatively charged amino acids.
As most animal cells contain sodium pumps, cellular studies of a specific pump can benefit from making it ouabain resistant to ensure that the activity is unaffected by ouabain at concentrations sufficient to abolish endogenous pumps. Rat α1 Na,K-ATPase is naturally ouabain resistant, and introduction of two of the rat residues on the extracellular part of the α subunit confers ouabain resistance (Price and Lingrel Citation1988). Human α1 with the ouabain resistance mutations and the C-terminal YY sequence altered was expressed in oocytes from Xenopus laevis and studied with electrophysiology. The extracellular sodium affinity was determined from the slow charge movement after a series of 20 mV voltage steps between −200 mV and 40 mV.
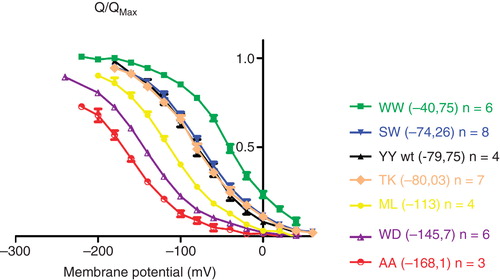
Due to the electrogenicity of binding and release of extracellular Na+, the occupancy of the Na+ binding sites is controlled by [Na+]ext and voltage. The amount of slow charge moved in response to a voltage step follows a Boltzmann function centered at a half-maximal voltage, the midpoint potential V0.5. The slow charge corresponds to a +1 charge moving through approximately 0.7 of the membrane field, and it is believed to reflect the movement of one of the three sodium ions transported (Holmgren et al. Citation2000). The charge movements were previously shown to be strongly left-shifted by mutations expected to impair C-terminal binding, i.e., the mutations lowered the sodium affinity (Poulsen et al. Citation2010). Of the C-terminal mutants tested here, there was a wide spreading in midpoint potentials from −168 mV to −41 mV ().
Compared to wild type, two of the constructs had clearly increased extracellular sodium affinities (WW and EW), and two had similar affinities (TK and SW). The crystal structures and mutational studies indicate that the aromatic systems of the tyrosines in the wild type form cation-π interactions with Arg940 and Arg1005 (Homo sapiens α1 numbering will be used throughout) (Morth et al. Citation2007, Poulsen et al. Citation2010). The higher affinity of WW compared to YY may therefore be due to stronger cation-π interactions with the more electron rich aromatic systems, but improved fitting within the binding pocket may also, at least partly, explain the apparently higher affinity of WW (). The other nine constructs tested all have midpoint potentials lower than the wild type, −113 mV to −168 mV. A left-shift of the Q-V curve by 25 mV generally reflects a two-fold reduction in external Na+ affinity (Holmgren et al. Citation2000, Holmgren and Rakowski Citation2006), indicating that the AA construct has a 12 times lower sodium affinity than the wild type (). The two alanines are likely to promote extension of the last α-helix all the way to the C-terminus, making it unable to enter the binding pocket. We propose that the mutants with the worst sodium affinities have C-termini with low likelihood for binding in the pocket.
Figure 1. The Na,K-ATPase C-terminal structure and the functional cycle. (A) The structural context of the two C-terminal tyrosines (in spheres) of the Na,K-ATPase α-subunit (brown). The β-subunit (purple), the γ-subunit (green), the two occluded potassium ions (red) and the approximate position of the membrane (grey) are also indicated. The Figure was made with pymol (www.pymol.org) using PDB ID 2ZXE (a shark Na,K-ATPase). (B) The Post-Albers scheme for the Na,K-ATPase functional cycle. In E1P (top left), three sodium ions (green circles) are bound in the occluded pump. With a conformational change to the E2P state (top middle), a proton (black circle) approaches site IIIb from the cytoplasm, the extracellular gate opens, and the sodium ions are released. With a proton at the aspartate in site IIIb (Asp933 in Homo sapiens α1), two potassium ions (red squares) can bind, which promotes dephosporylation to the occluded E2 state (top right). Release of potassium and proton depends on opening of the C-terminal channel between TM5, TM7 and TM8 (bottom right). Binding of sodium and ATP (bottom middle) leads to E1 (bottom left). Figure adapted from Poulsen et al. (Citation2010) with the author's permission. (C) ChemDraw illustration of the interactions of the C-terminal tyrosines in 2ZXE. The program Capture “http://capture.caltech.edu/” suggests a single significant cation-π interaction between Arg940 and Tyr1022. Furthermore, Arg1005 is only 4.3 Å from Tyr1023, and mutational studies show decreased sodium affinity for Arg1005Gln (Poulsen et al. Citation2010), indicating that this may also be a relevant cation-π interaction, at least in part of the catalytic cycle. (D) ChemDraw illustration of the interactions suggested by Vina of the tryptophane-tryptophane dipeptide. In both cases, the most important interactions are between arginines and aromatic systems. This Figure is reproduced in colour in Molecular Membrane Biology online
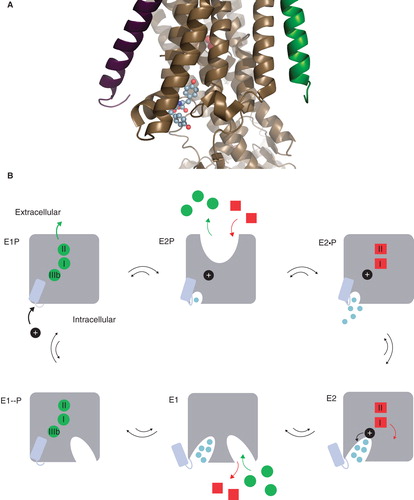
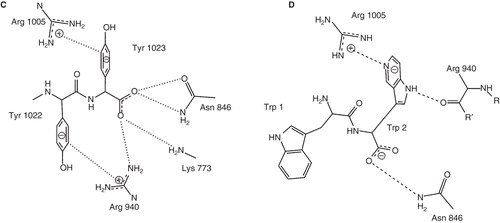
Figure 2. The voltage dependence of transient currents of C-terminally mutated Na,K-ATPases. The charge translocation (Q/Qmax) of C-terminal mutants was determined from the off pulses of a series of 20-mV voltage steps between −200 mV and 40 mV and Boltzmann fitted. Numbers in brackets indicate the V0,5 for extracellular Na+. A full list of V0,5 and slope for each of the tested constructs can be found in . This Figure is reproduced in colour in Molecular Membrane Biology online
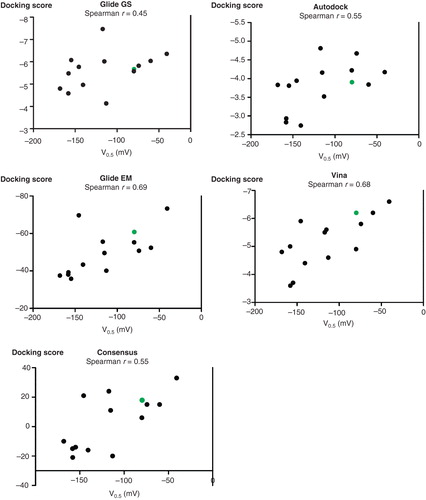
The results from the four docking methods varied significantly, so to determine if one of them was superior in predicting sodium affinity, we compared the V0.5 values for the C-terminal mutants tested to the corresponding dipeptide docking scores (). Spearman rank correlation suggests that Vina and Glide EM were useful for predicting C-terminal docking with coefficients of 0.68 and 0.69, respectively, while Glide GS and Autodock performed poorly. In a search for compounds that might bind the pocket, Vina or Glide EM would therefore be the preferred choice.
Figure 3. Comparisons of docking scores and sodium affinities. The scores for each of the four docking methods as well as the consensus score were plotted against the V0.5 values for the C-terminal mutants. The wild-type YY is green. For each of the plots, the Spearman correlation coefficient is indicated. There is a significant correlation for Vina, Glide EM and the consensus score (p < 0.05). The statistical analyses were done with GraphPad Prism. This Figure is reproduced in colour in Molecular Membrane Biology online
The Vina and Glide EM dockings both predict most of the top scoring dipeptides to have at least one tryptophan, and it is clear from the top ranked binding poses that cation-π interactions with arginines are favored, and various hydrogen bonds are also suggested.
According to Vina's prediction, the best WW pose shows a cation-π interaction between Arg1005 and the N-terminal tryptophan (). Similarly, Glide EM suggests that the N-terminal tryptophan forms a cation-π interaction, in this case with Arg837.
The rate constants of the slow charge movement of the sodium pump reach a minimum at positive membrane potentials and a maximum at negative membrane potentials, when voltage-insensitive reactions become limiting for sodium release and binding, respectively, and lowering of the extracellular sodium concentration causes a left-shift in the curve (Holmgren et al. Citation2000).
The rate constants at −180 mV for the WW, SW, YY, ML and DR constructs follow a trend that correlates with the measured sodium affinities; the construct with the highest affinity, WW, also has the highest rate constant at −180 mV, and the rate constants of the constructs decrease in accordance with the measured affinities ().
Figure 4. The voltage dependence of the relaxation rates of C-terminally mutated Na,K-ATPases. From single exponential fittings of the on pulses of a series of voltage steps between −180 mV and 40 mV, the relaxation rate constants of C-terminal mutants were determined. This Figure is reproduced in colour in Molecular Membrane Biology online
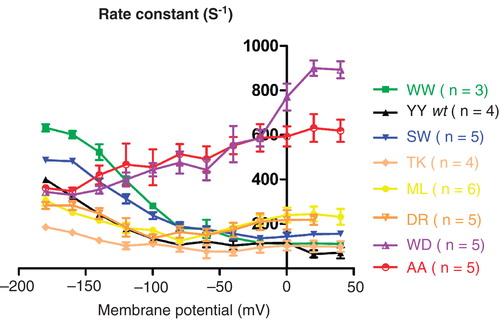
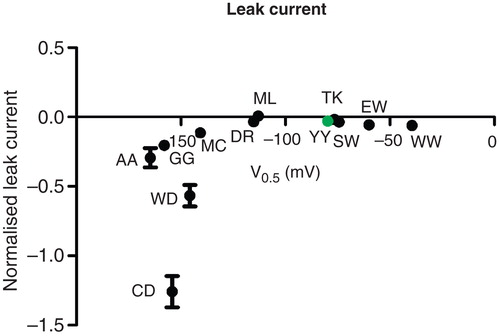
Figure 5. Leak versus sodium affinity. The inwardly rectifying leak current at −160 mV in a sodium containing, potassium-free extracellular environment compared to the affinity for extracellular sodium. The leak current was normalized to the potassium-induced pump current at +20 mV. This Figure is reproduced in colour in Molecular Membrane Biology online
In contrast, the rate constant of the very low affinity constructs WD and AA are lower at negative potentials and five- to 10-fold higher than wild type at positive membrane potentials as previously observed for C-terminal mutants (Poulsen et al. Citation2010). The very fast rates at positive potentials for the mutants suggest that rearrangement of the C-terminal tail may be a rate limiting step for sodium release in the wild type as previously noted (Poulsen et al. Citation2010).
TK has a sodium affinity comparable to the wild type, but very slow relaxation rates both at high and low membrane potentials. The slow rates cannot be caused by the basic nature of the last lysine alone, since the construct DR, which also has a basic residue at the last position, displays markedly higher rate constants. TK has wild type like sodium affinity, but the rate constants indicate that the binding of TK does differ.
The constructs ML and DR have V-shaped rate constant curves with maximum rate constants at positive and negative potentials and a minimum around −80 mV. With affinities in between wild type and the very low affinity constructs like AA, ML and DR, they share characteristics of constructs with both high and low sodium affinity.
The Na,K-ATPase is known to carry an inwardly rectifying leak current of protons, when both potassium and sodium are absent on the extracellular side. In C-terminal mutants, however, the leak current is generally observed to be enhanced rather than inhibited by physiological levels of extracellular sodium (Jespersen et al. Citation2002, Vedovato and Gadsby Citation2010).
We observed a marked leak in sodium buffers for some of the C-terminal mutants, but not for all. To examine how the C-terminus may influence the leak, we plotted the normalized proton leak current in sodium for the constructs examined as a function of their midpoint potentials (). Noteworthy, all constructs with midpoint potentials above −120 mV have little or no leak, and all constructs with lower midpoint potentials leak. The leak current is measured at −160 mV, so for pumps with high affinity, the majority will be sodium-saturated and adopting the occluded sodium bound E1P conformation, while this state is destabilized in the mutants with reduced affinity. The leak current has a temperature-dependence similar to the potassium induced pump current, and the leak was suggested to result from intracellular cation release from the E1P state (Meier et al. Citation2010). Alternatively, disturbing the C-terminal structure may allow sodium ions to promote the forward reaction from E2P, i.e., through dephosphorylation. In either case, poor docking of the C-terminus promotes sodium-induced occupancy of one or more states where the leak pathway becomes accessible for protons.
The constructs WD and CD have leak currents much larger than those observed for the other constructs with similar sodium affinities. The leak current was previously shown to depend on Asp933, a residue that may be involved in sodium ion binding. Mutating Asp933 to an asparagine, which mimics the protonated state of the aspartate, completely eliminates the leak current (Poulsen et al. Citation2010), suggesting that the residue is required as a ‘stepping stone' in the proton pathway. Asp933 is approximately 6 Å from the C-terminal Tyr1023 in the E2P structures, so an acidic C-terminal residue may facilitate the protonation and/or deprotonation of Asp933.
Discussion
The Na,K-ATPase is an exceptionally well-studied enzyme and one of the earliest drug targets to be used systematically, but all sodium pump inhibitors studied so far are believed to bind a common pocket on the extracellular side (Tobin et al. Citation1975, Yatime et al. Citation2011, Peluffo and Berlin Citation2012). In this study we have analyzed the properties of the C-terminal binding pocket of the Na,K-ATPase. By combining docking studies with electrophysiological measurements, we show that docking of dipeptides with the programs Vina and Glide EM reliably predict the midpoint potential of mutants with the corresponding sequences introduced into the in cis ligand, namely the C-terminus. The correlation between docking scores and midpoint potentials corroborates previous claims that the ability of the C-terminus to fit into the binding pocket directly influences the pump's affinity for sodium ions. It also indicates that regulation at the C-terminal pocket occurs through a simple binding mechanism and therefore would be druggable.
The P-type ATPases go through many states with different structures during their catalytic cycles, and for the Na,K-ATPase, only E2 states associated with potassium binding or extracellular exchange of ions (with or without inhibitors) have been solved. Our docking studies are based on the structure of the potassium-occluded enzyme stabilized by magnesium fluoride mimicking a [K2]E2·Pi product complex of dephosphorylation, where the C-terminus docks into the pocket. The electrophysiological studies examine either the whole catalytic cycle (pump currents) or the transition between E1P·3Na and E2P (transient currents). Previous studies had indicated that the C-terminal conformation is especially important for sodium binding and release, while it has little effect on the interactions with potassium (Toustrup-Jensen et al. Citation2009, Meier et al. Citation2010, Vedovato and Gadsby Citation2010). There are not many interactions between the C-terminal di-tyrosine and the binding pocket in the crystal structures (), and the correlation between the docking scores and the electrophysiological data suggests that it is reasonable to use the currently available structures to gain information about the pocket structure during the catalytic cycle, but sodium binding may, nonetheless, be associated with conformational changes that promote stronger interactions.
Biochemical studies of mutant pumps found that the hydroxyl groups on the tyrosines are not important, since introduction of phenylalanine on the ultimate or penultimate position had only negligible effects on sodium affinity (Toustrup-Jensen et al. Citation2009). From our data, it is also clear that π-systems are preferred, so it is likely that the binding depends to a large extent on cation-π interactions.
Several C-terminal sequences appear to bind the pocket more strongly than the wild-type YY. Why has nature selected a suboptimal binder? The C-terminal conformation influences not only the extracellular sodium affinity as measured here with electrophysiology, but also the intracellular sodium affinity as estimated from biochemical assays on purified membranes with mutant pumps (Toustrup-Jensen et al. Citation2009). Therefore, optimal pump function likely depends on a C-terminus that allows high affinity binding of intracellular sodium, yet also allows efficient extracellular sodium release, suggesting that YY is an appropriate compromise.
Two of the dipeptides predicted by the docking programs to bind better than the wild-type YY were EW and WW, and the corresponding sodium pump mutants did have higher affinity for sodium. This buttresses that it may be a feasible strategy to design a compound targeted at the C-terminal binding pocket to outcompete the endogenous ligand, and such a drug might even be able to serve as a pump activator, which would have the prospect of opening a whole new field of drug development. For it to be successful in vivo, the compound should have hydrophobic properties allowing it to cross the cellular membrane in substantial amounts or make use of already existing transporters, e.g., PEPT1 or PEPT2 that mediate the uptake of peptide substrates and is a well-known system for delivery of peptidomimetic drugs (Daniel and Kottra Citation2004).
As a next step, we intend to test the effect on ATP hydrolysis of different dipeptides using both wild-type and C-terminally deleted pumps under varying sodium and potassium concentrations. For any lead compound (inhibitory or stimulatory), further optimization with docking would be favorable, and the effect of the compound in a cellular setting with membrane potential and the individual isoforms could be tested using the oocyte system.
There may be several advantages of developing alternatives to the cardiotonic steroids for sodium pump inhibition, especially if the pharmacokinetic properties and subunit specificities were tunable. It is a major challenge that drugs like digoxin have a very low therapeutic index, so alternative inhibitors could be beneficial for the cardiac indications, where cardiotonic steroids are already used in treatment. Studies on transgenic mice where α1 and/or α2 are resistant to cardiotonic steroids clearly indicate that the effects of sodium pump inhibition on, e.g., hypertension and cardiac properties differ depending on the subunit targeted (Lorenz et al. Citation2008, Wansapura et al. Citation2011). Future experiments would be required to determine if it would be feasible to improve the subunit and tissue selectivity for a ligand targeted at the C-terminal binding pocket. The pocket is highly conserved between the isoforms and within an 8 Å radius from the tyrosines, there is only an isoleucine/leucine difference between α1, α2 and α3, while α4 is different at five positions compared to the other three. However, the structure and its accessibility will also be influenced by residues in a broader radius and by the relative kinetics of the steps in the catalytic cycle and tissue availability, so there may be subunit differences to exploit.
In addition to the indications known, drugs targeting the C-terminal pocket may be expected to have a wide range of physiological effects depending not only on their subunit specificity and pharmacokinetic properties, but also on which step in the catalytic cycle they stabilize. Clearly, the great difference in sodium affinities and rate constant profiles of the various C-terminal mutants indicate that the way a ligand binds in the pocket markedly affects the pump cycle. At nanomolar concentrations of cardiotonic steroids, the pump can function as a receptor that alters intracellular signaling events and proliferation rates (Aizman et al. Citation2001, Li et al. Citation2009b), and the Na,K-ATPase is clearly emerging as an important component in many cellular protein interaction networks (Tian and Xie Citation2008). It would be interesting from both a cellular and a physiological point of view to examine the effects of pumps stabilized in other conformations than E2P.
Supplementary Table I, available online
Download MS Word (1.4 MB)Acknowledgements
This study was funded by the European Drug Initiative on Channels and Transporters (EDICT) program. PUMPKIN is a Center of Excellence funded by the Danish National Research Foundation. PN was supported by the ERC Advanced program BIOMEMOS. HP was supported by grants from the Carlsberg Foundation, the Lundbeck Foundation and L'Oréal/UNESCO. RA was supported by ERC grant 209825. We are grateful to A. Skov Kristensen and M. H. Poulsen (University of Copenhagen) for supplying oocytes and to M. V. Clausen for discussions on electrogenic transport.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Aizman O, Uhlen P, Lal M, Brismar H, Aperia A. 2001. Ouabain, a steroid hormone that signals with slow calcium oscillations. Proc Natl Acad Sci USA 98(23):13420–13424.
- Axelsen KB, Palmgren MG. 2001. Inventory of the superfamily of P-type ion pumps in Arabidopsis. Plant Physiol 126:696–706.
- Daniel H, Kottra G. 2004. The proton oligopeptide cotransporter family SLC15 in physiology and pharmacology. Pflugers Arch 447(5):610–618.
- Ehle M, Patel C, Giugliano RP. 2011. Digoxin: Clinical highlights: A review of digoxin and its use in contemporary medicine. Crit Pathways Cardiol 10:93–98.
- Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, 2004. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem 47(7):1739–1749.
- Gheorhgiade M, van Veldhuisen D, Colucci W. 2006. Contemporary use of digoxin in the management of cardiovascular disorders. Circulation 113:2556–2564.
- Hess B, Kutzner C, van der Spoel D, Lindahl E. 2008. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J Chem Theory Computat 4(3):435–447.
- Holmgren M, Rakowski RF. 2006. Charge translocation by the Na+/K+ pump under Na+/Na+ exchange conditions: Intracellular Na+ dependence. Biophys J 90(5):1607–1616.
- Holmgren M, Wagg J, Bezanilla F, Rakowski RF, De Weer P, Gadsby DC. 2000. Three distinct and sequential steps in the release of sodium ions by the Na+/K+-ATPase. Nature 403(6772):898–901.
- Huey R, Morris GM, Olson AJ, Goodsell DS. 2007. A semiempirical free energy force field with charge-based desolvation. J Computat Chem 28(6):1145–1152.
- Hulthen UL, Bolli P, Kiowski W, Buhler FR. 1984. Forearm vasoconstrictor response to ouabain: Studies in patients with mild and moderate essential hypertension. J Cardiovasc Pharmacol 6(Suppl 1):S75–S81.
- Haas M, Askari A, Xie Z. 2000. Involvement of Src and epidermal growth factor receptor in the signal-transducing function of Na+/K+-ATPase. J Biol Chem 275(36):27832–27837.
- Jespersen T, Grunnet M, Angelo K, Klaerke DA, Olesen SP. 2002. Dual-function vector for protein expression in both mammalian cells and Xenopus laevis oocytes. Biotechniques 32(540):536–538.
- Jorgensen PL, Hakansson KO, Karlish SJD. 2003. Structure and mechanism of Na,K-ATPase: Functional sites and their interactions. Ann Rev Physiol 65:817–849.
- Kimura K, Manunta P, Hamilton BP, Hamlyn JM. 2000. Different effects of in vivo ouabain and digoxin on renal artery function and blood pressure in the rat. Hypertens Res 23(Suppl):S67–S76.
- Larre I, Lazaro A, Contreras RG, Balda MS, Matter K, Flores-Maldonado C, 2010. Ouabain modulates epithelial cell tight junction. Proc Natl Acad Sci USA 107(25):11387–11392.
- Levinsky RA, Lewis RM, Bynum TE, Hanley HG. 1975. Digoxin induced intestinal vasoconstriction. The effects of proximal arterial stenosis and glucagon administration. Circulation 52(1):130–136.
- Li Z, Cai T, Tian J, Xie JX, Zhao X, Liu L, 2009a. NaKtide, a Na/K-ATPase-derived peptide Src inhibitor, antagonizes ouabain-activated signal transduction in cultured cells. J Biol Chem 284(31):21066–21076.
- Li ZC, Cai T, Tian J, Xie JX, Zhao XC, Liu LJ, 2009b. NaKtide, a Na/K-ATPase-derived peptide Src inhibitor, antagonizes ouabain-activated signal transduction in cultured cells. J Biolog Chem 284(31):21066–21076.
- Libet B. 1948. Adenosinetriphosphatase (ATP-ASE) in nerve. Fed Proc 7:72.
- Lorenz JN, Loreaux EL, Dostanic-Larson I, Lasko V, Schnetzer JR, Paul RJ, 2008. ACTH-induced hypertension is dependent on the ouabain-binding site of the alpha(2)-Na+-K+-ATPase subunit. Am J Physiol – Heart Circulatory Physiol 295(1):H273–H280.
- Manunta P, Hamilton J, Rogowski AC, Hamilton BP, Hamlyn JM. 2000. Chronic hypertension induced by ouabain but not digoxin in the rat: Antihypertensive effect of digoxin and digitoxin. Hypertens Res 23(Suppl):S77–S85.
- Meier S, Tavraz NN, Durr KL, Friedrich T. 2010. Hyperpolarization-activated inward leakage currents caused by deletion or mutation of carboxy-terminal tyrosines of the Na+/K+-ATPase {alpha} subunit. J Gen Physiol 135(2):115–134.
- Mijatovic T, Van Quaquebeke E, Delest B, Debeir O, Darro F, Kiss R. 2007. Cardiotonic steroids on the road to anti-cancer therapy. Biochim Biophys Acta 1776(1):32–57.
- Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, 1998. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J Computat Chem 19(14):1639–1662.
- Morth JP, Pedersen BP, Toustrup-Jensen MS, Sorensen TL, Petersen J, Andersen JP, 2007. Crystal structure of the sodium-potassium pump. Nature 450(7172):1043–1049.
- Peluffo RD, Berlin JR. 2012. Membrane potential dependent inhibition of the Na,K-ATPase by para-nitrobenzyltriethylammonium bromide. Mol Pharmacol 82:1–8.
- Poulsen H, Khandelia H, Morth JP, Bublitz M, Mouritsen OG, Egebjerg J, 2010. Neurological disease mutations compromise a C-terminal ion pathway in the Na(+)/K(+)-ATPase. Nature 467(7311):99–102.
- Price EM, Lingrel JB. 1988. Structure-function relationships in the Na,K-ATPase a subunit: Site-directed mutagenesis of glutamine-111 to arginine and asparagine-122 to aspartic acid generates a ouabain-resistant enzyme. Biochemistry 7:8400–8408.
- Schatzmann HJ. 1953. Hertzglycoside als hemmstoffe fur den aktiven kalium- und natriumtransport durch die erythrocytenmembran [Cardiac glycosides as inhibitors of active potassium and sodium transport by erythrocyte membrane]. Helv Physiol Pharmacol Acta 11:346–354.
- Shapiro JI, Kennedy DJ, Malhotra D, Xie M, Vetteth S. 2006. Hypokalemia potentiates ouabain's effect on calcium cycling and cardiac growth. Cell Mol Biol (Noisy-le-grand) 52(8):87–91.
- Shinoda T, Ogawa H, Cornelius F, Toyoshima C. 2009. Crystal structure of the sodium-potassium pump at 2.4 A resolution. Nature 459(7245):446–450.
- Skou JC. 1957. The influence of some cation on an adenosine triphosphatase from peripheral nerves. Biochem Biophys Acta 23:394–401.
- Stenkvist B. 2001. Cardenolides and cancer. Anticancer Drugs 12(7):635–638.
- Tian J, Li X, Liang M, Liu L, Xie JX, Ye Q, 2009. Changes in sodium pump expression dictate the effects of ouabain on cell growth. J Biol Chem 284(22):14921–14929.
- Tian J, Xie ZJ. 2008. The Na-K-ATPase and calcium-signaling microdomains. Physiology 23(4):205–211.
- Tobin T, Akera T, Brody SL, Ku D, Brody TM. 1975. Cassaine: Mechanism of inhibition of Na+ +K+ -ATPase and relationship of this inhibition to cardiotonic actions. Eur J Pharmacol 32(02):133–145.
- Toustrup-Jensen MS, Holm R, Einholm AP, Schack VR, Morth JP, Nissen P, 2009. The C terminus of Na(+), K(+)-ATPase controls Na(+) affinity on both sides of the membrane through Arg(935). J Biolog Chem 284(28):18715–18725.
- Trott O, Olson AJ. 2010. Software news and update AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Computat Chem 31(2):455–461.
- Van Quaquebeke E, Simon G, Andre A, Dewelle J, El Yazidi M, Bruyneel F, 2005. Identification of a novel cardenolide (2′′-oxovoruscharin) from Calotropis procera and the hemisynthesis of novel derivatives displaying potent in vitro antitumor activities and high in vivo tolerance: Structure-activity relationship analyses. J Med Chem 48(3):849–856.
- Vedovato N, Gadsby DC. 2010. The two C-terminal tyrosines stabilize occluded Na/K pump conformations containing Na or K ions. J Gen Physiol 136(1):63–82.
- Wansapura AN, Lasko VM, Lingrel JB, Lorenz JN. 2011. Mice expressing ouabain-sensitive alpha1-Na,K-ATPase have increased susceptibility to pressure overload-induced cardiac hypertrophy. Am J Physiol Heart Circ Physiol 300(1):H347–H355.
- Yatime L, Laursen M, Morth JP, Esmann M, Nissen P, Fedosova NU. 2011. Structural insights into the high affinity binding of cardiotonic steroids to the Na+,K+-ATPase. J Struct Biol 174(2):296–306.