Abstract
Nucleosides play key roles in biology as precursors for salvage pathways of nucleotide synthesis. Prokaryotes import nucleosides across the cytoplasmic membrane by proton- or sodium-driven transporters belonging to the Concentrative Nucleoside Transporter (CNT) family or the Nucleoside:H+ Symporter (NHS) family of the Major Facilitator Superfamily. The high resolution structure of a CNT from Vibrio cholerae has recently been determined, but no similar structural information is available for the NHS family. To gain a better understanding of the molecular mechanism of nucleoside transport, in the present study the structures of two conformations of the archetypical NHS transporter NupG from Escherichia coli were modelled on the inward- and outward-facing conformations of the lactose transporter LacY from E. coli, a member of the Oligosaccharide:H+ Symporter (OHS) family. Sequence alignment of these distantly related proteins (∼ 10% sequence identity), was facilitated by comparison of the patterns of residue conservation within the NHS and OHS families. Despite the low sequence similarity, the accessibilities of endogenous and introduced cysteine residues to thiol reagents were found to be consistent with the predictions of the models, supporting their validity. For example C358, located within the predicted nucleoside binding site, was shown to be responsible for the sensitivity of NupG to inhibition by p-chloromercuribenzene sulphonate. Functional analysis of mutants in residues predicted by the models to be involved in the translocation mechanism, including Q261, E264 and N228, supported the hypothesis that they play important roles, and suggested that the transport mechanisms of NupG and LacY, while different, share common features.
Introduction
Many bacteria are able to scavenge low concentrations of nucleosides from the environment for synthesis of nucleotides and deoxynucleotides by salvage pathways of synthesis, and in Escherichia coli nucleosides can be used as the sole source of nitrogen and carbon for growth (Munch-Petersen and Mygind Citation1983, Neuhard and Nygaard Citation1987). Uptake across the cytoplasmic membrane is facilitated by cation-linked transporters from two evolutionarily unrelated protein families, the Concentrative Nucleoside Transporter (CNT) family (designated 2.A.41 in the transporter classification database [TCDB; http://www.tcdb.org/]) and the Nucleoside:H+ Symporter (NHS) family (TCDB family 2.A.1.10). In E. coli two members of these families, NupC and NupG respectively, are the predominant routes for nucleoside uptake; mutants defective in both the nupC and nupG genes are not capable of high-affinity nucleoside uptake or growth on nucleosides as single carbon source (Munch-Petersen and Mygind Citation1983). Both of these transporters are proton-linked but they differ in their permeant selectivity, NupG transporting a wide range of nucleosides and deoxynucleosides while NupC does not transport guanosine or deoxyguanosine.
The structure of a homologue of NupC, the sodium-linked transporter VcCNT from Vibrio cholerae, has recently been published (Johnson et al. Citation2012). However, to date no structural information is available on NupG or any other member of the NHS family. Thus little is currently known about the mechanisms of permeant recognition and transport in this widely-distributed bacterial transporter family. In the absence of a crystal structure, useful information on the likely mechanism of a transporter can sometimes be gained by modelling the structure on that of a homologue for which a structure is available. The NHS family belongs to the major facilitator superfamily (MFS) and so NupG probably exhibits a fold similar to those MFS proteins for which a crystal structure is available. However, homology modelling is difficult because NupG exhibits only about 10% sequence identity to the most closely related MFS transporter of known structure, the lactose transporter LacY from E. coli (Mirza et al. Citation2006). The latter belongs to the Oligosaccharide:H+ Symporter (OHS) family (TCDB family 2.A.1.5). In the present study we have exploited patterns of residue conservation and polarity in the NHS and OHS families to increase the confidence with which the NupG and LacY sequences can be aligned, thus enabling the LacY structure and a predicted alternative conformation of the protein to be used as templates for homology modelling of NupG. The reliability of the resultant models was assessed by probing the accessibility of endogenous and introduced cysteine residues to thiol reagents, and the roles of residues predicted from the model to be functionally important were investigated via site-directed mutagenesis and functional assays.
Methods
Materials
E. coli strains XL1-blue and BL21(DE3) were obtained from Stratagene, La Jolla, CA, USA. [5,6-3H]uridine (39.5 Ci/mmol) was obtained from PerkinElmer Life Sciences Ltd, Buckinghamshire, UK. p-Chloromercuribenzene sulphonate (pCMBS) was obtained from Toronto Research Chemicals Inc., Canada.
Generation of NupG mutants
The plasmid pGJL25, encoding NupG fused in frame to the C-terminal oligohistidine tag KLAAALEHHHHHH (Xie et al. Citation2004), was employed as a template for mutagenesis using the QuikChange® method (Stratagene). The entire NupG open reading frame of the resultant constructs was sequenced to confirm the presence of the desired mutation and the lack of other mutations.
Measurement of nucleoside uptake by bacterial cells
Measurements of [5,6-3H]uridine uptake by E. coli strain BL21(DE3) cells harbouring pGJL25 and its derivatives, or the non-recombinant parent vector pTTQ18 (Stark Citation1987), were performed at 25°C, essentially as described previously (Xie et al. Citation2004). For such experiments, cultures were performed at 37°C and with orbital shaking at 200 rpm in M9 minimal medium (50 mM Na2HPO4, 20 mM KH2PO4, 10 mM NaCl, 20 mM NH4Cl, 0.2 mM CaCl2, 2 mM MgSO4) containing carbenicillin (100 μg/ml) and supplemented with 0.2% (w/v) casamino acids plus 0.2% (w/v) glycerol. When the cultures had reached a D600nm value of 0.6, expression was induced by the addition of 0.4 mM isopropyl-β-D-thiogalactoside (IPTG) and incubation continued for 1 h unless otherwise stated. Cells were then washed three times in transport buffer (5 mM 2-(N-morpholino)ethanesulfonic acid (MES), 150 mM KCl, pH 6.6) before measurement of uridine transport. For inhibition experiments, cells were first incubated at 25°C in transport buffer for 5 min with N-ethylmaleimide (NEM) or pCMBS and then washed a further three times in transport buffer before uridine uptake assays were performed. Typically, uridine was used at a final concentration of 50 μM and at a specific radioactivity of 3–5 mCi/mmol. For estimation of the apparent Vmax and K M values for transport, permeant concentrations were varied between 6.25 and 200 μM and an uptake period of 15 s was employed to estimate initial velocities of transport. Data, expressed per mg dry cell mass, were corrected for uptake into IPTG-induced cells harbouring control vector (pTTQ18) and then kinetic parameters estimated by non-linear curve fitting to the Michaelis-Menten equation using OriginPro7 (OriginLab Corporation).
Quantification of NupG expression
To quantify the expression of NupG and mutants thereof in IPTG-induced cultures of E. coli, membranes were prepared by the water lysis procedure (Ward et al. Citation2000). Following SDS-polyacrylamide gel electrophoresis and transfer to nitrocellulose membranes by electroblotting, the His-tagged protein was stained with INDIA HisProbe™ (Pierce) followed by SuperSignal® West Pico chemiluminescent substrate (Perbio Science UK Ltd, Northumberland, UK). Signals were detected and quantified using a GeneGnome Detection system and GeneTools software respectively (Syngene Bio Imaging).
Modelling of NupG
The sequences of 111 NupG homologues exhibiting 24–95% identity to one another were obtained by BlastP searching the UniProt protein sequence database and then aligned using ClustalX (Thompson et al. Citation1997), followed by small manual adjustments using BioEdit (Hall Citation1999), guided by visual pattern recognition in alignments coloured according to residue type. The same approach was employed to obtain the sequences of 99 LacY homologues exhibiting 23–94% identity. The rate of evolutionary change at each position in the NupG and LacY sequences was then estimated from phylogenetic analysis of the alignments using the ConSeq algorithm (Berezin et al. Citation2004), and represented using the colour scale shown in .
Figure 1. Sequence alignment of NupG and LacY. The rate of evolutionary change (conservation, ‘c') and the frequency of residue type (polarity, ‘p') at each position in multiple sequence alignments of 111 NupG homologues and of 99 LacY homologues, calculated as described in the Methods section, are indicated using the colour scales shown. Residues completely or almost completely conserved in all 210 sequences are indicated by red asterisks. Conserved basic residues of the ‘R-X-G-R-R' motif are indicated by blue asterisks. Conserved positions containing aromatic residues (W, Y or F) or residues with small side chains (G, S, A, C, P or T), used to aid alignment of the two sequences, are indicated by magenta and black asterisks respectively.
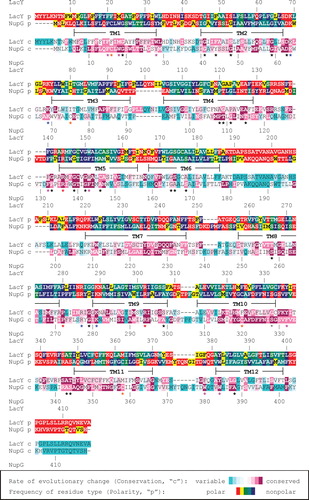
The pattern of residue polarity at each position in the alignments was also quantified and likewise represented as a colour scale (red and yellow, polar residues [E,D,N,Q,K,R,H,T,S] present in > 20% or ≤ 20% of the sequences respectively; green and turquoise, S or T the only polar residues, and present in > 20% or ≤ 20% of the sequences, respectively; blue, never occupied by a polar residue). The patterns of residue conservation and polarity were then used to guide the alignment of the NupG and LacY sequences, yielding the optimized alignment shown in . Modeller 8v2 (Fiser and Sali Citation2003) was then employed to model the structure of NupG on that of a permeant-free inward-facing conformation of the C154G mutant of LacY (PDB accession 2CFQ; (Mirza et al. Citation2006)), in which the substrate binding site is open to the cytoplasm. The NupG structure was also modelled on that of a predicted outward-facing conformation of LacY, produced by a molecular simulation approach (Pendse et al. Citation2010), in which the substrate binding site is open to the periplasm. In each case 100 models were made and the quality of the five of lowest energy was assessed using MolProbity (Davis et al. Citation2007). The model with the highest percentage of residues within the most favoured and allowed regions of the Ramachandran plot was selected for further study. The models were visualized using the PyMOL Molecular Graphics System, Version 0.99rev8, Schrödinger, LLC. The predicted locations of transmembrane helices were taken from analysis of the LacY structure using the Orientations of Proteins in Membranes (OPM) database (Lomize et al. Citation2006).
Results
Modelling of NupG
Despite exhibiting very low sequence identities, the overall folds of those MFS proteins for which structures are currently available are remarkably similar, and thus it has been proposed that the known structures can successfully be employed as templates for the modelling of distantly-related family members (Vardy et al. Citation2004). In the present study, LacY was employed as such a template for the modelling of the nucleoside transporter NupG. Although the proteins exhibit only about 10% sequence identity, they share similarities in their substrates, these being disaccharides and nucleosides respectively, and in their mechanisms, both being proton symporters (Abramson et al. Citation2003, Xie et al. Citation2004). More importantly, they also share some conserved sequence motifs. These include absolutely conserved arginine residues in transmembrane helix 5 (TM5) and TM9, an almost completely conserved proline residue in the cytoplasmic loop connecting TM8 and TM9, and an absolutely conserved aspartate residue in the cytoplasmic loop connecting TM2 and TM3 (indicated with red asterisks in ). The latter is immediately upstream of a short motif rich in basic residues (blue asterisks in ) that is repeated in modified form in the loop connecting TM8 and TM9. This motif, usually designated the R-X-G-R-R motif, is characteristic of the MFS family and has been shown to play a role in determining the topology of the proteins (Sato and Mueckler Citation1999). Such conserved motifs allow confident alignment of the adjacent regions of NupG and LacY, but lack of sequence similarity in other regions renders their alignment more difficult. To aid in the alignment of such regions, the patterns of residue conservation and side-chain polarity within the NHS and OHS families, determined as detailed in the Methods section, were employed to optimize the alignment. The rationale for this approach was to generate a model of NupG in which the exposure of variable positions on the membrane-facing surface of the model was maximized and the exposure of hydrophilic residues (other than serine and threonine, the side chains of which can hydrogen bond to backbone carbonyl groups in the same transmembrane helix) and of conserved positions was minimized. It proved possible to align numerous conserved positions in the OHS and NHS multiple sequence alignments, in particular ones in which either aromatic residues (W, Y or F; magenta asterisks in ) or residues with small side chains (G, S, A, C, P or T; black asterisks in ) predominated. Strengthening the validity of this approach, a similar alignment was obtained using the profile-profile sequence alignment approach of Jaroszewski et al. (Citation2011), the only differences being in the precise alignment of the loop regions connecting the TM helices. The alignment shown in was therefore used to generate a model of NupG based on the crystal structure of LacY in its inward-facing conformation, with the substrate binding site open to the cytoplasm, as detailed in Methods.
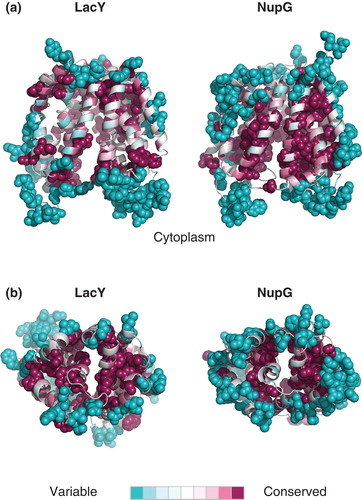
The backbone atoms of the five models of lowest energy could be superimposed with a root mean-square deviation (RMSD) of between 0.52 Å and 0.67Å, the major differences between the models being in the conformation of the longer loops connecting the predicted TM helices. Importantly, the backbone atoms of the residues chosen for subsequent mutagenesis (see below) could be superimposed with an average RMSD of 0.15Å, although in some cases they differed in side chain rotamer. In the model selected for further analysis, 94.2% of the residues were in the most favoured regions of the Ramachandran plot and 99% were in the allowed regions. The model exhibited a largely hydrophobic surface ‘belt' approximately 30Å thick, largely devoid of residues with polar side-chains (E, D, Q, N, K, R), consistent with exposure to the core of the lipid bilayer (). To evaluate the model further, the residue conservation scores calculated from the OHS and NHS family multiple sequence alignments using the ConSeq algorithm were mapped onto the LacY structure and the NupG model respectively. shows that in the LacY structure, the conserved residues are largely clustered at the interfaces between TM helices and at the surface of the hydrophilic substrate-binding cavity, whereas non-conserved residues predominate in the extramembranous loops and on the surface of the TM helices predicted to interact with the core of the bilayer. A very similar picture, with a conserved core, was evident for NupG, supporting the validity of the model ().
Figure 2. Distribution of polar residues on the surface of a model of NupG based on the inward-facing crystal structure of LacY. (a–d) Molecular surface representations of the model, viewed from the plane of the membrane, successively rotated by 90° around an axis normal to the plane of the membrane. The side chain nitrogen and oxygen atoms of aspartate, glutamate, asparagine, glutamine, lysine and arginine residues are shown in blue and red respectively. The brown rectangles indicate the likely location of the hydrophobic core of the lipid bilayer.
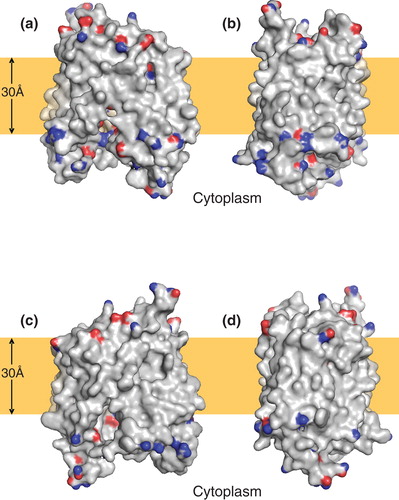
Figure 3. Pattern of residue conservation in LacY and NupG. The rate of evolutionary change at each position in the transporters was calculated using the ConSeq algorithm and mapped onto the crystal structure of LacY and the corresponding inward-facing model of NupG respectively using the colour scale indicated. The structures are shown in cartoon form, with the most variable and conserved positions in solid molecular representation. (a) View from the plane of the lipid bilayer and (b) view from the periplasm.
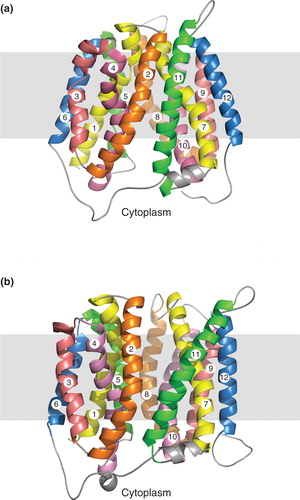
Currently, no crystal structure is available for the outward-facing conformation of LacY in which the substrate binding site is open to the periplasm, although the nature of this conformation has been probed in detail by biochemical and biophysical approaches (Smirnova et al. Citation2011). An outward-facing structure is available for the E. coli fucose transporter FucP (Dang et al. Citation2010), but this protein is more distantly related to NupG than LacY, and in particular lacks the highly conserved residues mentioned above. A model of the outward-facing structure of LacY based on FucP has been produced (Radestock and Forrest Citation2011), but exhibits some apparent anomalies, such as the orientation of the absolutely conserved residue R302, which plays a critical role in the transport mechanism, presumably reflecting the difficulty in aligning these distantly related proteins. Because of similar uncertainty in the alignment of NupG with FucP, the outward-facing conformation of NupG was instead modelled on the predicted outward facing-structure of LacY itself, produced by a molecular simulation approach (Pendse et al. Citation2010). While the results of such modelling are clearly more speculative than for modelling based on a crystal structure, they do allow predictions of residue accessibility and function to be made, which can be experimentally tested (see below). In the best model of the outward-facing conformation of NupG produced in this manner, 94.7% of the residues were in the most favoured regions of the Ramachandran plot and 99% were in the allowed regions. The model is compared with that of the inward-facing conformation in .
Figure 4. Comparison of (a) inward- and (b) outward-facing models of NupG. TM helices 1–12 are numbered and shown in cartoon form, with evolutionarily-related helices in the N- and C-terminal halves of the protein being given the same colour. The grey rectangles indicate the likely location of the hydrophobic core of the lipid bilayer.
Accessibility of the endogenous cysteine residues of NupG to the aqueous medium
While the use of profile-profile alignments and analysis of residue variability in the NHS and OHS families provided additional confidence for the alignment used for modelling, a model based on only ∼ 10% sequence identity between the target sequence and its structural template must be treated with caution in the absence of experimental verification. To obtain such verification, we next sought to examine the accessibility to water-soluble thiol reagents of residues predicted by the models to be exposed at the surface of the predicted permeant-binding cavity, identified by analogy with that shown to bind the lactose homologue β-D-galactopyranosyl-1-thio-beta-D-galactopyranoside (TDG) in LacY (Abramson et al. Citation2003, Mirza et al. Citation2006).
Wild-type NupG contains four cysteine residues, each located within the predicted membrane-spanning region of the protein (). To assess the accessibility of these cysteines to the aqueous medium, E. coli cells harbouring the expression plasmid pGJL25 were induced with IPTG to express NupG and then exposed for 5 min in transport buffer at pH 6.6 to the membrane-impermeable thiol-specific reagent pCMBS. shows that this reagent caused a concentration-dependent inhibition of transport, incubation with 100 μM pCMBS reducing transport to a level even less than that seen in uninduced cells (). Uridine uptake in the uninduced cells probably reflects the presence of endogenous NupC and other nucleoside transporters, because Western blotting revealed the absence of His-tagged NupG (data not shown and Xie et al. Citation2004). The inhibition of transport could be completely reversed by treatment with 100 mM DTT, demonstrating that the effect of pCMBS probably reflected its reaction with one or more cysteine residues, likely to be located in NupG ().
Figure 5. Locations of cysteine residues in NupG. Cross-sections of the transmembrane regions of (a) the inward-facing and (b) the outward-facing models of NupG are illustrated, viewed from the cytoplasmic side of the membrane, showing the locations of the four cysteine residues of the protein in space filling representation. TM helices are shown as α-carbon traces, coloured as in .
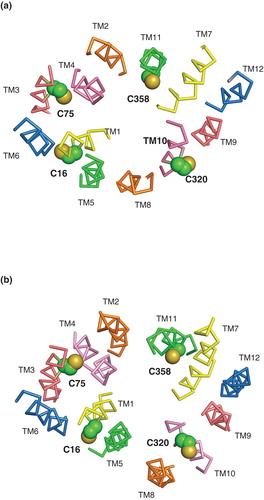
Figure 6. Effect of pCMBS treatment on uridine transport in E. coli cells induced to express wild-type NupG. (a) Concentration-dependence of the effect of pCMBS on uptake of 50 μM uridine, measured for 15 s or 2 min as indicated. Results shown are mean ± SD (n = 3) and have been corrected for uptake into non-induced cells. (b) Reversibility of pCMBS inhibition by DTT. Non-induced (control) or induced cells were treated with or without 100 μM pCMBS for 5 min, washed with transport buffer and then treated with or without 100 mM DTT for 5 min before subsequent measurement of 50 μM uridine uptake, over periods of 15 s or 2 min, as indicated. Results shown are mean ± SD (n = 3).
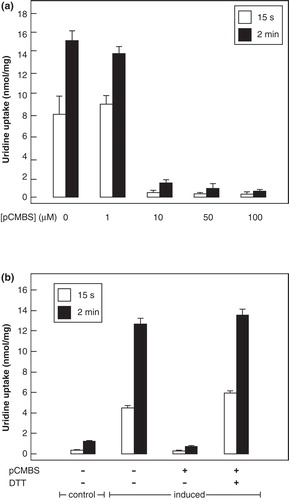
The side-chains of residues C16 in TM1 and C320 in TM10 are predicted by both the inward- and outward-facing models of NupG to be exposed on the lipid-facing surface of the transporter, while that of C75 in TM3 is predicted to be buried at the interface with TM4 and that of C358 is predicted to be exposed on the surface of the permeant-binding cavity (). The profound effect of pCMBS on the transport activity of NupG was therefore hypothesized to result from modification of the latter residue. To investigate this hypothesis further, C358 was mutated to serine and to alanine, both of which are found at the equivalent position in NupG homologues. Both mutants exhibited substantial uridine transport activity (), mutant C358S showing an apparent Vmax value approximately 80% that of the wild-type protein, and a similar K M value ( and ). The finding that the cysteine residue is not required for function is consistent with the fact that alanine is a much more frequent residue than cysteine at this position in NupG homologues. However, in contrast to the wild-type protein, the transport activity of the mutants was not inhibited by pCMBS treatment (). This finding indicates that the inhibition of uridine transport by pCMBS in bacteria expressing NupG stems from reaction with the transporter itself, rather than from an effect on bacterial metabolism and/or the proton gradient across the membrane, and that residue C358 is responsible for the sensitivity of NupG to thiol reagents. Inhibition likely stems from steric hindrance of nucleoside binding by the bulky reagent. No protection against inhibition was afforded by inclusion of 10 mM uridine during incubation with pCMBS (data not shown), but this may reflect the difficulty of competing with the covalent modification of the protein, when using a reversible ligand of modest affinity.
Figure 7. Uridine transport activity of NupG C358 mutants. (a) Uptake of 50 μM uridine, measured for 15 s or 2 min as indicated, in E. coli cells harbouring vectors encoding wild-type NupG or its C358A or C358S mutants, before and after induction of expression by treatment with IPTG for 1 h. Results shown are mean ± SD (n = 3). (b) Concentration dependence of uridine uptake, measured over a period of 15 s, in E. coli cells induced to express the C358S mutant of NupG. Data shown are mean ± SD (n = 3) and have been corrected for the endogenous uridine uptake activity found in non-induced cells. The line shows the fit of the data to the Michaelis-Menten equation, obtained by non linear regression.
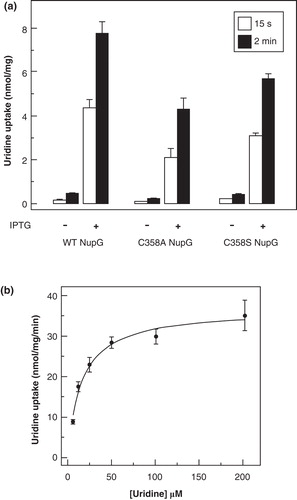
Table I. Kinetic properties of wild-type NupG and its mutants.
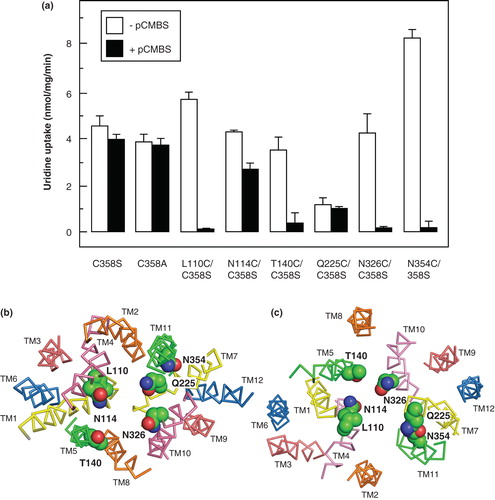
Figure 8. Effect of pCMBS treatment on uridine transport in E. coli cells induced to express cysteine mutants of NupG. (a) Cells induced to express the indicated mutants were treated with (solid bars) or without (open bars) 100 μM pCMBS for 5 min and then washed with transport buffer before subsequent measurement of 50 μM uridine uptake, over a period of 2 min. Results shown are mean ± SD (n = 3). (b) Cross-section of the transmembrane region of the inward-facing NupG model, viewed from the cytoplasmic side of the membrane. (c) Cross-section of the outward-facing NupG model, viewed from the periplasmic side of the membrane. The locations of the residues replaced by cysteine are shown in space filling representation. TM helices are shown as α-carbon traces, coloured as in .
Use of the substituted cysteine accessibility method (SCAM) to probe the structure and function of proteins ideally requires a cysteine-free template. In an attempt to achieve this in the present case, the C358S mutant was employed as a template to make double cysteine mutants involving the other three endogenous cysteine residues of NupG. These were mutated either to alanine (C16 and C320) or to serine (C75), these residue types being present at the corresponding locations in NupG homologues. The C358S/C320A mutant exhibited wild-type kinetic parameters (). Unfortunately, the double mutants C358S/C16A and C358S/C75S were expressed at substantially lower levels than the wild-type protein, and exhibited lower apparent Vmax values, even following correction for the reduced expression levels (). Creation of a cysteine-less template was therefore not pursued, and it was decided to employ the single cysteine mutant C358S as a template for further SCAM investigations because of its nearly wild-type expression levels and activity, plus insensitivity to inhibition by pCBMS.
Introduction of additional cysteine residues to test the model of NupG
Identification of C358 as the sole site involved in the inhibition of NupG by pCMBS was consistent with the prediction of the model that this position is accessible to the aqueous medium, being located on the surface of the putative nucleoside binding cavity. This finding thus supports the correctness of the alignment in the TM11 region used in model building. Similarly, the lack of involvement of the other three endogenous cysteine residues of NupG is consistent with their predicted exposure to the core of the lipid bilayer or burial at a helix-helix interface, and thus supports the correctness of the alignment in the TM1, TM3 and TM10 regions.
To confirm these conclusions, and further investigate the validity of the NupG model, cysteine residues were introduced into the C358S mutant in other regions where alignment with the LacY sequence was difficult, in place of residues predicted to be exposed on the surface of the putative ligand-binding cavity in the models both of the inward-facing and outward-facing conformations of NupG. These included TM4 residues L110 and N114, TM5 residue T140, TM7 residue Q225, TM10 residue N326 and TM11 residue N354 (). All of the mutants could be successfully expressed, and all of them exhibited uridine uptake activity. In most cases the measured kinetic parameters for transport were not more than three-fold different from those of the wild-type protein, indicative of a native or near-native conformation (). However, the activity of one mutant, Q225C/C358S, was too low for accurate determination of the Vmax and K M values. In all but the latter case the transport activity, measured over an uptake period of 2 min, differed from that of the parental C358S mutant in being susceptible to inhibition by pCMBS (), confirming the predicted accessibility of the residues on the surface of the ligand-binding cavity (). For the L110C, T140C, N326C and N354C mutants, pCMBS treatment led to essentially complete inhibition of transport, a situation resembling that seen for the wild-type protein. However, the N114C mutant was only partially inhibited (). While no significant inhibition by pCMBS of uptake was seen in the low activity Q225C/C358S mutant when transport was measured over a period of 2 min, pCMBS treatment did produce a significant decrease in the initial rate of uptake, as estimated using an uptake period of 15 sec. These rates, corrected for those seen in uninduced cells, were 4.48 ± 0.59 (n = 3) nmol/mg/min and 1.07 ± 0.41 nmol/mg/min (n = 3) before and after pCMBS treatment, respectively.
Probing the roles of conserved residues in the function of NupG
The results of investigating the accessibility of endogenous and introduced cysteine residues to modification by pCMBS supported the validity of the models of NupG created using the distantly related transporter LacY as a template. The models can therefore reasonably be used to provide testable hypotheses concerning the roles of specific residues in the translocation mechanism of NupG, either in nucleoside recognition or proton translocation. Clearly, retention of activity in most of the cysteine mutants described above indicates that none of the corresponding residues plays an essential role in nucleoside transport. In contrast the mutant Q225C/C358S, despite being expressed at a level greater than that of the wild-type protein, was severely impaired in transport activity (), suggesting that it plays an important role in transporter structure and/or function.
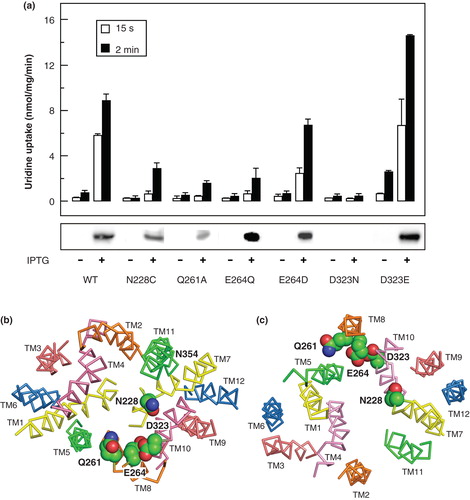
To identify other residues that might be functionally important in permeant recognition, a further set of mutants was made in the wild-type NupG protein and their transport functions investigated. Four sites were chosen for mutagenesis, in TM7 (N228), TM8 (Q261 and E264) and in TM10 (D323), because the side-chains of these residues were predicted by the models to be exposed on the surface of the putative nucleoside binding cavity () and because they are highly or absolutely conserved within the NHS family. In the case of the absolutely conserved residue D323, mutation to asparagine led to complete failure of protein expression, as revealed by Western blotting (), even when the expression period was increased from 1–3 h. Therefore, only background levels of uridine uptake were seen for this mutant. In contrast, mutation of this residue to glutamate yielded an expression level and uridine uptake activity somewhat greater than for the wild-type transporter, indicating the structural and/or functional importance of the presence of a negatively charged side-chain at this position (). Mutants N228C, Q261A and E264Q exhibited uridine uptake activities greater than those seen in uninduced cells, but substantially lower than that of the wild-type protein (), suggesting that the corresponding residues play significant roles in transport but are not individually essential for activity, despite the fact that Q261 and E264 are absolutely conserved positions in the NHS family. In the case of residue E264 in TM8, mutation to aspartate yielded a greater activity than that seen for the glutamine mutant (), consistent with a role for the negative charge of the side chain at this position. The functional importance of all four positions was confirmed by comparison of the kinetics of uridine transport by the mutants with that of the wild-type protein (). This revealed that the Vmax for transport, when corrected for the expression level, was substantially reduced (≥ 2.7-fold) in each case, while in the case of the N228C mutant, the apparent K M for transport was increased 3.4-fold.
Figure 9. Comparison of the uridine transport activity of wild-type NupG with that of variants bearing mutations of highly conserved residues in TM7, TM8 and TM9. (a) Uptake of 50 μM uridine, measured for 15 s or 2 min as indicated, in E. coli cells induced by treatment with IPTG for 1 h or, in the case of D323N, 3 h, to express WT NupG or the indicated mutants. Results shown are mean ± SD (n = 3). The panel below the histogram shows a western blot of membrane samples (20 μg) from the corresponding cultures, stained for the His-tagged NupG protein. (b) Cross-section of the transmembrane region of the inward-facing NupG model, viewed from the cytoplasmic side of the membrane, and (c) cross-section of the outward-facing NupG model, viewed from the periplasmic side of the membrane, showing the locations of the mutated residues, in space filling representation. TM helices are shown as α-carbon traces, coloured as in .
Discussion
Homologues of NupG from E. coli are found in a wide range of eubacteria, including human gut pathogens such as Salmonella typhimurium, organisms associated with periodontal disease such as Porphyromonas gingivalis and Prevotella intermedia, and plant pathogens in the genus Erwinia. In these organisms, the transporters are likely to play important roles in nucleoside scavenging from the host environment and also represent potential routes of uptake for cytotoxic nucleoside analogues that could be used for treatment of disease. Distantly related homologues have also been identified in humans and other eukaryotes, although their substrates have not yet been established (Xie et al. Citation2004). Gaining a greater understanding of the molecular mechanism of nucleoside recognition and translocation in NupG therefore has wide biological significance, and is potentially of therapeutic relevance.
The key to understanding the mechanism of membrane transporters is knowledge of their structures, ideally for each of the conformations associated with the translocation cycle. Unfortunately, no high resolution structural information is currently available for NupG, nor have structures been determined for any closely related members of the MFS of transporters. In the present study we therefore exploited bioinformatic approaches to increase the confidence with which the NupG sequence could be aligned with the sequence of the distantly related transporter LacY, allowing the crystal structure of the inward-facing conformation of the latter and, more speculatively, an outward-facing structure of LacY produced by molecular simulation, to be used as templates for homology modelling of the nucleoside transporter structure. We previously used a similar approach, involving bioinformatic analysis of the much smaller set of NHS and OHS family sequences then available, to generate a model of the inward-facing conformation of NupG based on the crystal structure of a complex of LacY with the substrate analogue β-D-galactopyranosyl-1-thio-β-D-galactopyranoside (Abramson et al. Citation2003, Holyoake et al. Citation2006). The plausibility of the resultant model was suggested by its stability during a 15-ns molecular dynamics simulation in a solvated dimyristoyl phosphatidylcholine bilayer, but the model was not tested biochemically (Holyoake et al. Citation2006). The validity of the models produced in the present study was supported by examining the accessibility of endogenous and introduced cysteine residues, predicted to be exposed on the surface of a hydrophilic nucleoside binding cavity, to the bulky water-soluble thiol reagent pCMBS. In most cases, accessibility to pCMBS, as revealed by inhibition of uridine transport, was consistent with the predictions of the model (). Apparent lack of accessibility of endogenous cysteines at positions 75, 16 and 320 is also consistent with the predictions of the models. In the case of mutant N114C, the observation that loss of activity produced by pCMBS treatment was only partial might reflect poor accessibility of this residue, located near the cytoplasmic end of TM4, to extracellular pCMBS. Alternatively, complete modification of this site by pCMBS might cause less steric hindrance to uridine binding than modification of L110C, because of the greater distance of the residue from the putative binding site. Such possibilities cannot at present be distinguished, given that modification by pCMBS cannot be directly quantified. In the case of the TM7 mutant Q225C/C358S, the low intrinsic activity of the mutant rendered assessment of accessibility to pCMBS difficult (), but the reagent was found to have a significant inhibitory effect on the initial rate of uridine uptake, consistent with the predicted exposure of the side chain on the surface of the putative nucleoside binding cavity.
The low transport activity of the Q225C/C358S mutant suggests that Q225 plays an important role in the structure and/or function of NupG. Such a role would be consistent with the finding that 76% of the 111 NupG homologues analyzed in the present study also contain glutamine at this position. The corresponding residue in LacY, D237 (), forms a salt-bridge with TM11 residue K358. While neither residue plays an essential role (mutation of both simultaneously to cysteine or alanine leads to retention of wild-type activity (Dunten et al. Citation1993)), K358 forms a hydrogen bond with the O4' hydroxyl of TDG, and D237 is likely to interact with the same atom via a water molecule (Abramson et al. Citation2003). The residue corresponding to LacY residue K358 in NupG is N354 (), but in contrast to the low activity resulting from mutation of Q225 to cysteine, mutant N354C/C358S exhibited near wild-type transport activity ( and ). This finding indicates that, like LacY residue K358, it does not play a key functional role, despite the fact that asparagine is found at this position in 75% of the NupG homologues investigated in the present study.
Additional residues predicted to be of functional importance from the models and subsequently investigated by mutagenesis were N228 in TM7, Q261 and E264 in TM8 and D323 in TM10. Although alignment in the TM7 region was difficult, that shown in suggests that the N228 position, occupied by asparagine in 71% of the NupG homologues, corresponds to the highly conserved residue D240 of LacY. The latter forms a salt-bridge with K319 and is suggested to be involved in the regulation and/or stabilization of the C-terminal salt-bridge/hydrogen bond network of LacY involved in proton translocation (Abramson et al. Citation2003), although the salt-bridge is not essential for transport activity (Sahin-Toth et al. Citation1992). An important, though not essential, functional role for N228 in NupG, is suggested by the profound effects of mutagenesis to cysteine on the transport activity (). While the nature of this role remains unclear, in both the inward- and outward-facing models the side chain of this residue is predicted to form a hydrogen bond with that of absolutely-conserved TM9 residue R294 (). The latter corresponds to LacY residue R302, which plays a key role in proton translocation but not in permeant recognition (Abramson et al. Citation2003).
Figure 10. Residues predicted to be important in the mechanism of nucleoside transport by NupG. (a) Cross-section of part of the transmembrane region of the inward-facing NupG model, viewed from the cytoplasmic side of the membrane and (b) cross-section of part of the transmembrane region of the outward-facing NupG model, viewed from the periplasmic side of the membrane. The locations of residues likely to play roles in nucleoside recognition and/or proton translocation are indicated. TM helices are shown as α-carbon traces, coloured as in . In (a) an adenosine molecule in space filling representation has been manually placed, for scale, at a position analogous to that occupied by TDG in the crystal structure of the inward-facing form of LacY.
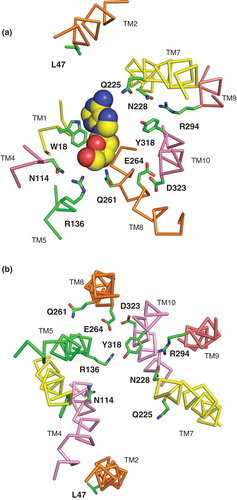
Residues Q261 and E264 in TM8 were investigated because they are completely conserved in the NHS family. In , Q261 aligns with LacY residue E269, which is proposed to play key roles in both permeant and proton translocation. In particular, it forms a salt-bridge with another essential residue, R144 in TM5, in the permeant-bound state of the transporter. The arginine residue in turn forms a bifurcated hydrogen bond with the O3 and O4 atoms of the galactopyranosyl ring. The corresponding residue in NupG, R136, is likewise absolutely conserved and may perhaps play a similar role in binding the ribose moiety of the nucleoside. The salt bridge between E269 and R144 in LacY represents a critical energetic link between the N-terminal domain of the transporter, which is largely responsible for permeant binding, and the C-terminal domain, where residues involved in proton translocation are located (Abramson et al. Citation2003). In contrast to LacY residue E269, mutation of which to any residue other than aspartate abolishes permeant binding and translocation (Abramson et al. Citation2003), mutation of NupG residue Q261 to alanine reduced the Vmax of uridine transport but did not abolish activity (). Clearly NupG residue Q261 could not act as a site of protonation analogous to LacY residue E269, nor be involved in salt bridge formation. However, such a role might be contributed by another absolutely conserved NupG residue, E264, which is located 1 helical turn away from Q261 in TM8. In support of such a role, mutation of E264 to glutamine decreased the Vmax for uridine transport more than 5-fold, a loss in activity that was partially reversed upon mutation of the residue to aspartate ( and ). Interestingly, the OHS family transporter CscB from E. coli, which is a sucrose-proton symporter, similarly lacks an ionizable residue at the position corresponding to LacY E269, but a glutamate residue at the position corresponding to E264 of NupG appears to play a role analogous to that of the LacY glutamate at position 269 (Vadyvaloo et al. Citation2006).
The role of a third absolutely conserved NupG residue, D323 in TM10, is unclear at this stage, because an asparagine mutant at this position could not be expressed, although the wild-type activity of the corresponding glutamate mutant suggests that the negative charge at this position is important (). The position aligns with residue P327 of LacY (), but it is possible that because of differences in the conformation of this helix, it is in fact equivalent to conserved position E325 of the lactose transporter. The latter plays a critical role in proton translocation, mutants bearing neutral replacements at this position lacking proton-coupled transport but retaining high-affinity permeant binding and disaccharide exchange without proton translocation (Abramson et al. Citation2003).
Conclusion
The objective of this study was to generate models of NupG that would be useful in the design and interpretation of experiments aimed at improving our understanding of nucleoside transport at the molecular level. The validity of the resultant models was established by experiment, although further improvements might be possible through an iterative cycle of experimental testing and re-modelling. Similarly, it would be beneficial in future to test the models further by molecular dynamics simulations (Holyoake et al. Citation2006). While only a small subset of conserved residues was investigated in the present study, the way is now open for further investigations of the transport mechanism, for example, to probe the roles of the absolutely conserved arginine residues at positions 136 and 294. By analogy with the corresponding residues in LacY, R144 and R302, these may be involved in nucleoside binding and proton translocation respectively (Abramson et al. Citation2003). Similarly, the potential role of TM10 residue Y318 in proton translocation could be investigated; this position is occupied by tyrosine in 75% of the NHS family members investigated in the present study and by histidine in the remainder. In LacY, the corresponding residue, H322, plays a key role in coupling proton translocation and substrate binding (Abramson et al. Citation2003). Conserved residues, including those experimentally investigated in the present study, which are predicted from the NupG model to be involved in permeant recognition and proton translocation are shown in .
Further light on the mechanism of permeant recognition may also be sought by comparative modelling of NupG homologues with differing permeant selectivities. For example, the E. coli xanthosine permease XapB, which exhibits 58% sequence identity to NupG, transports xanthosine but not guanosine, while NupG can transport guanosine but not, at least with high affinity, xanthosine (Norholm and Dandanell Citation2001). In contrast to other physiological nucleosides, xanthosine exists primarily as an anion at physiological pH values (Kulikowska et al. Citation2004), and we hypothesize that the presence of a lysine residue in XapB in place of L47 in NupG () may be responsible for the difference in selectivity. This and other hypotheses should now be addressable by mutagenesis in the light of models of NupG and its homologues.
Acknowledgements
This work was supported by a scholarship to HV from the Government of Iran, by the Wellcome Trust (ref. 019322/7/10/Z) and by the EU (FP7 grant 201924; European Drug Initiative for Channels and Transporters; EDICT). Additional support from the University of Leeds is acknowledged. JDY is an Alberta Heritage Foundation for Medical Research Senior Investigator. We thank Jean Ingram and Denise Ashworth for excellent technical assistance.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Abramson J, Smirnova I, Kasho V, Verner G, Kaback HR, Iwata S. 2003. Structure and mechanism of the lactose permease of Escherichia coli. Science 301:610–615.
- Berezin C, Glaser F, Rosenberg J, Paz I, Pupko T, Fariselli P, 2004. ConSeq: The identification of functionally and structurally important residues in protein sequences. Bioinformatics 20:1322–1324.
- Dang S, Sun L, Huang Y, Lu F, Liu Y, Gong H, 2010. Structure of a fucose transporter in an outward-open conformation. Nature 467:734–738.
- Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, 2007. MolProbity: All-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res 35:W375v383.
- Dunten RL, Sahin-Toth M, Kaback HR. 1993. Role of the charge pair aspartic acid-237-lysine-358 in the lactose permease of Escherichia coli. Biochemistry 32:3139–3145.
- Fiser A, Sali A. 2003. Modeller: Generation and refinement of homology-based protein structure models. Methods Enzymol 374:461–491.
- Hall TA. 1999. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser 41:95–98.
- Holyoake J, Caulfeild V, Baldwin SA, Sansom MSP. 2006. Modeling, docking, and simulation of the major facilitator superfamily. Biophys J 91:L84–L86.
- Jaroszewski L, Li Z, Cai XH, Weber C, Godzik A. 2011. FFAS server: Novel features and applications. Nucleic Acids Res 39:W38–W44.
- Johnson ZL, Cheong CG, Lee SY. 2012. Crystal structure of a concentrative nucleoside transporter from Vibrio cholerae at 2.4 Å. Nature 483:489–493.
- Kulikowska E, Kierdaszuk B, Shugar D. 2004. Xanthine, xanthosine and its nucleotides: Solution structures of neutral and ionic forms, and relevance to substrate properties in various enzyme systems and metabolic pathways. Acta Biochim Pol 51:493–531.
- Lomize MA, Lomize AL, Pogozheva ID, Mosberg HI. 2006. OPM: Orientations of proteins in membranes database. Bioinformatics 22:623–625.
- Mirza O, Guan L, Verner G, Iwata S, Kaback HR. 2006. Structural evidence for induced fit and a mechanism for sugar/H+ symport in LacY. EMBO J 25:1177–1183.
- Munch-Petersen A, Mygind B. 1983. Transport of nucleic acid precursors. In: Munch-Petersen A, editor. Metabolism of nucleotides, nucleosides and nucleobases in microorganisms. London: Academic Press. pp 259–305.
- Neuhard J, Nygaard P. 1987. Biosynthesis and conversion of nucleotides, purines and pyrimidines. In: Neidhardt FC, Ingraham JL, Low KB, Magasanik B, Schaechter M, Umbarger HE, editors. Escherichia coli and Salmonella typhimurium: Cellular and molecular biology. Washington DC: ASM Press. pp 445–473.
- Norholm MHH, Dandanell G. 2001. Specificity and topology of the Escherichia coli xanthosine permease, a representative of the NHS subfamily of the major facilitator superfamily. J Bacteriol 183:4900–4904.
- Pendse PY, Brooks BR, Klauda JB. 2010. Probing the periplasmic-open state of lactose permease in response to sugar binding and proton translocation. J Mol Biol 404:506–521.
- Radestock S, Forrest LR. 2011. The alternating-access mechanism of MFS transporters arises from inverted-topology repeats. J Mol Biol 407:698–715.
- Sahin-Toth M, Dunten RL, Gonzalez A, Kaback HR. 1992. Functional interactions between putative intramembrane charged residues in the lactose permease of Escherichia coli. Proc Natl Acad Sci USA 89:10547–10551.
- Sato M, Mueckler M. 1999. A conserved amino acid motif (R-X-G-R-R) in the GLUT1 glucose transporter is an important determinant of membrane topology. J Biol Chem 274:24721–24725.
- Smirnova I, Kasho V, Kaback HR. 2011. Lactose permease and the alternating access mechanism. Biochemistry 50:9684–9693.
- Stark MJR. 1987. Multicopy expression vectors carrying the lac repressor gene for regulated high-level expression of genes in Escherichia coli. Gene 51:255–267.
- Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. 1997. The CLUSTAL_X windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25:4876–4882.
- Vadyvaloo V, Smirnova IN, Kasho VN, Kaback HR. 2006. Conservation of residues involved in sugar/H+ symport by the sucrose permease of Escherichia coli relative to lactose permease. J Mol Biol 358:1051–1059.
- Vardy E, Arkin IT, Gottschalk KE, Kaback HR, Schuldiner S. 2004. Structural conservation in the major facilitator superfamily as revealed by comparative modeling. Protein Sci 13:1832–1840.
- Ward A, Sanderson NM, O'Reilly J, Rutherford NG, Poolman B, Henderson PJF. 2000. The amplified expression, identification, purification, assay and properties of histidine-tagged bacterial membrane transport proteins. In: Baldwin SA, editor. Membrane transport – a practical approach. Oxford: Oxford University Press. pp 141–166.
- Xie H, Patching SG, Gallagher MP, Litherland GJ, Brough AR, Venter H, 2004. Purification and properties of the Escherichia coli nucleoside transporter NupG, a paradigm for a major facilitator transporter sub-family. Mol Membr Biol 21:323–336.