Abstract
Transmembrane translocation of C60 fullerenes functionalized by the single amino-derivative in neutral and charged forms was studies by extensive all-atom molecular dynamics simulations. It is shown that these complexes exhibit very strong affinity to the membrane core, but their spontaneous translocation through the membrane is not possible at practical time scale. In contrast, free amino derivatives translocate through the membrane much easier than their complexes with fullerenes, but do not have pronounced affinity to the membrane interior. Our results suggest that monofunctionalized C60 could be extremely efficient membrane targeting agents, which facilitate accumulation of the water-soluble compounds in the hydrophobic core of lipid bilayer.
Introduction
Nanomedicine, a combined multidisciplinary approach using nanotechnology in biomedical studies, is becoming very popular in recent years (Caruthers et al. Citation2007, Jain Citation2009, Riehemann et al. Citation2009). A wide range of nanomaterials, such as liposomes (Gregoriadis Citation1988, Rolland Citation1993, Torchilin Citation2006), polymeric micelles (Nishiyama and Kataoka Citation2006), metallic or metal oxide nanoparticles (Shi et al. Citation2007) and carbon-based nanoparticles like functionalized nanotubes (Klumpp et al. Citation2006, Prato et al. Citation2008) and fullerenes (Brettreich and Hirsch Citation1998, Brettreich et al. Citation2000, Burghardt et al. Citation2005, Partha and Conyers Citation2009b) is used in nanomedicine mainly for therapeutic and diagnostic purposes. The fullerenes, and especially the most popular C60 cage made of 60 carbon atoms, are among the best studied nanoparticles nowadays. Their nanometer size and exceptional physical and chemical properties (Da Ros and Prato Citation1999) make them perfectly suitable for biomedical applications upon proper chemical modifications. The solubility, size, and charge of fullerene can be triggered by chemical modifications and, thus, create novel nanoparticles with specific chemical properties. For instance, C60, being hydrophobic, is poorly soluble in water (less than 0.1 ng.l-1) and displays a strong tendency for aggregation (Sivaraman et al. Citation1992, Ruoff et al. Citation1993). Various surface functionalizations (–OH, –COOH, –NH2) (Da Ros and Prato Citation1999) have been used to increase its solubility up to 34 mg.l-1 (Brettreich and Hirsch Citation1998). By changing their surface chemistry (Hirsch Citation2010), fullerene derivatives also become attractive for various biological applications, and are currently investigated as effective anti-HIV, antitumor and antimicrobial agents, as well as enzyme inhibitors (Bosi et al. Citation2003b, Bakry et al. Citation2007, Nielsen et al. Citation2008, Nakamura and Mashino Citation2009, Partha and Conyers Citation2009a). So far, it has been shown that water-soluble fullerene derivatives, namely carboxylated and hydroxylated C60 have antibiotic activity (Mashino et al. Citation1991, Bosi et al. Citation2003b, Aoshima et al. Citation2009, Nakamura and Mashino Citation2009).
Functionalization of fullerenes aimed at drug delivery application is another active field of research (Partha and Conyers Citation2009b). The hydrophobic nature of the fullerenes and their affinity to the membranes makes them promising drug carriers, which can transport polar or charged compounds into the cells. Particularly, amphiphilic fullerenes and their supramolecular complexes (‘buckysomes') are very promising vectors that can deliver drugs to specific sites (Partha et al. Citation2008). Carboxyfullerenes, functionalized with malonic acid, were shown to be effective neuroprotective antioxidants in vitro and in vivo (Dugan et al. Citation1997). The toxicity of fullerene-based nanoparticles is mostly caused by their affinity to the membranes, which may cause membrane disruption (Cusan et al. Citation2002). It is shown that monofunctionalized C60 derivatives are much less toxic than multifunctionalized compounds (Bosi et al. Citation2003a). Lower toxicity makes monofunctionalized fullerenes more attractive as drug vectors even if it comes at the cost of lower water solubility.
Although the interactions of the fullerenes with the membrane are simulated for a long time (Bedrov et al. Citation2008) only a few studies were devoted to characterize the molecular mechanisms of the cellular uptake of functionalized fullerenes. Recently, translocation of C60(OH)n from water to di-palmitoyl-phosphatidylcholine (DPPC) bilayer was studied by means of atomistic molecular dynamics (MD) simulations (Qiao et al. Citation2007, D'Rozario et al. Citation2009). It was shown that neat fullerene and its (OH)n derivatives with n < = 10 easily enter the membrane, while C60(OH)20 remains in the aqueous phase. In our previous works, we studied interactions of C60 functionalized by different number of cationic amino derivatives CCNCCOCCOCCNH3 + (referred as DR hereon, see ) with model lipid membranes (Kraszewski et al. Citation2011). Three main results issued from this work can be drawn. First, we showed that C60 appears to greatly enhance the uptake of the cationic groups by the membrane. Second, we argued that the amino derivatives should be deprotonated at the lipid headgroup level in order to fully translocate the membrane by passive diffusion. Finally, the fullerenes bearing too many cationic groups display mostly a hydrophilic character; thus, the lipophilic fullerene core is not anymore effective as an insertion enhancer.
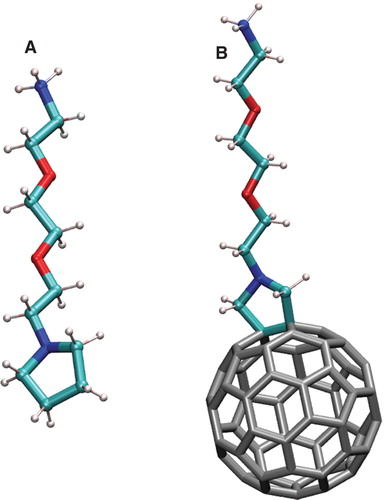
Figure 1. (A) Schematic representation of the CCNCCOCCOCCNH3 + group (amino derivative DR+). (B) The C60 fullerene functionalized by DR+ group. This Figure is reproduced in colour in Molecular Membrane Biology online.
Despite these efforts there are no reliable estimates of the rate of transmembrane translocation of functionalized fullerenes. Particularly, the energy profiles of such translocation were computed in our previous works on the semi-quantitative level only (Kraszewski et al. Citation2011) and needs to be improved. The influence of the charge of the functionalization groups on the fullerenes translocation was not studied systematically. However, this could have a high importance for their clinical and biomedical applications.
In this work, we performed a more comprehensive study of transmembrane translocation of the C60 fullerene functionalized by the single DR amino derivative, in order to reveal the role of each partners (the ‘drug' and the ‘vector') at the membrane interface. We consider translocation of free DR molecules in neutral (DR) and cationic (DR+) forms as well as respective C60-DR and C60-DR+ functionalized fullerenes. These compounds can serve as simple models of neutral and charged cargo molecules used in biomedical applications. It can also reveal the precise role of protonation/deprotonation mechanism at the membrane interface. This work aims at obtaining the energy profiles of translocation of C60-DR and C60-DR+ molecules through the model membrane, estimating their translocation rates and revealing an influence of functionalization charge on these parameters.
Methods
All MD simulations were performed in NAMD software version 2.9 (Phillips et al. Citation2005) using CHARMM 27 force field (MacKerell et al. Citation1998). A switching scheme for Van-der-Waals interactions was used with the switching distance of 8 Å and the cut-off distance of 11 Å. Electrostatic interactions were computed explicitly within the cut-off distance of 11 Å. For longer distances, the PME method was used (Darden et al. Citation1993). All simulations were performed in the NPT ensemble at 300 K using Langevin dynamics with damping coefficient of 0.1 ps and the Nosé-Hoover barostat with a period of 200 fs and a decay time of 100 fs. An integration time step of 2 fs was used.
The model bilayer consisted of 72 POPC lipids (36 in each leaflet) solvated by 3670 water molecules. This corresponds to ∼ 51 water molecules per lipid, which is enough for full hydration of the studied bilayer. Pre-equilibrated bilayer from our previous work (Kraszewski et al. Citation2011) was used.
The force field for C60 fullerene and the cationic amino derivative DR were developed in our previous papers (Kraszewski et al. Citation2011) (). The neutral form of DR was obtained by modifying atomic charges of the NH3 aminogroup in order to make it neutral without explicitly removing the proton.
The umbrella sampling technique was used to compute the potentials of mean force (PMFs) of translocating C60-DR and C60-DR+ complexes as well as DR and DR+ through the membrane. This method belongs to the family of ‘non-equilibrium MD' techniques, which allow observing the phenomena, which do not happen spontaneously on the time scale of simulation. Particularly in our case an external biasing potential is applied to keep the fullerene in particular positions inside the membrane. The harmonic biasing potential with the force constant of 2 kcal/mol was applied to the center of mass of C60 in C60-DR and C60-DR+ complexes and to the center of masses of the molecule in the case of DR and DR+. The biasing potential was applied only in Z dimension, i.e., the direction perpendicular to the membrane plane and centered at discrete points distributed along Z axis in 1 Å intervals. This produced 43 umbrella sampling windows spanning from the center of bilayer (Z = 0 Å) to bulk water through the upper membrane leaflet (Z > 40 Å). The PMFs were computed using the weighted histogram technique implemented in WHAM software (http://membrane.urmc.rochester.edu/content/wham).
The diffusion coefficients of the permeants in Z direction were computed as
where ΔF(z,t) is the difference between the instantaneous force and mean force ![]() acting on the permeant. Diffusion coefficients were computed in several selected umbrella sampling windows where the sampling was extended to 20 ns.
acting on the permeant. Diffusion coefficients were computed in several selected umbrella sampling windows where the sampling was extended to 20 ns.
Results
The PMFs for all four studied systems are shown in . In the case of free DR/DR+ molecules, the shape of the energy profiles depends strongly on the charge state of compound. Neutral DR molecules exhibit a strong affinity for the hydrophobic core of the membrane. There, a wide energy well located in the region of the hydrocarbon tails of the lipids occurs with the maximal depth of 6.1 kcal/mol. In contrast, charged DR+ molecules exhibit energy well of 7.3 kcal/mol in the region of the lipid carbonyl groups and display a large energy barrier in the center of bilayer. The height of this barrier respectively to the energy well was 14.2 kcal/mol. These results could be explained easily taking into account the amphiphilic nature of the charged DR+ molecule. The pyrrole group on one end of the molecule was hydrophobic while charged protonated aminogroup NH3 + on the other end was strongly hydrophilic. As a result, the translocation of DR+ through the hydrophobic core of bilayer implied moving charged aminogroup into unfavorable environment, which caused the appearance of a large energy barrier. In the case of neutral DR form, the small polar NH2 group does not produce any significant energy barrier during the translocation. As a result, almost flat energy well is formed in the center of bilayer.
Figure 2. Potentials of mean force for transmembrane translocation of C60 functionalized by amino derivative in charged (C60-DR+) and neutral (C60-DR) states and free amino derivative in charged (DR+) and neutral (DR) states. This Figure is reproduced in colour in Molecular Membrane Biology online.
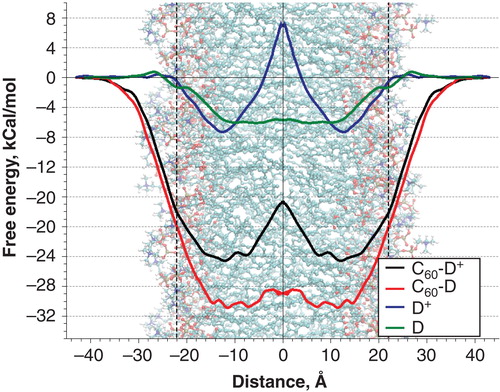
The PMFs of C60-DR and C60-DR+ complexes show that the fullerene leads to dramatic increase of the energy well depth in the center of bilayer. However, the overall shape of the PMFs for functionalized fullerenes follow the shape of the curves for corresponding free DR and DR+ groups in the hydrophobic regions of the membrane. In the case of neutral C60-DR the bottom of the energy well was rather rugged but the largest energy barriers did not exceed 3 kcal/mol. Maximal depth of the energy well reached 31 kcal/mol. In the case of charged C60-DR+ the maximal depth of the energy well was much smaller (24.6 kcal/mol) while there was a pronounced barrier in the center of bilayer with the height of 7.9 kcal/mol (measured from the bottom of the energy well).
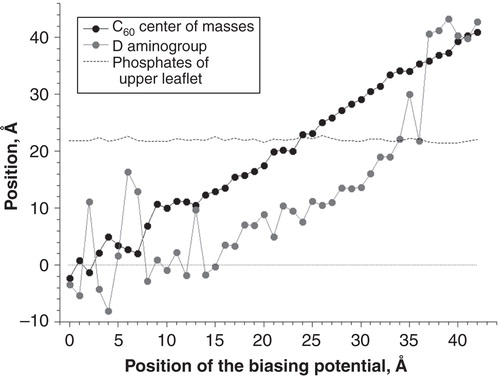
Since the DR group was rather extended and can span the distance from the membrane center to the lipid head groups easily, it was interesting to know the orientation of DR and DR+ groups at different positions of C60 inside the membrane. and show average positions of the centers of mass of C60 and the aminogroup of DR+ and DR in the course of umbrella sampling simulations.
Figure 3. Positions of C60 and the aminogroup of DR+ in different umbrella sampling windows. Average positions of the phosphate groups of the upper membrane leaflet are shown.
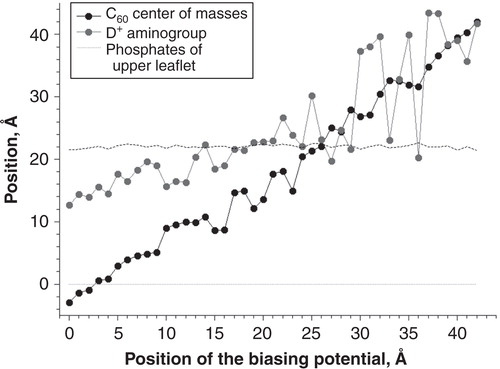
Figure 4. Positions of C60 and the aminogroup of DR in different umbrella sampling windows. Average positions of the phosphate groups of the upper membrane leaflet are shown.
In the case of C60-DR+ () the position of charged NH3 + group fluctuated randomly until the fullerene moves from the bulk water towards the membrane. When C60 reached the level of lipid phosphate groups (Z = 22 Å) the NH3 + group of DR+ binds to lipid heads. During subsequent motion of C60 toward the bilayer center DR+ was oriented outwards toward the polar lipid head groups.
In the case of C60-DR (), the position of neutral NH2 group also fluctuated randomly in bulk water. However, it binds to the lipid head groups early at Z = 36 Å while C60 was still located in bulk solvent. Subsequent motion of C60 toward the membrane was accompanied by simultaneous penetration of NH2 group deeper into the membrane core. When NH2 reached the center of bilayer at Z = 0 Å it began to fluctuate randomly within the region of lipid hydrocarbon tails.
It is necessary to note that the functional group may change its orientation in the membrane by spontaneous flipping between ‘inward' and ‘outward' states. Only few such transitions were observed in several umbrella sampling windows during our simulations. Thus the characteristic time of such flipping was larger than the simulation time (∼v 20 ns) and likely to be of the order of hundreds of nanoseconds. Precise estimate of the flipping time requires much longer dedicated simulations. It was shown that the equilibrium in respect to orientation of extended molecule in the lipid membrane was unlikely to be reachable in all-atom simulations providing limited computational power and time (Yesylevskyy et al. Citation2009). In principle, accounting flipping may increase the height of the central energy barrier for charged moieties C60-DR+ and DR+, but the qualitative picture of the translocation and the depths of the energy wells were unlikely to change.
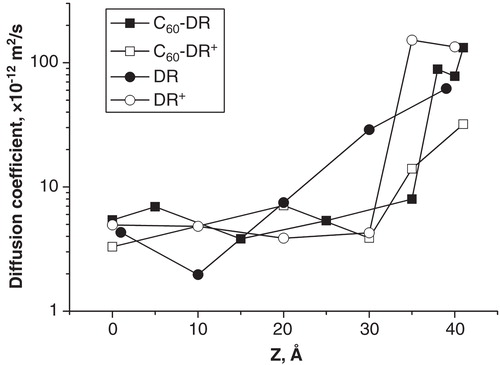
The diffusion coefficients of all studied species are shown in . In the hydrophobic region of bilayer (Z = 0..20 Å) all diffusion coefficients are surprisingly similar and fall into the range 3·10-12 to 7·10-12 m2/s. It is remarkable that large and bulky C60 particles and small DR and DR+ molecules have almost identical diffusion coefficients in this region. In the region of the lipid head groups (Z = 20..30 Å) the diffusion coefficient of DR increases up to 3·10-11 m2/s while the diffusion coefficients of all other species remain the same as in the hydrophobic part of the membrane. An abrupt increase of diffusion coefficients for C60-DR+ and DR occurs in the headgroup-water interface at Z = 35 Å and Z = 30 Å, respectively. In contrast, the diffusion coefficients of C60-DR and DR+ show gradual increase when moving toward the water phase. Thus there was no visible correlation between the charge and size of the permeating molecule and the behavior of its diffusion coefficient in our simulations.
Figure 5. Diffusion coefficients of the studied compounds in selected umbrella sampling windows.
The permeability coefficient P is usually used to characterize the transmembrane diffusion of various compounds (Marrink and Berendsen Citation1994, Citation1996). In the case of MD simulations the solubility-diffusion model (Marrink and Berendsen Citation1994, Citation1996) is usually applied to compute P. This model presumes that the diffusion coefficient changes smoothly across the membrane from one bulk solution to the other and leads to the following expression for P
where d is the effective membrane thickness, R(z) is the membrane resistance computed as
where R is the gas constant, T is the absolute temperature.
The solubility-diffusion model works very well for small hydrophilic moieties like ions, water or small charged molecules (Marrink and Berendsen Citation1994, Citation1996). The membrane imposes significant free energy barrier for such molecules, which means that the resistance R could be considered negligible in water phase. Thus the integral in Equation 1 converges to well-defined value, which is insensitive to the choice of effective membrane thickness d. However, the solubility-diffusion model fails to give reliable results for hydrophobic molecules, particularly for fullerenes and their derivatives (Fiedler and Violi Citation2010). For such moieties the resistance inside the membrane is smaller than in the water phase and the integral in Equation (1) becomes divergent and highly sensitive to the integration limit d (Fiedler and Violi Citation2010). Another difficulty comes from the very steep change of the diffusion coefficient at the water-membrane interface, which make the solubility-diffusion approach not applicable in this region.
In our case the solubility-diffusion model could be applied only in the hydrophobic region of the membrane (Z = 0..30 Å), where the diffusion coefficient changes smoothly for all permeants. The following permeability coefficients were obtained in this region: C60-DR+ – 5575 m/s, C60-DR – 45459 m/s, DR+ – 5.8·10-5, DR – 1.65 m/s. These permeability values reflect only the mobility of the permeants inside the membrane. High energy barriers, which should be surmounted in order to escape from the membrane interior, are not taken into account due to the limitations of the solubility-diffusion model discussed above. Thus, in order to estimate realistic relative permeability for our compounds, another approach was needed.
The problem of diffusion in highly inhomogeneous media with rapidly changing diffusion coefficient was addressed recently in several theoretical works. Particularly Monte-Carlo simulations with fixed steplength were shown to be a promising computational method for such systems (Ruiz Barlett et al. Citation2013). In contrast to the solubility-diffusion model, this approach can give convergent values of permeability despite abrupt changes of diffusion coefficient at the membrane-water interface. Unfortunately this technique does not lead to simple analytical formulas and requires numerical solution of the stochastic equations of motion with specific treatment of the microscopic transition probabilities, thus its application is beyond the scope of this work.
It is evident from and that the energy wells in the PMFs are very high and the diffusion coefficients of all compounds are of the same order of magnitude up to the water-membrane interface. This means that the translocation rates of all studied compounds are limited by the highest energy barrier H, which is present on the translocation pathway. This allows using Arrhenius reaction rate theory as a reasonable approximation. According to this approximation the characteristic translocation time is
where A is an Arrhenius prefactor, kB is the Boltzmann constant, T is the absolute temperature. The choice of the Arrhenius prefactor is the most challenging problem. In our case, A is proportional to diffusion coefficients of particular permeants, which are all of the same order of magnitude inside the membrane (). Thus we can assume that Arrhenius prefactors for all studied compounds are also of the same order of magnitude. This allows us obtaining the ratio of characteristic translocation times as:
where H are the highest energy barriers on the translocation pathway for corresponding compound. The following values of H were obtained: 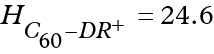 Kcal/mol,
Kcal/mol, 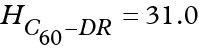 Kcal/mol,
Kcal/mol, ![]() Kcal/mol,
Kcal/mol, ![]() Kcal/mol. Corresponding ratio of the translocation times is:
Kcal/mol. Corresponding ratio of the translocation times is:
These numbers can vary by several orders of magnitude due to various factors, which are neglected in very rough Arrhenius approximation, however the differences between them are so large that the qualitative figure will likely to be captured correctly.
Discussion
In this work, we obtained the PMFs of translocation of mono-functionalized fullerenes as well as free functional groups through the membrane. This allows estimating efficiency of the fullerenes as the drug delivery vectors or as targeting agent for the lipid bilayers. The ratio of the characteristic translocation times of the studied compounds is
It is clearly seen that both neutral (D) and charged (D+) functional groups cross the membrane much easier than the corresponding complexes with fullerenes. Neutral DR has the translocation time, which is three orders of magnitude smaller than the charged DR+. Complexes with the fullerenes have so high affinity to the hydrophobic membrane core that they are likely to stay inside the membrane forever on the physiological time scale (translocation times are 108–1010 times larger than for DR). The probability of spontaneous escape from the membrane core for C60-DR and C60-DR+ are so small, that the fullerenes functionalized by single charged or polar group cannot be viewed as conventional drug delivery vectors, which simplify membrane penetration by water-soluble compounds. They appear to be very efficient membrane targeting agents instead, which accumulate in the membrane hydrocarbon region and pull water soluble cargo into the membrane core.
It is necessary to note that accumulation of the fullerenes in the membrane core may destabilize the membrane and lead to formation of the pores or other types of local defects. It was shown in experiments with pristine fullerenes that such defects may allow passage of water and redistribution of the lipids between the leaflets of the membrane (Prylutska et al. Citation2012). Investigation of such effects for our monofunctionalized fullerene derivatives is beyond the scope of this work because it requires study of multiple aggregating fullerenes. Meanwhile, no significant deformation of the membrane was observed for single fullerene in our simulations.
Another interesting feature concerns the protonation/deprotonation of the aminoderivative, which can occur at the region of lipid head group. Indeed, from an energetic point of view, the energy profiles given in reveal that it would be favorable for DR+ to deprotonate in order to cross the membrane. This may occur at the entry of the first membrane leaflet at the lipid head group region. The possibility of such deprotonation event is governed by the relative position of ![]() with regard to pH. Hence, a change of the environment of aminogroup can modify the equilibrium concentrations [DR+] and [DR] in favor to either charged or neutral form. pKa
of the amino derivative DR+ is not known experimentally. However, one can consider the monoethanolammmonium ion (HO(CH2)2NH3
+) as a reasonable model for this cationic group. The monoethanolammmonium ion dissociation constant in water has a defined experimentally pKa
value of 9.50 (Hall Citation1957). This pKa
value is large when compared to pH = 7 and explains why only the charged HO(CH2)2NH3
+ (and though DR+) should be considered outside the membrane. To the best of our knowledge, there is also no experimental value of pKa
for the POPC molecule. Since DR+ was observed to display a large energy well at the lipid carbonyl groups, we investigated the possibility of deprotonation at this specific location only. As a first approximation, we consider here that phosphocholine is a good representation of the POPC head group. We use here a predicted value of 6.18 for phosphocholine molecule (see supplementary material in Kraszewski et al. Citation2011). The resulting pKa
for such chemical equilibrium is given by the difference of pKa
of monoethanolammmonium and phosphocholine, namely 9.50–6.18 and hence equals to 3.32. It indicates a substantial negative shift of the pKa
between aqueous solvent and the lipid head group region and consequently that the DR form will dominate and deprotonation will be favored in the lipid head groups region. As a result, the amino derivative would have to ‘jump' from the energy profile for DR+ to the profile of DR during its translocation across the membrane but the validation of such mechanism requires additional dedicated studies.
with regard to pH. Hence, a change of the environment of aminogroup can modify the equilibrium concentrations [DR+] and [DR] in favor to either charged or neutral form. pKa
of the amino derivative DR+ is not known experimentally. However, one can consider the monoethanolammmonium ion (HO(CH2)2NH3
+) as a reasonable model for this cationic group. The monoethanolammmonium ion dissociation constant in water has a defined experimentally pKa
value of 9.50 (Hall Citation1957). This pKa
value is large when compared to pH = 7 and explains why only the charged HO(CH2)2NH3
+ (and though DR+) should be considered outside the membrane. To the best of our knowledge, there is also no experimental value of pKa
for the POPC molecule. Since DR+ was observed to display a large energy well at the lipid carbonyl groups, we investigated the possibility of deprotonation at this specific location only. As a first approximation, we consider here that phosphocholine is a good representation of the POPC head group. We use here a predicted value of 6.18 for phosphocholine molecule (see supplementary material in Kraszewski et al. Citation2011). The resulting pKa
for such chemical equilibrium is given by the difference of pKa
of monoethanolammmonium and phosphocholine, namely 9.50–6.18 and hence equals to 3.32. It indicates a substantial negative shift of the pKa
between aqueous solvent and the lipid head group region and consequently that the DR form will dominate and deprotonation will be favored in the lipid head groups region. As a result, the amino derivative would have to ‘jump' from the energy profile for DR+ to the profile of DR during its translocation across the membrane but the validation of such mechanism requires additional dedicated studies.
Finally, it is necessary to compare obtained PMFs with those calculated previously using the Adaptative Biasing Forces (ABF) approach (Kraszewski et al. Citation2011). Although the shapes of the corresponding energy profiles for DR, DR+, C60-DR and C60-DR+ look quite different in the present study and in our previous work (Kraszewski et al. Citation2011), the same qualitative results are obtained. Following the results from both studies, C60 increases the membrane affinity for single amino-derivative either in their neutral or charged forms. The differences in locations of the energy minima and their corresponding depths are likely to appear due to the different choices of the reference groups. Using ABF method obtained energy profile was referred to the center of mass of C60 while in the current umbrella sampling simulations the energy profile applies to the center of the whole C60-DR or C60-DR+. This leads to an apparent systematic shift of the energy well position associated with the length and conformation of the functional group. An influence on the absolute values of the free energies appears to be non-trivial and rather hard to interpret. We can, however, speculate that the flipping of C60-DR/C60-DR+ molecule between ‘inward' to ‘outward' orientations modifies the local values of free energy. Particularly, there is an 8 Kcal/mol energy barrier for C60-DR at the lipid-water interface in ABF simulations, which is absent in the umbrella sampling simulations. Another probable reason for this discrepancy could be an undersampling in our previous ABF simulations in the regions, where diffusion coefficient changes significantly. Thus the PMFs computed in the present study should be considered as more precise and more reliable than reported previously.
Conclusions
The potentials of means force for transmembrane translocation of C60 fullerene functionalized by the single amino-derivative in neutral and charged forms were computed. It is shown that these complexes exhibit strong affinity to the membrane core, which makes their translocation through the membrane impossible at practical time scale. Free amino derivatives translocate through the membrane much easier than their complexes with fullerenes, which suggests that monofunctionalized C60 cannot be used as a drug delivery vector, which penetrates the membranes spontaneously. However monofunctionalized C60 could be extremely efficient membrane targeting agents, which facilitate accumulation of water-soluble compounds in the hydrophobic core of lipid bilayer.
Acknowledgments
Computations have been performed on the supercomputer facilities of the Mésocentre de calcul de Franche-Comté. S.O. Yesylevskyy thanks the Universite de Franche-Comté for providing the Visiting Professor grant, which stimulated this collaboration.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Aoshima H, Kokubo K, Shirakawa S, Ito M, Yamana S, Oshima T. 2009. Antimicrobial activity of fullerenes and their hydroxylated derivatives. Biocontrol Sci 14(2):69–72.
- Bakry R, Vallant RM, Najam-ul-Haq M, Rainer M, Szabo Z, Huck CW, et al. 2007. Medicinal applications of fullerenes. Int J Nanomed 2:639–649.
- Bedrov D, Smith GD, Davande H, Li L. 2008. Passive transport of C60 fullerenes into lipid membrane. A molecular dynamics simulation study. J Phys Chem B 112:2078–2084.
- Bosi S, Da Ros T, Spalluto G, Balzarini J, Prato M. 2003a. Synthesis and anti-HIV properties of new water-soluble bis-functionalized 60 fullerene derivatives. Bioorg Med Chem Lett 13(24):4437–4440.
- Bosi S, Da Ros T, Spalluto G, Prato M. 2003b. Fullerenes derivatives: An attractive tool for biological applications. Eur J Med Chem 38:913–923.
- Brettreich M, Burghardt S, Bottcher C, Bayerl T, Bayerl S, Hirsch A. 2000. Globular amphiphiles: Membrane-forming hexaadducts of C-60. Angew Chem Int Ed 39(10):1845–1848.
- Brettreich M, Hirsch A. 1998. A highly water-soluble dendro 60 fullerene. Tetrahedron Lett 39(18):2731–2734.
- Burghardt S, Hirsch A, Schade B, Ludwig K, Bottcher C. 2005. Switchable supramolecular organization of structurally defined micelles based on an amphiphilic fullerene. Angew Chem Int Ed 44(19):2976–2979.
- Caruthers SD, Wickline SA, Lanza GM. 2007. Nanotechnological applications in medicine. Curr Opin Biotechnol 18(1):26–30.
- Cusan C, Da Ros T, Spalluto G, Foley S, Janto JM, Seta P, et al. 2002. A new multi-charged C-60 derivative: Synthesis and biological properties. Eur J Org Chem 17:2928–2934.
- D'Rozario RSG, Wee CL, Wallace EJ, Sansom MSP. 2009. The interaction of C-60 and its derivatives with a lipid bilayer via molecular dynamics simulations. Nanotechnology 20(11):115102–115107.
- Da Ros T, Prato M. 1999. Medicinal chemistry with fullerenes and fullerene derivatives. Chem Commun 8:663–669.
- Darden T, York D, Pedersen L. 1993. Particle mesh Ewald: An N.log(N) method for Ewald sums in large systems. J Chem Phys 98(12):10089–10092.
- Dugan LL, Turetsky DM, Du C, Lobner D, Wheeler M, Almli CR, et al. 1997. Carboxyfullerenes as neuroprotective agents. Proc Natl Acad Sci USA 94(17):9434–9439.
- Fiedler SL, Violi A. 2010. Simulation of nanoparticle permeation through a lipid membrane. Biophys J 99(1):144–152.
- Gregoriadis G. 1988. Liposomes as drug carriers: Recent trends and progress. New York: John Wiley & Sons.
- Hall HK. 1957. Correlation of the base strengths of amines. J Am Chem Soc 79(20):5441–5444.
- Hirsch A. 2010. The era of carbon allotropes. Nat Mat 9:868–871.
- Jain KK. 2009. Role of nanobiotechnology in the development of personalized medicine. Nanomedicine 4(3):249–252.
- Klumpp C, Kostarelos K, Prato M, Bianco A. 2006. Functionalized carbon nanotubes as emerging nanovectors for the delivery of therapeutics. Biochim Biophys Acta Biomembr 1758(3):404–412.
- Kraszewski S, Tarek M, Ramseyer C. 2011. Uptake and translocation mechanisms of cationic amino derivatives functionalized on pristine C60 by lipid membranes: A molecular dynamics simulation study. ACS Nano 5(11):8571–8578.
- MacKerell AD, Bashford D, Bellott Dunbrack RL, Evanseck JD, Field MJ, et al. 1998. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B 102:3586–3616.
- Marrink SJ, Berendsen HJC. 1994. Simulation of water transport through a lipid membrane. J Phys Chem 98:4155–4168.
- Marrink SJ, Berendsen HJC. 1996. Permeation process of small molecules across lipid membranes studied by molecular dynamics simulations. J Phys Chem 100:16729–16738.
- Mashino T, Okuda K, Hirota T, Hirobe M. 1991. Inhibition of E. coli growth by fullerene derivatives and inhibition mechanism. Bioorg Med Chem Lett 9:2959–2962.
- Nakamura S, Mashino T. 2009. Biological activities of water- soluble fullerene derivatives. J Phys: Conf Ser 159:012003.
- Nielsen GD, Roursgaard M, Jensen KA, Poulsen SS, Larsen ST. 2008. In vivo biology and toxicology of fullerenes and their derivatives. Basic Clin Pharmacol Toxicol 103:197–208.
- Nishiyama N, Kataoka K. 2006. Current state, achievements, and future prospects of polymeric micelles as nanocarriers for drug and gene delivery. Pharmacol Ther 112(3):630–648.
- Partha R, Conyers JL. 2009a. Biomedical applications of functionalized fullerene-based nanomaterials. Int J Nanomed 4:261–275.
- Partha R, Conyers JL. 2009b. Biomedical applications of functionalized fullerene-based nanomaterials. Int J Nanomed 4:261–275.
- Partha R, Mitchell LR, Lyon JL, Joshi PP, Conyers JL. 2008. Buckysomes: Fullerene-based nanocarriers for hydrophobic molecule delivery. ACS Nano 2(9):1950–1958.
- Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, et al. 2005. Scalable molecular dynamics with NAMD. J Comput Chem 26(16):1781–1802.
- Prato M, Kostarelos K, Bianco A. 2008. Functionalized carbon nanotubes in drug design and discovery. Acc Chem Res 41(1):60–68.
- Prylutska S, Bilyy R, Overchuk M, Bychko A, Andreichenko K, Stoika R, et al. 2012. Water-soluble pristine fullerenes C60 increase the specific conductivity and capacity of lipid model membrane and form the channels in cellular plasma Membrane. J Biomed Nanotechnol 8(3):522–527.
- Qiao R, Roberts AP, Mount AS, Klaine SJ, Ke PC. 2007. Translocation of C-60 and its derivatives across a lipid bilayer. Nano Lett 7(3):614–619.
- Riehemann K, Schneider SW, Luger TA, Godin B, Ferrari M, Fuchs H. 2009. Nanomedicine – challenge and perspectives. Angew Chem Int Ed 48(5):872–897.
- Rolland A. 1993. Pharmaceutical particulate carriers. New York: Marcel Dekker.
- Ruiz Barlett V, Hoyuelos M, Mártin HO. 2013. Monte Carlo simulation with fixed steplength for diffusion processes in nonhomogeneous media. J Computat Phys 239(0):51–56.
- Ruoff RS, Tse DS, Malhotra R, Lorents DC. 1993. Solubility of C60 in a variety of solvents. J Phys Chem 97(13):3379–3383.
- Shi XY, Thomas TP, Myc LA, Kotlyar A, Baker JR. 2007. Synthesis, characterization, and intracellular uptake of carboxyl-terminated poly(amidoamine) dendrimer-stabilized iron oxide nanoparticles. Phys Chem Chem Phys 9(42):5712–5720.
- Sivaraman N, Dhamodaran R, Kaliappan I, Srinivasan TG, Rao PRV, Mathews CK. 1992. Solubility of C60 in organic solvents. J Org Chem 57(22):6077–6079.
- Torchilin VP. 2006. Multifunctional nanocarriers. Adv Drug Delivery Rev 58(14):1532–1555.
- Yesylevskyy SO, Marrink S-J, Mark AE. 2009. Alternative mechanisms for the interaction of the cell-penetrating peptides penetratin and the TAT peptide with lipid bilayers. Biophys J 97(1):40–49.