Abstract
The Klotho gene was identified as an ‘aging suppressor' in mice. Overexpression of the Klotho gene extends lifespan and defective Klotho results in rapid aging and early death. Both the membrane and secreted forms of Klotho have biological activity that include regulatory effects on general metabolism and a more specific effect on mineral metabolism that correlates with its effect on aging. Klotho serves as a co-receptor for fibroblast growth factor (FGF), but it also functions as a humoral factor that regulates cell survival and proliferation, vitamin D metabolism, and calcium and phosphate homeostasis and may serve as a potential tumor suppressor. Moreover, Klotho protects against several pathogenic processes in a FGF23-independent manner. These processes include cancer metastasis, vascular calcification, and renal fibrosis. This review covers the recent advances in Klotho research and discusses novel Klotho-dependent mechanisms that are clinically relevant in aging and age-related diseases.
Introduction
Aging is a complex phenomenon resulting from the interaction between multiple genetic and environmental factors (Tsujikawa et al. Citation2003, Razzaque et al. Citation2006). The complex relationship between aging-related genes remains poorly understood. Aging is associated with increased incidence of diabetes, hypertension, and chronic kidney disease (CKD) with impairment of endothelial function (Rakugi et al. Citation2007, Maekawa et al. Citation2009) and increased risk of cardiovascular morbidity and mortality (Semba et al. Citation2011a).
Klotho is a novel anti-aging gene encoding a protein with multiple pleiotropic effects, fortuitously discovered in 1997 by Kuro-o and colleagues, it was named after one of the three Fates in Greek mythology, the goddess who spins the thread of life (Kuro-o M et al. Citation1997). The Klotho family of proteins consists of three members: αKlotho, βKlotho and γKlotho. All three are single-pass transmembrane proteins (Kuro-o M et al. Citation1997). The founder Klotho is designated αKlotho to distinguish it from the other two members.
A defect in mouse Klotho gene expression leads to a syndrome resembling aging, including substantially decreased lifespan, organ atrophy, infertility, vascular calcification, arteriosclerosis, mitral annular calcification, osteomalacia, hypercalcemia, hyperphosphatemia, osteoporosis, elevated circulating vitamin D hormone, increased peripheral insulin sensitivity and emphysema (Kuro-o M et al. Citation1997, Utsugi et al. Citation2000, Yoshida et al. Citation2002, Manya et al. Citation2010), whereas overexpression of the gene in mice extends lifespan (Kurosu et al. Citation2005).
The αKlotho gene is composed of five exons, in humans, mice and rats (Wang and Sun Citation2009a) expressed predominantly in the distal convoluted tubule (DCT) cells in addition to a lesser expression in the proximal convoluted tubule (PCT) cells of the kidney (Kuro-o M et al. Citation1997, Hu et al. Citation2010b) and in the choroid plexus epithelial cells of the brain (John et al. Citation2011, Zacchia and Capasso Citation2011). In addition, but to a lesser extent, αKlotho is also expressed in the pituitary gland, placenta, skeletal muscle, urinary bladder, aorta, pancreas, testis, ovary, colon and thyroid gland (Kuro-o M et al. Citation1997, Manya et al. Citation2010, Kuro Citation2012). Human αKlotho gene (hKL) encodes a single-pass transmembrane protein consisting of 1012 amino acids, whereas mouse Klotho (mKL) consists of 1014 amino acids (Kuro-o M et al. Citation1997, Tsujikawa et al. Citation2003). Klotho is composed of three domains with a molecular weight of about 130 kDa: A large extracellular domain with two internal repeats (KL1 and KL2), a transmembrane domain; and a very short intracellular domain with 10 amino acids (). Klotho protein exists in two forms: A membrane and a secreted form (Kuro-o M et al. Citation1997, Yamamoto et al. Citation2005). Soluble (secreted) Klotho can arise either by proteolytic cleavage of the Klotho extracellular domain just above the plasma membrane of the transmembrane form operated by the membrane-anchored proteases ADAM10 and ADAM17, two members of the ADAM (A Disintegrin and Metalloproteinase) family of peptidase proteins (Chen et al. Citation2007), or as a result of alternative mRNA splicing (isoform 70 kDa) of increased Klotho gene transcription (Matsumura et al. Citation1998). Secreted Klotho protein is then released from the cell into the extracellular space and is detectable in blood, urine, and cerebrospinal fluid (CSF) (Kuro-o M et al. Citation1997, Yamamoto et al. Citation2005) as well as possibly functioning as an endocrine factor targeting distant organs (). The specific role of ectodomain shedding and alternative splicing to the secreted Klotho protein still remains uncertain. Nonetheless, recent studies have revealed multiple biological functions for membrane and secreted Klotho, these being of importance in several physiology and pathological conditions.
Figure 1. Schematic diagram showing the domain structure, membrane topology and main functions of membrane and secreted Klotho(Jones et al. Citation2012, Kuro Citation2012). (A)Human Klotho protein, composed of 1012 amino acids in length, possesses a putative signal sequence (SS) at its N-terminus, two internal repeats (KL1 and KL2) of the extracellular domain that shares sequence homology to the β-glucosidase, linker (L), and a putative transmembrane domain (TM) with a short cytoplasmic domain (CD) at the C-terminus (Not drawn to scale). (B)The transmembrane Klotho forms a complex with the FGFR to create a co-receptor binding site for FGF23 (1), and plays an essential role in renal regulation of phosphate and vitamin D metabolism. Secreted Klotho is created by α- and β-secretases ectodomain shedding of membrane Klotho (2) and released into urine, blood, or cerebrospinal fluid (3).The FGF23/Klotho complex in turn reduces circulating calcitriol (1,25(OH)2D3) levels, either by inhibiting the expression levels of enzyme (1-α-hydroxylase (Cyp27b1)) that participates in calcitriol synthesis(4) or by inducing of another enzyme 24-hydroxylase (Cyp24a1) (5) which catalyze the catabolism of calcitriol into inactive form of calcitroic acid (6). Additionally, FGF23/Klotho complex decreases sodium phosphate cotransporters cell membrane expression, either by downregulating their gene expression (7) or by inducing cell membrane protein internalization (8) resulting in their degradation. See text for details. This Figure is to be reproduced in colour in the online version of Molecular Membrane Biology.
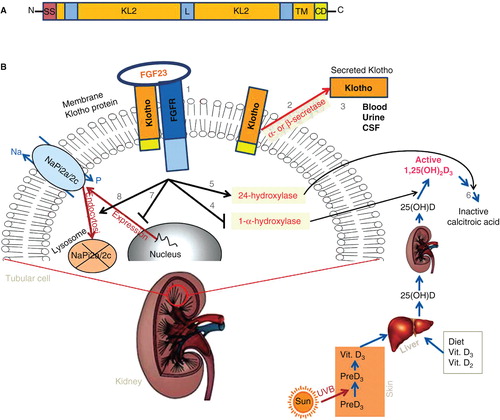
Functions of membrane and secreted Klotho
Studies have shown that the membrane and secreted Klotho have distinct functions (Chen et al. Citation2007, Cha et al. Citation2008, Alexander et al. Citation2009, Camilli et al. Citation2011, Dermaku-Sopjani et al. Citation2011, Hu et al. Citation2011, Kuro Citation2011, Andrukhova et al. Citation2012, Anour et al. Citation2012). The systemic effects of Klotho protein appear to be primarily due to its circulating form of secreted soluble Klotho protein, which operates as a humoral factor (Chang et al. Citation2005) – potentially through binding to its putative, yet still unknown, cell-surface receptor – and as an enzyme regulating the biological activity of several cell surface glycoproteins and receptors in multiple tissues (Chang et al. Citation2005, Cha et al. Citation2008, Hu et al. Citation2010a, Sopjani et al. Citation2011, Tang et al. Citation2011).
The circulating form of Klotho protein in a FGF independent mode regulates, by means of deglycosylation, the activity and membrane abundance of the epithelial calcium (Ca2+) channel, the transient receptor potential vanilloid subtype 5 (TRPV5) in distal tubules of kidney (Chang et al. Citation2005, Cha et al. Citation2008), the function of the Na+-phosphate transporter type 2 (NaPi2a) in proximal renal tubules (Hu et al. Citation2010a, Dermaku-Sopjani et al. Citation2011), the activity of NaPi2b in intestinal brush border membrane (Dermaku-Sopjani et al. Citation2011), and additionally interferes with insulin/insulin-like growth factor-1 (IGF-1) signalling (Wolf et al. Citation2008). In vitro studies have shown circulating αKlotho to downregulate insulin and IGF-I-induced autophosphorylation of insulin and IGF-1 receptor (Wolf et al. Citation2008) and subsequently decreasing intracellular oxidative stress (Yamamoto et al. Citation2005). Inhibiting insulin/IGF-related signaling pathways extend lifespan in a wide variety of species (Yamamoto et al. Citation2005, Wolf et al. Citation2008, Li et al. Citation2012), i.e., in accordance with an anti-aging role for Klotho (Kurosu et al. Citation2005).
In addition, there are reports suggesting Klotho mediation in parathyroid hormone (PTH) secretion (Yoshida et al. Citation2002). Studies in mice have shown that isolated parathyroid glands from Klotho-deficient (Klotho -/-) mice secrete less PTH than wild-type mice in response to hypocalcemic stimuli in organ culture. This study also provided molecular evidence of αKlotho stimulating cell membrane abundance enhancements of Na+/K+-ATPase in response to hypocalcemic stimuli, indicating involvement of Klotho in PTH secretion in Na+/K+-ATPase-dependent manner. Increased Na+ gradient by Na+/K+-ATPase might drive the transepithelial transport of Ca2+ even as decreased levels of extracellular free calcium rapidly stimulate PTH secretion (Imura et al. Citation2007). In contrast, another recent in vivo study cast doubt on a physiologically relevant role for Klotho, independent of vitamin D and FGF23, in the regulation of glucose metabolism, bone turnover, and steady-state PTH secretion (Anour et al. Citation2012), suggesting no influence by either the secreted or the transmembrane Klotho on steady-state PTH secretion. However, Klotho role in PTH secretion remains still open, and further studies are needed to clarify this process.
Indeed, secreted αKlotho regulates multiple activities, including suppression of growth factor signaling, regulation of several ion channels, carriers and pumps, and suppression of oxidative stress (Chen et al. Citation2007, Cha et al. Citation2008, Wolf et al. Citation2008, Hsieh et al. Citation2010, Dermaku-Sopjani et al. Citation2011, Sopjani et al. Citation2011, Woudenberg-Vrenken et al. Citation2012). Some of these effects will be discussed below in detail.
The other form of Klotho, i.e., transmembrane Klotho protein, forms a constitutive binary complex with multiple FGF receptors and works as an obligatory co-receptor for FGF23, a bone-derived hormone (Kurosu et al. Citation2006, Urakawa et al. Citation2006), that regulates phosphate excretion into urine (Dermaku-Sopjani et al. Citation2011) as well as synthesis of the active form of vitamin D in the kidney (Imai et al. Citation2004). The fact that FGF23 requires cell membrane Klotho to bind for activating FGF receptors explains why Klotho -/- mice and FGF23 deficient (FGF23 -/-) mice develop many identical phenotypes (Tsujikawa et al. Citation2003, Razzaque et al. Citation2006). In addition, Klotho -/- mice express higher levels of serum FGF23, hence the inability to promote any phosphaturic effect in the absence of Klotho (Kurosu et al. Citation2006). Furthermore, increased sodium-dependent phosphate cotransporters activity in both FGF23 and Klotho-mutant mice enhanced phosphate reabsorption in the kidney, thus facilitating calcification. The observed hyperphosphatemia is caused by hypervitaminosis D and increased renal expression/activity of sodium phosphate cotransporters (Kurosu et al. Citation2006). Vitamin D restriction partially reverses the accelerated aging and early death of Klotho -/- mice (Dermaku-Sopjani et al. Citation2011).
Soluble Klotho also inhibits the highly homologous and ubiquitously expressed transmembrane type III sodium dependent phosphate cotransporters (Pit1 and Pit2) that mediate phosphate uptake (Hu et al. Citation2010a). Klotho decreases the maximal transport rate of both carriers NaPi2a (SLC34A1), expressed on the brush border membrane of renal tubular cells, and NaPi2b (SLC34A2), expressed in intestinal cells. The effect was mimicked by the Klotho protein, which is known to be effective as protease (Kuro-o M Citation2010). Klotho resembles β-glycosidase protein in terms of β-glucuronidase activity (Kuro-o M et al. Citation1997, Tsujikawa et al. Citation2003), and its effect on NaPi2a could be reversed by a β-glucuronidase inhibitor (Hu et al. Citation2010a, Dermaku-Sopjani et al. Citation2011).
It has been shown that Klotho/FGF23 complex signaling induces phosphaturia by suppressing the expression of NaPi2a. Moreover, soluble circulating Klotho protein has also been found to promote phosphaturia independently of FGF23, this by directly downregulating renal phosphate transport via deglycosylation of NaPi2a cotransporters in the proximal tubule (Hu et al. Citation2010a). In this line, Klotho deficiency promotes a state of hyperphosphatemia-induced accelerated aging.
Klotho and intracellular signaling pathways
Several Klotho biological roles have been determined, but the underlying molecular mechanisms are not fully understood and have yet to be clarified. However, many studies have revealed lots of Klotho novel effects to be involved in different intracellular signaling pathways, including: insulin/IGF-1 (Yamamoto et al. Citation2005), cAMP (Rakugi et al. Citation2007), PKC (Imai et al. Citation2004), p53/p21 (Ikushima et al. Citation2006), and Wnt (Liu et al. Citation2007) signaling pathway. Most of these signaling pathways involving Klotho are reviewed elsewhere (Wang and Sun Citation2009a).
FGF23-Klotho complex
FGF23 is a member of the FGF19 subfamily of endocrine fibroblast growth factors, its gene (which exceeds over 8.5 kb) is located in chromosome 12 in humans, and chromosome 6 in mice, encodes a protein composed of 251-amino acids (approx. 32 kDa) with a N-terminal fragment binding domain for the FGF receptor (FGFR) and a novel C-terminal end binding domain for Klotho (Kurosu et al. Citation2006, Urakawa et al. Citation2006, Faul et al. Citation2011).
In humans and mice are found four distinct genes encoding FGFRs (FGFR1-4) (Itoh and Ornitz Citation2004). The ectodomain of prototypical FGFRs consists of three immunoglobulin (Ig)-like domains (D1, D2, and D3). Alternative mRNA splicing occurs within the D3 of FGFR 1-3, generating ‘b' and ‘c' receptor isoforms (FGFR2b and FGFR2c, respectively) with distinct FGF-binding specificities. The unique features of FGF23 require a binary complex of Klotho co-receptor proteins and FGFRs for FGFR activation, thus increasing their affinity to bind circulating FGF23. The αKlotho has different FGFRs binding abilities, including lower affinity to FGFR2 than to the other FGFRs. The FGF23 COOH-terminal domain forms a tertiary complex with αKlotho and the ‘c' isoforms of FGFR 1, 3, and 4, reaching its maximum cellular signaling when FGF23-FGFR1c-αKlotho complex is formed (Bai et al. Citation2004, Kurosu et al. Citation2006, Urakawa et al. Citation2006, Ho et al. Citation2009). As part of a tertiary complex, FGF23 initiates downstream signaling pathways through a variety of intracellular signal transduction proteins, including ERK1/2-SGK1, FRS2, Gab1, Shc, PLC, or STAT1, leading to a potential mechanism for differential and/or gene expression (Urakawa et al. Citation2006, Sitara et al. Citation2008, Wolf et al. Citation2008, Ho et al. Citation2009, Andrukhova et al. Citation2012).
FGF23 is predominantly expressed by osteocytes and osteoblasts in bone (which are main sources of FGF23), but it is also expressed in lower concentrations by other tissues, including the thymus, brain, heart, skeletal muscle, spleen, skin, lung, testes, liver, and the mammary and salivary glands (Liu et al. Citation2003, Urakawa et al. Citation2006, Sitara et al. Citation2008, Martin et al. Citation2012). Synthesis and secretion of FGF23 by osteocytes is upregulated by PTH, 1,25(OH)(2)D as well as dietary and serum phosphate levels (Liu et al. Citation2006). PGF23 is also downregulated, via still unclear mechanisms, by the dentin matrix protein 1 (DMP1) as well as by a phosphate-regulating gene with homologies to endopeptidases on the X chromosome (PHEX) () (Liu et al. Citation2008). However, studies using PHEX-deficient mice have demonstrated that PHEX deficiency is necessary, albeit insufficient, to induce FGF23 gene transcription in osteocytes (Sitara et al. Citation2004, Liu et al. Citation2006).
Figure 2. Multi-organ interaction in regulatory mechanisms of phosphate homeostasis(Fukumoto and Yamashita Citation2002, Wang and Sun Citation2009a,Blaine et al. Citation2011, Kuro Citation2012, Sopjani et al. Citation2011).Bone proteins, phosphate-regulating neutral endopeptidase (PHEX)(1) and dentin matrix protein-1 (DMP1)(2), downregulateFGF23 activity via still unclear mechanisms. FGF23is produced in the bone cells and influences bone mineralization. The emerged FGF23-Klotho system is of great importance as the principal phosphate-regulating endocrine axis. FGF23, via FGF23/Klotho pathway, suppresses renal cotransporters NaPi2a and NaPi2c (3) resulting in decreasedreabsorption and therefore increased urinary phosphate. PTH, synthesized and secreted by parathyroid glands, stimulates renal synthesis of vitamin D (1,25-dihydroxyvitamin D3) mediated through a PTH receptor (PTHR1) (4). PTH promotes renal phosphate excretion and mobilizes phosphate from the bone. Calcitriol in turn decreases PTH production (5), thus closing a negative feedback loops. FGF23 is secreted from bone cells and acts, independently of PTH, to suppress renal vitamin D synthesis (6). Since vitamin D increases FGF23 expression in bone (7), another endocrine negative feedback loop exists between bone (FGF23) and kidney (calcitriol). FGF23's effect on vitamin D metabolism results in decreased entry of phosphate into the circulation from intestine. Vitamin D induces both FGF23and Klotho. Likewise, vitamin D can also induce transcriptional upregulation of NaPi2b and Pit2 to increase intestinal phosphate absorption, this leads to increased serum phosphate levels. FGF23 acts directly on the parathyroid to reduce serum PTH levels (8). Because PTH stimulates FGF23 gene expression (9), another negative feedback loop exists between PTH and FGF23. Pi diet results in upregulationof NaPi2b. See text for further details.This Figure is to be reproduced in colour in the online version of Molecular Membrane Biology.
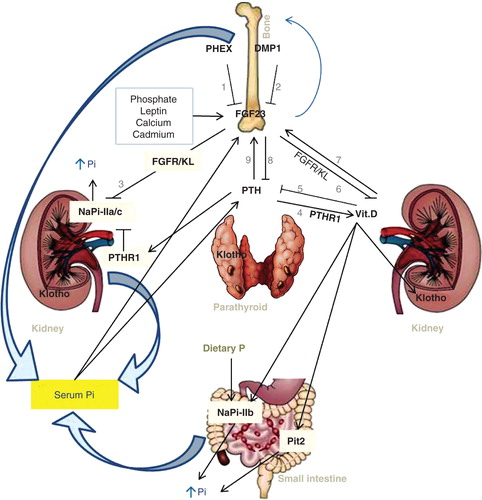
FGF23 mutant animals were used to identify the causal role of FGF23. In rodents, FGF23 overexpression or its parenteral administration suppresses both biosynthesis of 1,25(OH)2D and the phosphate reabsorption in the proximal tubules of the kidney (Bai et al. Citation2004, Shimada et al. Citation2004b). Conversely, FGF23 -/- mice or homozygous missense mutation in human FGF23 have shown significantly higher levels of serum 1,25(OH)2D3, leading to severe hyperphosphatemia and causing soft-tissue calcification (Kurosu et al. Citation2006, Liu et al. Citation2006, Razzaque et al. Citation2006, Sitara et al. Citation2006) – in effect indicating numerous FGF23-dependent biological functions.
Membrane expression of both FGFR and Klotho form a higher affinity FGFR-αKlotho necessary cofactor complex for FGF23 and defines the target organs for FGF23 (Kurosu et al. Citation2006). The important role of αKlotho membrane protein in FGF23 signaling has been confirmed in both human and mouse genetic disorders where the loss of αKlotho results in end-organ resistance to FGF23 and in abnormalities resembling those of FGF23 deficiency (Kuro-o M et al. Citation1997, Ichikawa et al. Citation2007, Imura et al. Citation2007, Segawa et al. Citation2007).
Several findings confirmed a role for putative humoral factors, known as phosphatonins, in regulating of circulating phosphate concentrations independently of calcium (Bai et al. Citation2004, Sitara et al. Citation2004). The best characterized of these is phosphatonin FGF23, known to be causally implicated in developing of severe hypophosphatemic diseases, including X-linked dominant hypophosphatemic rickets/osteomalacia (XLH), autosomal dominant hypophosphatemic rickets/osteomalacia (ADHR) and tumor-induced rickets/osteomalacia (TIO) (Fukumoto and Yamashita Citation2002, Shimada et al. Citation2004b, Sitara et al. Citation2004). In this line, many studies have revealed that FGF23 excess is characterized by aberrant vitamin D metabolism, hypophosphatemia, severe growth retardation, and rickets/osteomalacia (Fukumoto and Yamashita Citation2002, Bai et al. Citation2004, Larsson et al. Citation2004), whereas FGF23 ablation in mice results in higher serum phosphate, excess 1,25(OH)2D activity, and extensive soft tissue calcification (Liu et al. Citation2006, Razzaque et al. Citation2006). The FGF23 act as counter regulatory phosphaturic hormone for vitamin D; hence its excess leads to a reduction in circulating 1,25(OH)2D levels by regulating expression levels of vitamin D-synthesis/degradation renal enzymes, downregulating renal enzyme 25-hydroxyvitamin D 1-α-hydroxylase (Cyp27b1) participating in the production of 1,25(OH)2D, and upregulating 1,25-dihydroxyvitamin D 24-hydroxylase (Cyp24a1) catalyzing the catabolism of 1,25(OH)2D into calcitroic acid in the kidney tubules (Perwad et al. Citation2007), as shown in . Hypophosphatemia resulting from the FGF23 excess is mediated by downregulating expression of sodium-dependent phosphate cotransporters, NaPi2a and NaPi2c, in the kidney-proximal tubules (Gattineni et al. Citation2009), consequently increasing phosphate excretion in the urine. The Klotho/FGFR complex is necessary for eliciting the FGF23-mediated signaling (Bergwitz and Juppner Citation2010).
Klotho in the regulation of calcium and phosphate homeostasis and toxicity
Klotho in calcium homeostasis
Blood calcium concentration is maintained within a very narrow range at all times despite large variations in episodic food intake and body demand (Chang et al. Citation2005). Deregulated calcium homeostasis, as one of the observed aging-like phenotypes of Klotho -/- mice, has been attributed due to altering of the vitamin D metabolism, but recent studies have indicated alternative possibilities (Imura et al. Citation2007) with Klotho being a potential candidate. It has been reported that, in Klotho -/- mice, raised Ca2+ serum levels, hypercalciuria, and the reduction of vitamin D serum levels by dietary restriction reverts most, but not all, of their abnormal phenotypes, suggesting that they are downstream signaling events resulting from increased vitamin D (Tsujikawa et al. Citation2003). The role of Klotho, as a necessary cofactor for transducing the FGF23 signal through the FGFR in vitamin D homeostasis regulation, is one explanation. As illustrated in , circulating levels of 25(OH)D are directly related to dietary vitamin D intake and exposure of the skin to sunlight. In response to ultraviolet B sunlight exposure, pro-vitamin D3 (7-dehydrocholesterol) in the skin epidermal basal layers is converted to pre-vitamin D3 (pre-D3), and then to vitamin D3 (cholecalciferol, D3) (Alexander et al. Citation2009, Haussler et al. Citation2012). Vitamin D3 is transported to the liver where it is hydroxylated by liver 25-hydroxylases (25-OHase). The 25-hydroxycholecalciferol (25(OH) D3) is 1α-hydroxylated in the kidney by 25-hydroxyvitamin D3-1-hydroxylase (1α-OHase). The resulting active 1,25(OH)2D3 (calcitriol) form has different biological effects on various target tissues (Razzaque et al. Citation2006, Haussler et al. Citation2012, Woudenberg-Vrenken et al. Citation2012). Hence it is that Klotho, by suppressing cyp1b enzyme expression-which mediates the conversion of 25-hydroxyvitamin D into 1,25(OH)2D3 (calcitriol, active form of vitamin D) apart from stimulating another renal enzyme, cyp27b1 (Perwad et al. Citation2007, Cha et al. Citation2008, Woudenberg-Vrenken et al. Citation2012), that catalyzes the calcitriol hydrolysis-particularly inhibits vitamin D synthesis, and induce its degradation respectively (Yoshida et al. Citation2002, Tsujikawa et al. Citation2003). The latter enzyme is increased in Klotho -//- mice (Woudenberg-Vrenken et al. Citation2012). Conversely, the absence of Klotho results in modulation of calciotropic hormones, specifically increased serum levels of vitamin D, and decreased serum concentrations of PTH (Yoshida et al. Citation2002, Ohnishi et al. Citation2009). This would cause intestinal hyperabsorption of calcium, raised resorption of Ca2+ from bone, resulting in higher Ca2+ serum levels and therefore increased renal Ca2+ excretion in urine.
Several earlier studies on experimental animals confirmed that both Ca2+ and vitamin D can reduce PTH secretion via calcium-sensing receptors (CaSR) (Mizobuchi et al. Citation2004) as well as by interacting with vitamin D receptor (VDR) (Rodriguez et al. Citation2007). A complementary hypothesis, involving calcium channels regulated by Klotho, is consistent with this idea (Cha et al. Citation2008). Moreover, Klotho act as a β-glucuronidase that activates TRPV5 through hydrolyzing extracellular N-linked oligosaccharides residues on TRPV5, entrapping the channel in the cell membrane and therefore increasing Ca2+ influx in renal cells (Chang et al. Citation2005, Lu et al. Citation2008). Klotho upregulation of the cell membrane TRPV5 expression was confirmed also in in vitro studies (Cha et al. Citation2008). The TRPV5 channel is mainly expressed at the apical membrane of renal epithelial cells in the DCT, and is the main channel for transcellular absorption of calcium (Lu et al. Citation2008). Thus, deregulation in tubular Ca2+ handling might cause calcitriol hypervitaminosis and renal Ca2+ wasting, due to disturbed calcium reabsorption as displayed in Klotho knockout mice (Woudenberg-Vrenken et al. Citation2012). The renal Ca2+ wasting in Klotho -/- mice was determined by modulating the diet, and was found not to be secondary to hypercalcemia and/or hypervitaminosis, a result that underlines the relevance of Klotho to the prevention of renal Ca2+ loss, secondary hypervitaminosis D, osteopenia, and nephrocalcinosis (Alexander et al. Citation2009). Similar to its effect on TRPV5, Klotho significantly increases the activity of TRPV6, which predominantly mediate Ca2+ absorption in the intestine (Lu et al. Citation2008), further substantiating the role of Klotho in maintaining Ca2+ homeostasis.
Another very recent study, carried out in double-knockout mice for Klotho and for cyp27b1, showed that Klotho -/- cyp27b1-/- mice have phenotypes resembling cyp27b1 -/- mice, including significantly decreased serum total Ca2+ concentrations and normal urinary total Ca2+ excretion as compared to wild-type mice. Due to the fact that calcitriol is absent in these mice, the results imply that Ca2+ homeostasis in Klotho -/- mice is affected by their excessive calcitriol levels (Woudenberg-Vrenken et al. Citation2012). Thus, demonstrated interplay between Klotho and vitamin D and their relative role to the Ca2+ homeostasis. Klotho is also an important regulator of phosphate metabolism, and when phosphate dysregulation occurs that is primarily responsible for the aging-related phenotypes.
Klotho in phosphate metabolism
Phosphate is a widely distributed mineral ion in the body, being found mainly (app. 80%) in bone and teeth as apatite. To a lesser extent, phosphate is also present in the viscera and skeletal muscle; but also, in very small amounts (<0.1%), in extracellular fluids (Murer and Biber Citation2010, Ohnishi and Razzaque Citation2010, Quarles Citation2012). Proper balance is an essential prerequisite for basic cellular functions and is, moreover, necessary for several processes, including cell signaling, energy metabolism, membrane integrity and transport, lipid metabolism, enzyme activity, nucleic acid synthesis, muscle function and bone mineralization (Berndt and Kumar Citation2009, Gattineni et al. Citation2009, Kempe et al. Citation2010, Ohnishi and Razzaque Citation2010). Intracellular phosphate ions are stored mainly in the mitochondria (app. 20%), and ER (endoplasmic reticulum, app. 30%), and are used for oxidative phosphorylation, and for phosphorylation of various proteins. The remaining part of cellular phosphate is located in the nucleus, Golgi complex and lysosomes (Di Marco et al. Citation2008, Kempe et al. Citation2010). Not yet fully understood, however, are the exact molecular mechanisms behind the transporting of phosphate in and out of the cell according to the body's needs.
Phosphate is kept in a relatively narrow serum range, with a tightly controlled phosphate balance maintained through hormonal and complex cross-organ communication including the intestine, kidneys, parathyroid glands, and the skeleton, as shown in (Mizobuchi et al. Citation2004, Liu et al. Citation2006, Jin et al. Citation2009, Suyama et al. Citation2012). Despite nearly 70% of dietary phosphate being absorbed by the intestine, causing transient postprandial increases in the serum concentration of phosphate (Sabbagh et al. Citation2009), there are many factors influencing phosphate intestinal absorption or its renal reabsorption that are presented elsewhere (Berndt and Kumar Citation2009).
In general, the body's daily phosphate needs are ensured through intestinal tract absorption from consumed foods, whereas the delicate maintenance of serum phosphate levels is mainly regulated by renal reabsorption/excretion of phosphate through renal phosphate transporters. In addition, bone can also be implicated in phosphate homeostasis by providing phosphate all-out promoting skeletal resorption (Sitara et al. Citation2008, Sabbagh et al. Citation2009, Kempe et al. Citation2010, Dermaku-Sopjani et al. Citation2011, Foller et al. Citation2011).
Phosphate absorption
Studies suggest that intestinal phosphate absorption occurs via two pathways: A paracellular transport mechanism driven by passive diffusion that predominates under standard dietary conditions; and a transcellular transport mechanism driven by an active transport through the sodium-dependent phosphate cotransporters (Kempe et al. Citation2010, Bhandaru et al. Citation2011, Dermaku-Sopjani et al. Citation2011, Sabbagh et al. Citation2011) that constitute the predominant pathway in the intestine (Sabbagh et al. Citation2009). Sodium-dependent phosphate cotransporters, NaPi2b, Pit1 and Pit2 are localized in the intestine; however, NaPi2b predominates in active transport and, based on in vitro analysis performed on ileum segments isolated from wild-type or NaPi2b -/- mice, accounts for more than 90% of sodium-phosphate absorption in ileum (Segawa et al. Citation2007, Sabbagh et al. Citation2009). Although NaPi2b reaches maximal expression in the ileum, it is also present in duodenum and jejunum (Sabbagh et al. Citation2009).
Previous studies have found that both dietary phosphate and calcitriol are involved in NaPi2b activities, thereby regulating intestinal phosphate absorption (Hattenhauer et al. Citation1999, Katai et al. Citation1999). For instance, low-phosphate diet, by inducing the 1α(OH)ase renal expression, can upregulate circulating levels of calcitriol, which can, in turn, induce intestinal NaPi2b protein expression in the epithelial cells of the small intestine, resulting in increased intestinal phosphate absorption (Katai et al. Citation1999). In that regard, NaPi2b -/- mice were observed to exhibit approx. 50% less phosphate absorption, compared with wild-type mice, after being fed a low phosphate diet following acute administration of phosphate, thus demonstrating the significant role of NaPi2b in both intestinal phosphate absorption and systemic phosphate homeostasis (Sabbagh et al. Citation2009). Nevertheless, studies in humans demonstrated that mutations inactivating the human gene SLC34A2, encoding NaPi2b, did not show any significant decline in serum phosphate levels (Corut et al. Citation2006). Thus, further studies are needed to clarify the exact role of NaPi2b in systemic phosphate homeostasis in humans. Despite the importance of phosphate intestinal absorption, its renal reabsorption is critically implicated in maintaining appropriate phosphate homeostasis.
Phosphate reabsorption
The kidney plays a key role in ensuring phosphate balance through renal excretion and reabsorption of phosphate. Its function is to regulate phosphate excretion in the urine and phosphate reabsorption mainly in the kidney proximal tubules via a transcellular pathway, hence the relatively narrow serum phosphate concentration range (Segawa et al. Citation2007, Andrukhova et al. Citation2012) that is physiologically important in numerous biological processes, including aging. Three types of sodium-dependent phosphate cotransporters (Bergwitz and Juppner Citation2010) in proximal tubular cells have been identified: type 1 Na+-dependent phosphate transport protein 1, NPT1 (encoded by the SLC17A1 gene), which is a non-specific phosphate carriers that mediates transport of phosphate and other anions; type 2 family, NPT2, which includes three carriers: NPT2a [(NaPi2a) encoded by the SLC34A1 gene], NPT2b [(NaPi2b) encoded by the SLC34A2 gene] (Xu et al. Citation1999, Nowik et al. Citation2008), and NPT2c [(NaPi2c) encoded by the SLC34A3 gene] (Villa-Bellosta and Sorribas Citation2010); and the type 3 family which comprises of two transporters, PiT1 (SLC20A1) (Nowik et al. Citation2008) and PiT2 (SLC20A2) (Villa-Bellosta and Sorribas Citation2010). NaPi2a is almost exclusively located in the apical membrane of renal proximal tubule cells, and plays a central role in the reabsorption and phosphate balance, while its highly homologous isoform NaPi2c is almost exclusively expressed in the brush-border membrane of the kidney-proximal tubular cells (Sitara et al. Citation2008). NaPi-dependent phosphate reabsorption is regulated by several molecules. For instance, PTH is a strong inhibitor of NaPi transporters, causing increased renal phosphate excretion in urine (Zhao and Tenenhouse Citation2000). In the same line, studies have shown that FGF23 and Klotho can also decrease NaPi activities (Kurosu et al. Citation2006, Hattenhauer et al. Citation1999, Dermaku-Sopjani et al. Citation2011, Gattineni et al. Citation2009). Renal phosphate reabsorption is dependent on phosphate transporters expression and activities. Many regulators play a significant role in transporters (Bhandaru et al. Citation2011, Foller et al. Citation2011), including the amount of dietary phosphate content (Gattineni and Baum Citation2010). For instance, acute administration of low dietary phosphate stimulates NaPi cotransport activity as well as a protein abundance that occurs independently of de novo protein synthesis, this is a result of cellular mechanism mediated by microtubules (tubulin)-facilitating translocation of presynthesized NaPi2a cotransporter protein from intracellular compartments to the apical cell membrane (Lotscher et al. Citation1997). The most recent data confirmed upregulation of NaPi2b in both the renal cortex and medulla, in response to the active excretion of phosphate in high phosphorus diet-fed rats (Suyama et al. Citation2012). These results though are not thought to be necessarily contradictory. In fact, the kidney NaPi2b is mainly expressed on the basolateral side of epithelial cells, while phosphate is transported from the outside of the cell to the interior in both cases, therefore increasing levels of basal phosphate excretion in the kidney (Suyama et al. Citation2012). Within this context, the FGF23-Klotho system provides important mechanistic insights into phosphate homoeostatic control (Kurosu et al. Citation2006), and its manipulation may be used in developing novel strategies to treat diseases associated with abnormal mineral ion metabolism. In addition, an important factor for phosphorus balance is the bone as a major store of both phosphorus and calcium.
Bone in phosphate homeostasis
The skeleton serves multiple functions: Aside from being production site for the major phosphate regulatory hormone FGF23, it is also major storage pool for phosphate (Sitara et al. Citation2008). Presence of phosphate is essential for normal bone growth, development and mineralization, while an insufficiency thereof inhibits bone formation and induces rickets and osteomalacia, i.e., undermineralized soft bones (Liu et al. Citation2003, Citation2006, Citation2008, Shimada et al. Citation2004b, Perwad et al. Citation2007). Maintenance of adequate inorganic phosphorus concentrations is essential for the osteoblasts and osteocytes activity in the process of extracellular matrix mineralization (Sitara et al. Citation2008). In that process, phosphorus along with calcium are the main ionic components required for hydroxyapatite formation (Kuznetsov et al. Citation2000). The enzyme alkaline phosphatase, located at the osteoblast cell membrane with its catalytic unit oriented towards the outside of the plasma membrane, can generate sufficient free minerals by cleaving a phosphorus from beta-glycerol phosphate, allowing free phosphorus via Na+-dependent phosphate cotransporters to enter the cell, where its intracellular concentrations is then maintained within a narrow range (Murshed et al. Citation2005). Lowered serum phosphate levels join with rising alkaline phosphatase activity in bone to impart more phosphate into the bone cells, and may initiate mineralization; moreover, when appropriate phosphate levels are reached the enzyme's activity lessens, as a result of which monitoring bone-specific alkaline phosphatase activity can also be used as a marker indicator of bone phosphate homeostasis and for obtaining information about bone metabolism and bone diseases (Haraikawa et al. Citation2012). Increased vitamin D levels induce FGF23 production by osteoblasts, which, in turn, protects those cells from higher phosphorus concentrations that may, in practise, lead to cell death (Murshed et al. Citation2005, Sitara et al. Citation2008). However, the detailed mechanisms by which these effects on bone are mediated are unknown.
Klotho in phosphate toxicity
Maintaining the physiological balance of phosphate in the body is of key biological significance (Sitara et al. Citation2008). Excessive retention of phosphate in the body can lead to numerous cellular and tissue injuries and disease. Among these are: Vascular calcification in patients with CKD (Koh et al. Citation2001), independent risk factors for renal function weakening and high mortality in CKD patients (Voormolen et al. Citation2007), cardiovascular calcification in Klotho -/- mice (Kurosu et al. Citation2006), premature infertility (Ohnishi and Razzaque Citation2010), growth of lung tumors in humans mediated by Akt-signalling network, therefore stimulating lung tumorigenesis (Jin et al. Citation2009), reduced bone mineralization in rats (Amato et al. Citation1998), a stimulated apoptosis rate in various tissues (Sugiura et al. Citation2005, Citation2010, Ohnishi and Razzaque Citation2010), impaired cell signalling pathways leading to cellular and tissue damage (Jin et al. Citation2009, Sugiura et al. Citation2012), accelerating the aging process in mammalia by causing tissue damage and reducing their cell survival (Ohnishi and Razzaque Citation2010). Furthermore, phosphate-containing enema can also cause health complications in human patients and animals due to phosphate toxicity from higher serum phosphate levels (Grosskopf et al. Citation1991).
The exact mechanism of phosphate toxicity in mammalian aging is not clearly defined. However, Klotho-/- mice have been shown to increase renal activity of NaPi2a, causing phosphate toxicity to develop that induced premature aging-like features, whereas genetically modified NaPi2a/Klotho double -/- mice reduces phosphate toxicity and extends their survival (Ohnishi and Razzaque Citation2010). In addition, phosphate toxicity can exert cytotoxic effects on various organ systems, compromising their functional ability and ending in dysfunction (Osuka and Razzaque Citation2012). Thus, identifying factors that regulate phosphate homeostasis can provide a potential therapeutic tool for the controlling of phosphate homeostasis misregulation-dependent diseases (and so of mammalian aging). In this line-up, Klotho is an important regulator of phosphate balance.
Clinical relevance of Klotho
Current information suggests that Klotho protein has pleiotropic functions, including an emerging role in cardio-renal disease. The expression of the Klotho gene is influenced by many physiological and pathological factors (Aizawa et al. Citation1998). Decreased plasma, urinary and renal Klotho levels and increased mortality risk are associated with normal aging (Semba et al. Citation2011a, Citation2011b), kidney disease (Satoh et al. Citation2012), animal models of diabetes, and hypertension diseases (Semba et al. Citation2011a). Conversely, overexpression of Klotho in animals leads to reversal of the aging process, hence to a substantial extension of lifespan, and additionally provides cardiovascular and renal protection (Wang and Sun Citation2009b), possibly by protecting tissues from oxidative stress damage and by inducing resistance to oxidative stress (Mitobe et al. Citation2005). Klotho clearly has an emerging clinical relevance, especially in nephrology. The expression of the Klotho gene, as a key player for phosphate balance in the kidney, is decreased in the distal tubules, blood, and urine of rats subjected to bilateral renal ischemia (Hu et al. Citation2010b). Animal studies indicate that the renoprotective effects of Klotho are mostly due to the antioxidant effects of its secreted form (Yamamoto et al. Citation2005). Studies of Klotho conducted on humans have reported a significant reduction in soluble Klotho expression in the kidneys of patients with CKD, as compared to healthy control (Koh et al. Citation2001). In the same type of patients, Klotho expression is also decreased in several tissues, contributing to the calcification of soft tissues (Hu et al. Citation2011). Similarly, a reduction of Klotho protein levels in urine has been observed in acute kidney injury (AKI), while Klotho replacement has protective effects on kidney damage and promotes recovery in AKI (Hu et al. Citation2010b). Decreased urinary (Hu et al. Citation2011) and serum levels of Klotho is an early clinical biomarker for CKDs (Shimamura et al. Citation2012) and for AKI induced by ischemia reperfusion injury (IRI) (Hu et al. Citation2010b). In experimental ischaemic AKI mice, Klotho reduces apoptosis via stimulating the expression of heat shock protein 70 (HSP70) in mouse kidneys and HK2 cells, and therefore involved in the pathophysiology of IRI (Sugiura et al. Citation2010). Recently, it has been claimed that replacement of Klotho may potentially be a therapeutic agent for CKD (Hu et al. Citation2011), since its renal expression is markedly suppressed in CKDs. Experiments performed on mice (Sugiura et al. Citation2012) and cultured renal cell lines (Doi et al. Citation2011) suggest that decreased renal Klotho expression is the result of induced renal fibrosis (RF), a final common pathology of CKD, induced by enhancing fibrosis marker activity via, e.g, transforming growth factor-β(1) [TGF-β(1)], which, in turn, reduces renal Klotho expression (Sugiura et al. Citation2012). Moreover, when overexpressed, Klotho protein binds directly to the type II receptor (TGFβR2) and subsequently inhibits type I receptor (TGFβR1) activation that phosphorylates Smad2/3 transcription factors, which then regulates expression of TGF-β1 target genes in nucleus (Doi et al. Citation2011, Sugiura et al. Citation2012). Hence, by inhibiting TGFβ1 signalling (Doi et al. Citation2011), Klotho overexpression increases resistance to RF in mice, even as it suppresses RF caused by chronic glomerulonephritis (Haruna et al. Citation2007). The addition of Klotho protein decreased mitochondrial DNA damage as well as oxidative stress in renal tissues. Importantly, ameliorations were observed in both the renal tubules and glomerulus, although Klotho exclusively expresses to the tubules, so confirming the Klotho protein effect as a circulating hormone (Haruna et al. Citation2007). Another study in mice confirmed that soluble Klotho protects against RF on the obstructed kidney model by critically inhibiting Wnt signalling (Satoh et al. Citation2012). Taken together, RF is associated with Klotho expression, which inhibits multiple growth factor signaling pathways and is additionally involved in the pathophysiology of CKD progression. However, further prospective studies are needed to prove exact diagnostic values of Klotho as an early biomarker of both acute and chronic renal diseases.
Klotho may provide some partial explanation as between inflammation and diseases characterized by accelerated aging of organs, including CKD. Proinflammatory cytokines, such as tumor necrosis factor-alpha (TNF-α) and TNF-like weak inducer of apoptosis (TWEAK), are involved in renal injury by inhibiting the expression of downstream effector Klotho within kidney cells via a NFκB-dependent mechanism (Moreno et al. Citation2011). A summary of the Klotho contribution in partial conditions and diseases is given in .
Table I. Summary of the partial diseases/conditions owing to dysregulation of Klotho.
Klotho-//- mice also display osteoporosis, a skeletal disease, and formation of phenotypic and skeletal abnormalities associated with osteoporosis is prevented by Klotho overexpression (Kuro-o M et al. Citation1997). This suggests at least partial Klotho involvement in osteoporosis, via FGF23/Klotho pathway that is physiologically needed for maintaining normal bone metabolism, such as has been reported in experiments with FGF23-/-/NaPi2a-/- double mutant mice, in which bone mineralization seems to be as a phosphate-independent homeostasis effect of FGF23 (Sitara et al. Citation2008). In addition, among patients with chronic kidney disease-mineral bone disease (CKD-MBD), osteoporosis and adynamic bone are also highly evident. Even though many active antiosteoporotic drugs are now in use, the experience with CKD patient treatment is limited; therefore, its prevention is now accounted of greater importance (Cannata-Andia et al. Citation2012). Further, maintenance of physiologically optimal phosphate balance, influenced by Klotho, is necessary for normal skeletal growth, development and maintenance. Nonetheless, appropriate phosphate level maintenance constitutes of clinical challenge in various osteo-renal diseases, due to the harmful effects of phosphate toxicity and to therapeutic control difficulties with phosphate patients, particularly in those with CKD (Torres et al. Citation2007, Cannata-Andia et al. Citation2012). In addition, patients with osteoporosis frequently suffer from vascular calcification, also reported as a Klotho-mediated effect as discussed below.
Ectopic vasculature calcification is an age-related characteristic highly prevalent in patients with CKD and deficient in Klotho protein (Hu et al. Citation2011). Reports indicating that Klotho -//- animals also develop severe medial vascular calcification, including arteriosclerosis (Kuro-o M et al. Citation1997), suggest that a deficiency thereof may be a key factor in initiating calcification. Secreted Klotho has a particular role in promoting the health of the vasculature, maintaining endothelial wall homeostasis and participating in calcium homeostasis (Hu et al. Citation2011). In vitro, Klotho acts as a direct inhibitor of vascular smooth muscle cells (VSMCs) mediated calcification, induced by high phosphate levels through suppressing Na+-dependent uptake of phosphate and mineralization by sodium-phosphate cotransporters Pit1 and Pit2 (Hu et al. Citation2011). Moreover, soluble Klotho protects the vascular endothelium (Quyyumi Citation1998, Nagai et al. Citation2000, Saito et al. Citation2000), consequently decreased levels of Klotho protein induce endothelial dysfunction, and diffuse vascular calcification (Saito et al. Citation2000, Ikushima et al. Citation2006, Rakugi et al. Citation2007, Di Marco et al. Citation2008). Endothelial dysfunction, or the abnormal function of the blood vessels endothelium, is an early condition in the development and progressing of cardiovascular disease (CVD). The cause here is an imbalance between vasodilatation and vasoconstriction factors, due primarily to enhanced inactivation of nitric oxide (NO) through reactive oxygen species (ROS) formation, and resulting, in turn, in decreased NO bioavailability (Deanfield et al. Citation2007). In human umbilical vein endothelial cells (HUVECs), Klotho suppressed TNF-α induced expression of adhesion molecules, such as vascular cell adhesion molecule-1 (VCAM-1) and intracellular adhesion molecule-1 (ICAM-1), and it also reversed TNF-α inhibition of endothelial nitric oxide synthase (eNOS) phosphorylation. Moreover, NF-κB activation and IκB phosphorylation induced by TNF-α were also inhibited by Klotho protein administration (Maekawa et al. Citation2009). In a rat model of atherosclerosis, adenovirus-mediated Klotho gene delivery increased endothelium-derived NO production and ameliorated vascular endothelial dysfunction (Saito et al. Citation2000), suggesting a therapeutic potential of Klotho for atherosclerotic disease. Furthermore, Klotho protein inhibits H2O2-induced apoptosis and cellular senescence in vascular endothelial cells relevant to the progression of atherosclerosis, therefore Klotho participates in atherosclerosis development reduction (Ikushima et al. Citation2006). Even though Klotho is not expressed in blood vessels (Hu et al. Citation2011), it is reported that its gene delivery decreased elevated blood pressure in rats (Saito et al. Citation2000). Both angiogenesis (new blood vessel creation by pre-existing mature endothelial cells) and vasculogenesis (blood vessel formation through differentiation of endothelial precursor cells) are impaired with age, and were also impaired in the case of Klotho heterozygous mutant mice induced-ischemia, at least partly by endothelium-dependent NO synthesis (Shimada et al. Citation2004a). Therefore, Klotho may serve as a novel target in the therapy of age-related impairment of neovascularization. Moreover, in erythrocytes, Klotho is involved in the regulation of suicidal erythrocyte death, or eryptosis, a critical step toward anemia (Sopjani et al. Citation2008). Observations shown higher eryptosis in Klotho -/- mice erythrocytes, as compared to their Klotho wild-type littermate erythrocytes (Kempe et al. Citation2009). However, under vitamin D-deficient diet, Klotho -/- mice erythrocytes reversed some of the hallmarks of eryptosis, thus indication a role here for Klotho in at least partially vitamin D-dependent regulation in eryptosis (Kempe et al. Citation2009).
There is a correlation between the level of plasma Klotho and CVD in humans. In a population-based study conducted on adults, it was shown that higher plasma Klotho concentrations are independently related with lower risk of CVD (Semba et al. Citation2011a). Moreover, it was reported that plasma Klotho can be used as an independent predictor of all-cause mortality risk in older community-dwelling human adults (Semba et al. Citation2011b). The development of accelerated artery calcification in humans is potentiated in terms of vascular Klotho deficiency through a Runx2 and myocardin-serum response factor-dependent pathway (Lim et al. Citation2012). Vascular Klotho was shown to have bifunctional role: As a vascular calcification endogenous inhibitor and as a cofactor required for FGF23-dependent vascular signaling (Lim et al. Citation2012). Taken together, Klotho deficiency causes vascular calcification, itself a predictor of cardiovascular risk, which is a common complication in CKD (Wang and Sun Citation2009b, Hu et al. Citation2010b). However, further studies are required to evaluate Klotho as a predictor and novel early clinical biomarker for the diagnosis of CVDs in humans, also, for the better understanding the Klotho's role in vascular diseases and its relation with FGF23 in vascular cells.
Experiments on animals confirmed that suppressing Klotho gene expression and protein production in spontaneous hypertensive rats (SHRs) resulted in progression of spontaneous hypertension and renal damage, while Klotho gene delivery in SHRs prevented progression of spontaneous hypertension and renal damage in rats by increasing plasma IL-10 levels, decreasing renal and aortas superoxide production, and inhibiting Nox2 (NADPH oxidase) activity and expression (Wang and Sun Citation2009b) – which is the main source for ROS formation involved in the renal disease pathogenesis (Wang and Sun Citation2009b, Wang et al. Citation2012). Increased levels of ROS are very common in the pathogenesis of hypertension (Yamamoto et al. Citation2005, Shimizu et al. Citation2011). Even though Klotho treatment prevents progression of SHR, its renoprotective effects point, at least in part, to an acute-independent effect of Klotho on blood pressure regulation (Wang and Sun Citation2009b). Klotho gene deficiency is also associated in progression of heart damage in SHRs. Conversely, its expression can prevent the increase of blood pressure while protecting against myocardial hypertrophy and myocardial fibrosis in rats, an effect attributed to increased Klotho protein and decreased insulin/IGF-1 mediated signaling pathways resulting from inhibition of protein kinase B (PKB) phosphorylation (Li et al. Citation2012). Thus, Klotho control expression may offer a new therapeutic approach for the long-term handling of hypertension and heart damage, an approach aimed at CVD prevention and renoprotection.
In addition to the Klotho/FGF23 axis, recent studies clearly suggest emerging roles for βKlotho, a homologous single-pass 130 kDa transmembrane protein primarily expressed in the liver, pancreas and adipose tissue. The βKlotho protein shares about 41% amino acid structural identity and trait similarity with αKlotho, and determines the target organs for FGF15/19 (Kurosu et al. Citation2007, Lin et al. Citation2007). A key regulator In the FGFR4 pathway is its co-receptor βKlotho, both expressed in mature hepatocytes (Lin et al. Citation2007, Poh et al. Citation2012). Increased βKlotho expression in hepatocellular carcinoma (HCC) tissues play an important role in malignant progression; it represents one of the mechanisms of aberrant FGFR4 signaling in HCC, as well as being a novel biomarker (Poh et al. Citation2012). Therefore, controlling the expression of βKlotho may be of clinical relevance and a potent molecular therapeutic target for HCC patients.
Studies have confirmed a correlation between αKlotho and cancer in humans. New evidence suggests that Klotho have tumor-suppressor activities, either by antagonising Wnt signalling of importance for melanoma metastasis and cervical carcinoma tumorigenesis (Lee et al. Citation2010, Camilli et al. Citation2011) or by inhibiting calpain (Camilli et al. Citation2011) – thus decreasing the filamin cleavage critical for melanoma cell motility. Therefore, very low levels of Klotho expression were revealed during the carcinogenesis process (Lee et al. Citation2010, Camilli et al. Citation2011) due to Klotho epigenetic silencing and its resultant functional loss during tumorigenesis. In addition, many studies have reported that oxidative stress can also modulate DNA methylation through both by influencing DNA methyltransferase (DNMT) expression and by altering epigenetic regulation of gene expression involved in carcinogenesis (Franco et al. Citation2008). DNMTs are the key enzymes for inactivating transcription by DNA hypermethylation (Arai et al. Citation2006), a key mechanism for epigenetic regulation in critical tumor suppressor genes. An animal study showed that the Klotho gene promoter CpG hypermethylation was associated with decreased Klotho expression in cervical cancer cells (Lee et al. Citation2010). Recent studies have shown that DNMT 1 inhibitor (5-aza-20-deoxycytidine [5Aza-2dc]) significantly decreased the CpG hypermethylation, resulting in Klotho gene demethylation and therefore increased Klotho expression in vivo in mice and in vitro in HK2 cells (Sun et al. Citation2012).
Klotho has been found to act as a tumor suppressor in various other human tumors types (Chen et al. Citation2013, Xie et al. Citation2013, Zhu et al. Citation2013). In a recent study, Klotho was identified as a tumor suppressor gene that is epigenetically silenced by gene promoter methylation in gastric cancer (GC) cells; Klotho gene restoration stimulated apoptosis, autophagy, while inhibiting GC cell proliferation by downregulating of IGF-1 receptor phosphorylation and subsequent stimulation of IRS-1/PI3K/Akt/mTOR signaling cascade (Xie et al. Citation2013), thus highlighting the relevance of Klotho in GC cell survival. Furthermore, the overall survival rate of the renal cell carcinoma (RCC) patients has been reported as significantly higher in cases of high Klotho expression compared to cases of low Klotho expression. Experiments confirmed Kotho-mediated suppression of RCC progression through PI3K/Akt/GSK3β/Snail signaling (Zhu et al. Citation2013), indicating its therapeutic potential especially for advanced RCC patients. Klotho is endogenously expressed in lung cancer cells, but its expression is lower compared to lung normal cells. The overexpression of Klotho induces apoptosis and inhibits lung cancer cells proliferation and motility by suppressing the activation of Wnt-TCF/β-catenin signaling, thus participating in Klotho-induced growth inhibition (Chen et al. Citation2012). This makes Klotho a candidate target for developing novel therapies against cancers. However, another (and most recent) study reported the opposite results in HCC patients: Significant lower overall survival rate in patients with high Klotho expression compared to those with low Klotho expression. Moreover, this study revealed an anoikis resistance induced by Klotho by activating VEGFR2/PAK1 signaling in HCC, which may function as a putative molecular mechanism for tumor metastasis (Chen et al. Citation2013). Whether this could be used in therapeutics of HCC patients must awaits further clinical studies. Although many Klotho effects have been identified in tumor suppression, findings of a Klotho-mediated role in hepatoma resistance to anoikis in hepatocarcinogenesis suggest various Klotho effects are at work in tumor cells, hence the need for further validation in prospective studies addressing more fully the molecular mechanisms.
Conclusions and perspectives
It seems that anti-aging Klotho can influence various biological processes; in particular, it appears to impact development of several age-related diseases. Klotho functions as a cofactor for FGF23 in FGF23 signaling or, in an independent FGF23 mode, downregulates the expression of vitamin D and phosphate reabsorption (Imai et al. Citation2004, Anour et al. Citation2012, Woudenberg-Vrenken et al. Citation2012). Potential Klotho renoprotective effects suggest an interlinkage between Klotho functions and mitochondria, i.e., a relationship of biological aging with oxidative stress (Yamamoto et al. Citation2005, Di Marco et al. Citation2008, Hsieh et al. Citation2010, Wang et al. Citation2012). The latter is known to cause mtDNA mutations that induce mitochondrial dysfunction which appear to promote apoptotic cell death, and possibly induces mtDNA fragmentation (Wu et al. Citation2011) involved in the pathophysiology of many types of chronic renal disease. Klotho increases oxidative stress resistance, and its protein overexpression, by activating the forkhead transcription factors (FoxO) (Yamamoto et al. Citation2005), has been found to induce expression of manganese superoxide dismutase (Kops et al. Citation2002), a mitochondrial enzyme that facilitates removal of ROS. Thus Klotho protects cells and tissues from oxidative damage by inducing the inhibition of insulin/IGF-1 signaling (Chen et al. Citation2007, Wolf et al. Citation2008, Doi et al. Citation2011). Furthermore, Klotho reduces DNA fragmentation (Hu et al. Citation2010b), oxidative stress, apoptosis, β-galactosidase activity, even as it normalizes cytochrome C oxidase activity; in this way, Klotho ameliorates renal injury by modulating mitochondrial functions (Sugiura et al. Citation2005, Citation2010, Hu et al. Citation2010b). Klotho decreases mitochondrial oxidative stress and so modulates kidney injury (Chen et al. Citation2007). By acting as a β-glucuronidase Klotho activates TRPV5 and TRPV6 cation channels thus increasing of Ca2+ reabsorption and absorption, thus emphasizing the role of Klottho in maintaining Ca2+ homeostasis (Chang et al. Citation2005, Cha et al. Citation2008, Lu et al. Citation2008). Tumor-suppressive properties of Klotho have been determined in various malignant transformations (Chen et al. Citation2013, Xie et al. Citation2013, Zhu et al. Citation2013). Endothelial dysfunction as an early condition in progression and development of CVD results primarily from increased inactivation of NO by ROS, leading to decreased in NO bioavailability (Deanfield et al. Citation2007). Klotho inhibits eNOS phosphorylation (Maekawa et al. Citation2009), therefore unregulated endothelium-derived NO production ameliorates vascular endothelial dysfunction (Saito et al. Citation2000), suggesting a therapeutic potential of Klotho in atherosclerotic disease that remains to be clarified. The discovery of Klotho has not (yet) extended our lifespan, but it has opened up new avenues especially in research into the aging process, mineral homeostasis, cancer progression, apoptosis, and cardiovascular and renal diseases.
Acknowledgements
This work was supported by the University of Prishtina of the Republic of Kosova.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Aizawa H, Saito Y, Nakamura T, et al. 1998. Downregulation of the Klotho gene in the kidney under sustained circulatory stress in rats. Biochem Biophys Res Commun 249:865–871.
- Alexander RT, Woudenberg-Vrenken TE, Buurman J, et al. 2009. Klotho prevents renal calcium loss. J Am Soc Nephrol 20:2371–2379.
- Amato D, Maravilla A, Montoya C, et al. 1998. Acute effects of soft drink intake on calcium and phosphate metabolism in immature and adult rats. Rev Invest Clin 50:185–189.
- Andrukhova O, Zeitz U, Goetz R, et al. 2012. FGF23 acts directly on renal proximal tubules to induce phosphaturia through activation of the ERK1/2-SGK1 signaling pathway. Bone 51:621–628.
- Anour R, Andrukhova O, Ritter E, et al. 2012. Klotho lacks a vitamin D independent physiological role in glucose homeostasis, bone turnover, and steady-state PTH secretion in vivo. PLoS ONE 7:e31376.
- Arai E, Kanai Y, Ushijima S, et al. 2006. Regional DNA hypermethylation and DNA methyltransferase (DNMT) 1 protein overexpression in both renal tumors and corresponding nontumorous renal tissues. Int J Cancer 119:288–296.
- Bai X, Miao D, Li J, et al. 2004. Transgenic mice overexpressing human fibroblast growth factor 23 (R176Q) delineate a putative role for parathyroid hormone in renal phosphate wasting disorders. Endocrinology 145:5269–5279.
- Bergwitz C, Juppner H. 2010. Regulation of phosphate homeostasis by PTH, vitamin D, and FGF23. Annu Rev Med 61:91–104.
- Berndt T, Kumar R. 2009. Novel mechanisms in the regulation of phosphorus homeostasis. Physiology (Bethesda) 24:17–25.
- Bhandaru M, Kempe DS, Rotte A, et al. 2011. Decreased bone density and increased phosphaturia in gene-targeted mice lacking functional serum- and glucocorticoid-inducible kinase 3. Kidney Int 80:61–67.
- Blaine J, Weinman EJ, Cunningham R. 2011. The regulation of renal phosphate transport. Adv Chronic Kidney Dis 18:77–84.
- Camilli TC, Xu M, O'Connell MP, et al. 2011. Loss of Klotho during melanoma progression leads to increased filamin cleavage, increased Wnt5A expression, and enhanced melanoma cell motility. Pigment Cell Melanoma Res 24:175–186.
- Cannata-Andia JB, Rodriguez GM, Gomez AC. 2012. Osteoporosis and adynamic bone in chronic kidney disease. J Nephrol 26(1):73–80.
- Cha SK, Ortega B, Kurosu H, et al. 2008. Removal of sialic acid involving Klotho causes cell-surface retention of TRPV5 channel via binding to galectin-1. Proc Natl Acad Sci USA 105:9805–9810.
- Chang Q, Hoefs S, van der Kemp AW, et al. 2005. The beta-glucuronidase klotho hydrolyzes and activates the TRPV5 channel. Science 310:490–493.
- Chen B, Ma X, Liu S, et al. 2012. Inhibition of lung cancer cells growth, motility and induction of apoptosis by Klotho, a novel secreted Wnt antagonist, in a dose-dependent manner. Cancer Biol Ther 13:1221–1228.
- Chen CD, Podvin S, Gillespie E, et al. 2007. Insulin stimulates the cleavage and release of the extracellular domain of Klotho by ADAM10 and ADAM17. Proc Natl Acad Sci USA 104:19796–19801.
- Chen L, Liu H, Liu J, et al. 2013. Klotho endows hepatoma cells with resistance to anoikis via VEGFR2/PAK1 activation in hepatocellular carcinoma. PLoS ONE 8:e58413.
- Corut A, Senyigit A, Ugur SA, et al. 2006. Mutations in SLC34A2 cause pulmonary alveolar microlithiasis and are possibly associated with testicular microlithiasis. Am J Hum Genet 79:650–656.
- Deanfield JE, Halcox JP, Rabelink TJ. 2007. Endothelial function and dysfunction: Testing and clinical relevance. Circulation 115:1285–1295.
- Dermaku-Sopjani M, Sopjani M, Saxena A, et al. 2011. Downregulation of NaPi-IIa and NaPi-IIb Na-coupled phosphate transporters by coexpression of Klotho. Cell Physiol Biochem 28:251–258.
- Di Marco GS, Hausberg M, Hillebrand U, et al. 2008. Increased inorganic phosphate induces human endothelial cell apoptosis in vitro. Am J Physiol Renal Physiol 294:F1381–F1387.
- Doi S, Zou Y, Togao O, et al. 2011. Klotho inhibits transforming growth factor-beta1 (TGF-beta1) signaling and suppresses renal fibrosis and cancer metastasis in mice. J Biol Chem 286:8655–8665.
- Faul C, Amaral AP, Oskouei B, et al. 2011. FGF23 induces left ventricular hypertrophy. J Clin Invest 121:4393–4408.
- Foller M, Kempe DS, Boini KM, et al. 2011. PKB/SGK-resistant GSK3 enhances phosphaturia and calciuria. J Am Soc Nephrol 22:873–880.
- Franco R, Schoneveld O, Georgakilas AG, et al. 2008. Oxidative stress, DNA methylation and carcinogenesis. Cancer Lett 266:6–11.
- Fukumoto S, Yamashita T. 2002. Fibroblast growth factor-23 is the phosphaturic factor in tumor-induced osteomalacia and may be phosphatonin. Curr Opin Nephrol Hypertens 11:385–389.
- Gattineni J, Bates C, Twombley K, et al. 2009. FGF23 decreases renal NaPi-2a and NaPi-2c expression and induces hypophosphatemia in vivo predominantly via FGF receptor 1. Am J Physiol Renal Physiol 297:F282–F291.
- Gattineni J, Baum M. 2010. Regulation of phosphate transport by fibroblast growth factor 23 (FGF23): Implications for disorders of phosphate metabolism. Pediatr Nephrol 25:591–601.
- Grosskopf I, Graff E, Charach G, et al. 1991. Hyperphosphataemia and hypocalcaemia induced by hypertonic phosphate enema – an experimental study and review of the literature. Hum Exp Toxicol 10:351–355.
- Haraikawa M, Tanabe R, Sogabe N, et al. 2012. A study of the association between serum bone-specific alkaline phosphatase and serum phosphorus concentration or dietary phosphorus intake. J Nutr Sci Vitaminol (Tokyo) 58:442–445.
- Haruna Y, Kashihara N, Satoh M, et al. 2007. Amelioration of progressive renal injury by genetic manipulation of Klotho gene. Proc Natl Acad Sci USA 104:2331–2336.
- Hattenhauer O, Traebert M, Murer H, et al. 1999. Regulation of small intestinal Na-P(i) type IIb cotransporter by dietary phosphate intake. Am J Physiol 277:G756–G762.
- Haussler MR, Whitfield GK, Kaneko I, et al. 2012. The role of vitamin D in the FGF23, klotho, and phosphate bone-kidney endocrine axis. Rev Endocr Metab Disord 13:57–69.
- Ho HK, Pok S, Streit S, et al. 2009. Fibroblast growth factor receptor 4 regulates proliferation, anti-apoptosis and alpha-fetoprotein secretion during hepatocellular carcinoma progression and represents a potential target for therapeutic intervention. J Hepatol 50:118–127.
- Hsieh CC, Kuro-o M, Rosenblatt KP, et al. 2010. The ASK1-Signalosome regulates p38 MAPK activity in response to levels of endogenous oxidative stress in the Klotho mouse models of aging. Aging (Albany NY) 2:597–611.
- Hu MC, Shi M, Zhang J, et al. 2010a. Klotho: A novel phosphaturic substance acting as an autocrine enzyme in the renal proximal tubule. FASEB J 24:3438–3450.
- Hu MC, Shi M, Zhang J, et al. 2011. Klotho deficiency causes vascular calcification in chronic kidney disease. J Am Soc Nephrol 22:124–136.
- Hu MC, Shi M, Zhang J, et al. 2010b. Klotho deficiency is an early biomarker of renal ischemia-reperfusion injury and its replacement is protective. Kidney Int 78:1240–1251.
- Ichikawa S, Imel EA, Kreiter ML, et al. 2007. A homozygous missense mutation in human KLOTHO causes severe tumoral calcinosis. J Clin Invest 117:2684–2691.
- Ikushima M, Rakugi H, Ishikawa K, et al. 2006. Anti-apoptotic and anti-senescence effects of Klotho on vascular endothelial cells. Biochem Biophys Res Commun 339:827–832.
- Imai M, Ishikawa K, Matsukawa N, et al. 2004. Klotho protein activates the PKC pathway in the kidney and testis and suppresses 25-hydroxyvitamin D3 1alpha-hydroxylase gene expression. Endocrine 25:229–234.
- Imura A, Tsuji Y, Murata M, et al. 2007. Alpha-Klotho as a regulator of calcium homeostasis. Science 316:1615–1618.
- Itoh N, Ornitz DM. 2004. Evolution of the Fgf and Fgfr gene families. Trends Genet 20:563–569.
- Jin H, Xu CX, Lim HT, et al. 2009. High dietary inorganic phosphate increases lung tumorigenesis and alters Akt signaling. Am J Respir Crit Care Med 179:59–68.
- John GB, Cheng CY, Kuro-o M. 2011. Role of Klotho in aging, phosphate metabolism, and CKD. Am J Kidney Dis 58:127–134.
- Jones G, Prosser DE, Kaufmann M. 2012. 25-Hydroxyvitamin D-24-hydroxylase (CYP24A1): Its important role in the degradation of vitamin D. Arch Biochem Biophys 523:9–18.
- Katai K, Miyamoto K, Kishida S, et al. 1999. Regulation of intestinal Na+-dependent phosphate co-transporters by a low-phosphate diet and 1,25-dihydroxyvitamin D3. Biochem J 343(Pt 3):705–712.
- Kempe DS, Ackermann TF, Fischer SS, et al. 2009. Accelerated suicidal erythrocyte death in Klotho-deficient mice. Pflugers Arch 458:503–512.
- Kempe DS, Dermaku-Sopjani M, Frohlich H, et al. 2010. Rapamycin-induced phosphaturia. Nephrol Dial Transplant 25:2938–2944.
- Koh N, Fujimori T, Nishiguchi S, et al. 2001. Severely reduced production of klotho in human chronic renal failure kidney. Biochem Biophys Res Commun 280:1015–1020.
- Kops GJ, Dansen TB, Polderman PE, et al. 2002. Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature 419:316–321.
- Kuro O. 2011. Phosphate and Klotho. Kidney Int Suppl S20–S23.
- Kuro O. 2012. Klotho in health and disease. Curr Opin Nephrol Hypertens 21:362–368.
- Kuro-o M. 2010. Klotho. Pflugers Arch 459:333–343.
- Kuro-o M, Matsumura Y, Aizawa H, et al. 1997. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature 390:45–51.
- Kurosu H, Choi M, Ogawa Y, et al. 2007. Tissue-specific expression of betaKlotho and fibroblast growth factor (FGF) receptor isoforms determines metabolic activity of FGF19 and FGF21. J Biol Chem 282:26687–26695.
- Kurosu H, Ogawa Y, Miyoshi M, et al. 2006. Regulation of fibroblast growth factor-23 signaling by klotho. J Biol Chem 281:6120–6123.
- Kurosu H, Yamamoto M, Clark JD, et al. 2005. Suppression of aging in mice by the hormone Klotho. Science 309:1829–1833.
- Kuznetsov SA, Mankani MH, Robey PG. 2000. Effect of serum on human bone marrow stromal cells: ex vivo expansion and in vivo bone formation. Transplantation 70:1780–1787.
- Larsson T, Marsell R, Schipani E, et al. 2004. Transgenic mice expressing fibroblast growth factor 23 under the control of the alpha1(I) collagen promoter exhibit growth retardation, osteomalacia, and disturbed phosphate homeostasis. Endocrinology 145:3087–3094.
- Lee J, Jeong DJ, Kim J, et al. 2010. The anti-aging gene KLOTHO is a novel target for epigenetic silencing in human cervical carcinoma. Mol Cancer 9:109.
- Li BS, Ma HX, Wang YJ, et al. 2012. [Klotho gene attenuates the progression of hypertension and heart damage in spontaneous hypertensive rats]. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 29:662–668.
- Lim K, Lu TS, Molostvov G, et al. 2012. Vascular Klotho deficiency potentiates the development of human artery calcification and mediates resistance to fibroblast growth factor 23. Circulation 125:2243–2255.
- Lin BC, Wang M, Blackmore C, et al. 2007. Liver-specific activities of FGF19 require Klotho beta. J Biol Chem 282:27277–27284.
- Liu H, Fergusson MM, Castilho RM, et al. 2007. Augmented Wnt signaling in a mammalian model of accelerated aging. Science 317:803–806.
- Liu S, Guo R, Simpson LG, et al. 2003. Regulation of fibroblastic growth factor 23 expression but not degradation by PHEX. J Biol Chem 278:37419–37426.
- Liu S, Zhou J, Tang W, et al. 2006. Pathogenic role of Fgf23 in Hyp mice. Am J Physiol Endocrinol Metab 291:E38–E49.
- Liu S, Zhou J, Tang W, et al. 2008. Pathogenic role of Fgf23 in Dmp1-null mice. Am J Physiol Endocrinol Metab 295:E254–E261.
- Lotscher M, Kaissling B, Biber J, et al. 1997. Role of microtubules in the rapid regulation of renal phosphate transport in response to acute alterations in dietary phosphate content. J Clin Invest 99:1302–1312.
- Lu P, Boros S, Chang Q, et al. 2008. The beta-glucuronidase klotho exclusively activates the epithelial Ca2+ channels TRPV5 and TRPV6. Nephrol Dial Transplant 23:3397–3402.
- Maekawa Y, Ishikawa K, Yasuda O, et al. 2009. Klotho suppresses TNF-alpha-induced expression of adhesion molecules in the endothelium and attenuates NF-kappaB activation. Endocrine 35:341–346.
- Manya H, Akasaka-Manya K, Endo T. 2010. Klotho protein deficiency and aging. Geriatr Gerontol Int 10(Suppl 1):S80–S87.
- Martin A, David V, Quarles LD. 2012. Regulation and function of the FGF23/klotho endocrine pathways. Physiol Rev 92:131–155.
- Matsumura Y, Aizawa H, Shiraki-Iida T, et al. 1998. Identification of the human klotho gene and its two transcripts encoding membrane and secreted klotho protein. Biochem Biophys Res Commun 242:626–630.
- Mitobe M, Yoshida T, Sugiura H, et al. 2005. Oxidative stress decreases klotho expression in a mouse kidney cell line. Nephron Exp Nephrol 101:e67–e74.
- Mizobuchi M, Hatamura I, Ogata H, et al. 2004. Calcimimetic compound upregulates decreased calcium-sensing receptor expression level in parathyroid glands of rats with chronic renal insufficiency. J Am Soc Nephrol 15:2579–2587.
- Moreno JA, Izquierdo MC, Sanchez-Nino MD, et al. 2011. The inflammatory cytokines TWEAK and TNFalpha reduce renal klotho expression through NFkappaB. J Am Soc Nephrol 22:1315–1325.
- Murer H, Biber J. 2010. Phosphate transport in the kidney. J Nephrol 23(Suppl 16):S145–S151.
- Murshed M, Harmey D, Millan JL, et al. 2005. Unique coexpression in osteoblasts of broadly expressed genes accounts for the spatial restriction of ECM mineralization to bone. Genes Dev 19:1093–1104.
- Nagai R, Saito Y, Ohyama Y, et al. 2000. Endothelial dysfunction in the klotho mouse and downregulation of klotho gene expression in various animal models of vascular and metabolic diseases. Cell Mol Life Sci 57:738–746.
- Nowik M, Picard N, Stange G, et al. 2008. Renal phosphaturia during metabolic acidosis revisited: Molecular mechanisms for decreased renal phosphate reabsorption. Pflugers Arch 457:539–549.
- Ohnishi M, Nakatani T, Lanske B, et al. 2009. Reversal of mineral ion homeostasis and soft-tissue calcification of klotho knockout mice by deletion of vitamin D 1alpha-hydroxylase. Kidney Int 75:1166–1172.
- Ohnishi M, Razzaque MS. 2010. Dietary and genetic evidence for phosphate toxicity accelerating mammalian aging. FASEB J 24:3562–3571.
- Osuka S, Razzaque MS. 2012. Can features of phosphate toxicity appear in normophosphatemia? J Bone Miner Metab 30:10–18.
- Perwad F, Zhang MY, Tenenhouse HS, et al. 2007. Fibroblast growth factor 23 impairs phosphorus and vitamin D metabolism in vivo and suppresses 25-hydroxyvitamin D-1alpha-hydroxylase expression in vitro. Am J Physiol Renal Physiol 293:F1577–F1583.
- Poh W, Wong W, Ong H, et al. 2012. Klotho-beta overexpression as a novel target for suppressing proliferation and fibroblast growth receptor-4 signaling in hepatocellular carcinoma. Mol Cancer 11:14.
- Quarles LD. 2012. Role of FGF23 in vitamin D and phosphate metabolism: Implications in chronic kidney disease. Exp Cell Res 318:1040–1048.
- Quyyumi AA. 1998. Endothelial function in health and disease: New insights into the genesis of cardiovascular disease. Am J Med 105:32S–39S.
- Rakugi H, Matsukawa N, Ishikawa K, et al. 2007. Anti-oxidative effect of Klotho on endothelial cells through cAMP activation. Endocrine 31:82–87.
- Razzaque MS, Sitara D, Taguchi T, et al. 2006. Premature aging-like phenotype in fibroblast growth factor 23 null mice is a vitamin D-mediated process. FASEB J 20:720–722.
- Rodriguez ME, Almaden Y, Canadillas S, et al. 2007. The calcimimetic R-568 increases vitamin D receptor expression in rat parathyroid glands. Am J Physiol Renal Physiol 292:F1390–F1395.
- Sabbagh Y, Giral H, Caldas Y, et al. 2011. Intestinal phosphate transport. Adv Chronic Kidney Dis 18:85–90.
- Sabbagh Y, O'Brien SP, Song W, et al. 2009. Intestinal npt2b plays a major role in phosphate absorption and homeostasis. J Am Soc Nephrol 20:2348–2358.
- Saito Y, Nakamura T, Ohyama Y, et al. 2000. In vivo klotho gene delivery protects against endothelial dysfunction in multiple risk factor syndrome. Biochem Biophys Res Commun 276:767–772.
- Satoh M, Nagasu H, Morita Y, et al. 2012. Klotho protects against mouse renal fibrosis by inhibiting Wnt signaling. Am J Physiol Renal Physiol 303:F1641–F1651.
- Segawa H, Yamanaka S, Ohno Y, et al. 2007. Correlation between hyperphosphatemia and type II Na-Pi cotransporter activity in klotho mice. Am J Physiol Renal Physiol 292:F769–F779.
- Semba RD, Cappola AR, Sun K, et al. 2011a. Plasma klotho and cardiovascular disease in adults. J Am Geriatr Soc 59:1596–1601.
- Semba RD, Cappola AR, Sun K, et al. 2011b. Plasma klotho and mortality risk in older community-dwelling adults. J Gerontol A Biol Sci Med Sci 66:794–800.
- Shimada T, Takeshita Y, Murohara T, et al. 2004a. Angiogenesis and vasculogenesis are impaired in the precocious-aging klotho mouse. Circulation 110:1148–1155.
- Shimada T, Urakawa I, Yamazaki Y, et al. 2004b. FGF-23 transgenic mice demonstrate hypophosphatemic rickets with reduced expression of sodium phosphate cotransporter type IIa. Biochem Biophys Res Commun 314:409–414.
- Shimamura Y, Hamada K, Inoue K, et al. 2012. Serum levels of soluble secreted alpha-Klotho are decreased in the early stages of chronic kidney disease, making it a probable novel biomarker for early diagnosis. Clin Exp Nephrol 16(5):722–729.
- Shimizu H, Bolati D, Adijiang A, et al. 2011. Indoxyl sulfate downregulates renal expression of Klotho through production of ROS and activation of nuclear factor-kB. Am J Nephrol 33:319–324.
- Sitara D, Kim S, Razzaque MS, et al. 2008. Genetic evidence of serum phosphate-independent functions of FGF-23 on bone. PLoS Genet 4:e1000154.
- Sitara D, Razzaque MS, Hesse M, et al. 2004. Homozygous ablation of fibroblast growth factor-23 results in hyperphosphatemia and impaired skeletogenesis, and reverses hypophosphatemia in Phex-deficient mice. Matrix Biol 23:421–432.
- Sitara D, Razzaque MS, St Arnaud R, et al. 2006. Genetic ablation of vitamin D activation pathway reverses biochemical and skeletal anomalies in Fgf-23-null animals. Am J Pathol 169:2161–2170.
- Sopjani M, Alesutan I, Dermaku-Sopjani M, et al. 2011. Regulation of the Na+/K+ ATPase by Klotho. FEBS Lett 585:1759–1764.
- Sopjani M, Foller M, Dreischer P, et al. 2008. Stimulation of eryptosis by cadmium ions. Cell Physiol Biochem 22:245–252.
- Sugiura H, Yoshida T, Mitobe M, et al. 2010. Klotho reduces apoptosis in experimental ischaemic acute kidney injury via HSP-70. Nephrol Dial Transplant 25:60–68.
- Sugiura H, Yoshida T, Shiohira S, et al. 2012. Reduced Klotho expression level in kidney aggravates renal interstitial fibrosis. Am J Physiol Renal Physiol 302:F1252–F1264.
- Sugiura H, Yoshida T, Tsuchiya K, et al. 2005. Klotho reduces apoptosis in experimental ischaemic acute renal failure. Nephrol Dial Transplant 20:2636–2645.
- Sun CY, Chang SC, Wu MS. 2012. Suppression of Klotho expression by protein-bound uremic toxins is associated with increased DNA methyltransferase expression and DNA hypermethylation. Kidney Int 81:640–650.
- Suyama T, Okada S, Ishijima T, et al. 2012. High phosphorus diet-induced changes in NaPi-IIb phosphate transporter expression in the rat kidney: DNA microarray analysis. PLoS ONE 7:e29483.
- Tang C, Pathare G, Michael D, et al. 2011. Downregulation of Klotho expression by dehydration. Am J Physiol Renal Physiol 301:F745–F750.
- Torres PU, Prie D, Molina-Bletry V, et al. 2007. Klotho: An anti-aging protein involved in mineral and vitamin D metabolism. Kidney Int 71:730–737.
- Tsujikawa H, Kurotaki Y, Fujimori T, et al. 2003. Klotho, a gene related to a syndrome resembling human premature aging, functions in a negative regulatory circuit of vitamin D endocrine system. Mol Endocrinol 17:2393–2403.
- Urakawa I, Yamazaki Y, Shimada T, et al. 2006. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature 444:770–774.
- Utsugi T, Ohno T, Ohyama Y, et al. 2000. Decreased insulin production and increased insulin sensitivity in the klotho mutant mouse, a novel animal model for human aging. Metabolism 49:1118–1123.
- Villa-Bellosta R, Sorribas V. 2010. Compensatory regulation of the sodium/phosphate cotransporters NaPi-IIc (SCL34A3) and Pit-2 (SLC20A2) during Pi deprivation and acidosis. Pflugers Arch 459:499–508.
- Voormolen N, Noordzij M, Grootendorst DC, et al. 2007. High plasma phosphate as a risk factor for decline in renal function and mortality in pre-dialysis patients. Nephrol Dial Transplant 22:2909–2916.
- Wang Y, Kuro O, Sun Z. 2012. Klotho gene delivery suppresses Nox2 expression and attenuates oxidative stress in rat aortic smooth muscle cells via the cAMP-PKA pathway. Aging Cell 11(3):410–417.
- Wang Y, Sun Z. 2009a. Current understanding of klotho. Ageing Res Rev 8:43–51.
- Wang Y, Sun Z. 2009b. Klotho gene delivery prevents the progression of spontaneous hypertension and renal damage. Hypertension 54:810–817.
- Watanabe, et al. 2012. p-Cresyl sulfate causes renal tubular cell damage by inducing oxidative stress by activation of NADPH oxidase. Kidney Int 83(4):582–592.
- Wolf I, Levanon-Cohen S, Bose S, et al. 2008. Klotho: A tumor suppressor and a modulator of the IGF-1 and FGF pathways in human breast cancer. Oncogene 27:7094–7105.
- Woudenberg-Vrenken TE, van der Eerden BC, van der Kemp AW, et al. 2012. Characterization of vitamin D-deficient klotho-/- mice: do increased levels of serum 1,25(OH)2D3 cause disturbed calcium and phosphate homeostasis in klotho-/- mice? Nephrol Dial Transplant 27(11):4061–4068.
- Wu S, Zhou F, Zhang Z, et al. 2011. Mitochondrial oxidative stress causes mitochondrial fragmentation via differential modulation of mitochondrial fission-fusion proteins. FEBS J 278:941–954.
- Xie B, Zhou J, Shu G, et al. 2013. Restoration of klotho gene expression induces apoptosis and autophagy in gastric cancer cells: Tumor suppressive role of klotho in gastric cancer. Cancer Cell Int 13:18.
- Xu H, Bai L, Collins JF, et al. 1999. Molecular cloning, functional characterization, tissue distribution, and chromosomal localization of a human, small intestinal sodium-phosphate (Na+-Pi) transporter (SLC34A2). Genomics 62:281–284.
- Yamamoto M, Clark JD, Pastor JV, et al. 2005. Regulation of oxidative stress by the anti-aging hormone klotho. J Biol Chem 280:38029–38034.
- Yoshida T, Fujimori T, Nabeshima Y. 2002. Mediation of unusually high concentrations of 1,25-dihydroxyvitamin D in homozygous klotho mutant mice by increased expression of renal 1alpha-hydroxylase gene. Endocrinology 143:683–689.
- Zacchia M, Capasso G. 2011. Dehydration: A new modulator of klotho expression. Am J Physiol Renal Physiol 301:F743–F744.
- Zhao N, Tenenhouse HS. 2000. Npt2 gene disruption confers resistance to the inhibitory action of parathyroid hormone on renal sodium-phosphate cotransport. Endocrinology 141:2159–2165.
- Zhu Y, Xu L, Zhang J, et al. 2013. Klotho suppresses tumor progression via inhibiting PI3K/Akt/GSK3beta/Snail signaling in renal cell carcinoma. Cancer Sci 104:663–671.