
Abstract
System is an antiporter belonging to the hetero(di)meric amino acid transporter family. It is located on astrocytes as well as on blood-brain barrier within the CNS. It plays a pivotal role in free radical neutralization as well as neuronal signalling by regulating the glutathione production which occurs via the exchange of intracellular glutamate with extracellular cystine at 1:1 molar ratio. Understandably, it is a vital component responsible for the maintenance of neuronal homeostasis (e.g. redox state). Hence, it could be postulated that any perturbation in system
function may contribute, directly or indirectly, to the pathophysiology of a variety of CNS disorders like Alzheimer’s disease, schizophrenia, drug addiction, depression, multiple sclerosis, hypoglycemic neuronal cell death, glioma, and excitotoxicity, making system
a promising target for treating CNS disorders. In recent times, recognizing the potential of this target, variety of inhibitors has been synthesized by modifying commercially available potent inhibitors including sulfasalazine, erastin, and sorafenib. Although, they have demonstrated efficacy, the in-depth data is still lacking to warrant their use for the treatment of aforementioned CNS disorders. In this review, we discuss the in-depth role of system
transporter in maintaining normal physiology as well as in the pathophysiology of CNS diseases. Additionally, we have also listed some of the potent inhibitors of system
. In conclusion, the critical role of system
in multiple CNS disorders and advanced research on its inhibitors have promising future prospects for better management of the CNS ailments.
Introduction
The origin of neuronal diseases is very challenging as it involves understanding of the complex networks of receptors, membrane-expressed enzymes, transporters, and molecular mechanisms of neurotransmitters (Bridges et al., Citation2012a; DiNunzio & Williams, Citation2008). The central nervous system (CNS) is the most complicated system which controls almost all of the complex activities including sensory inputs, their interpretation and appropriate response in human body (Alazne et al., Citation2013). Hence, any disturbances in this regulatory system can result in multiple complications and disorders. Although difficult, it can be envisaged that, if possible, modification of one target could serve as a suitable treatment strategy for amelioration of several related CNS disorders.
System transporter belongs to the family of heteromeric amino acid transporters (HAT) (Bridges et al., Citation2012b) that plays a role in regulating intracellular cystine level and thus intracellular glutathione (GSH) production in mouse and human brain (Burdo et al., Citation2006). It was identified three decades ago as a Na+-independent exchanger (Bannai & Kitamura, Citation1980) that plays a role as a cystine-glutamate antiporter or exchanger. The exact role was to promote the uptake of one molecule of cystine with release of one molecule of glutamate dictated by the relative concentration gradients of the substrates across the cell membrane (Bannai et al., Citation1986; Bridges et al., Citation2012a; Lo et al., Citation2008). The expression of system
was found to be higher in astrocytes as compared to other body tissues known to release non-vesicular glutamate for the regulation of synaptic activity (Baker et al., Citation2002b; Kupchik et al., Citation2012; Moran et al., Citation2005; Pow, Citation2001). Owing to the pivotal role of system
in the maintenance of neuronal plasticity as well as glutathione production, any aberration in this system could contribute to pathophysiological changes in cystine and glutamate related functions in brain (Moussawi et al., Citation2011). This assumption has been corroborated with the research findings documented in various investigations carried out on epileptic seizures and glioma (De Bundel et al., Citation2011; Lewerenz et al., Citation2014; Takeuchi et al., Citation2013), neurodegenerative diseases like Alzheimer’s disease (AD) (Barger, Citation2004; Qin et al., Citation2006; Schallier et al., Citation2011), Parkinson’s disease (PD) (Massie et al., Citation2011), and multiple sclerosis (MS) (Pampliega et al., Citation2011). Besides this, there are reports which suggest indirect involvement of system
in the psychological disorders such as schizophrenia, mania, depression, and drug addiction (Lutgen, Citation2012). Based on these observations, it can be proposed that system
could serve as a potential common therapeutic target for numerous neurological disorders.
Recently, in the inhibition studies conducted on system , sulfasalazine () was identified as a potent inhibitor. Although, it was a potent molecule, it lacked the desired pharmacokinetic profile. There had been several attempts to modify sulfasalazine to improve its pharmacokinetic profile (Shukla et al., Citation2011). These resulted in no further improvement in the parent structure. In recent time two small molecules, Erastin and Sorafenib have been studied for their in vitro system
inhibition activity (Dixon et al., Citation2014; Singer et al., Citation2015). Amongst these two potent inhibitors, Erastin and its analogues are still under their in vitro pharmacological evaluation (Dixon et al., Citation2014; Singer et al., Citation2015) while Sorafenib is currently being investigated for its clinical efficacy in glioblastoma (National Cancer Institute, Citation2015). Considering the relevance of this target, this paper reviews the current understanding of the target, system
antiporter, in regulating neuronal physiology and pathophysiology. In addition to this, the paper also provides insights about the research outcomes on the novel inhibitors of system
.
Table 1. Amino acid transport systems in different mammalian cells.
Structure of system 
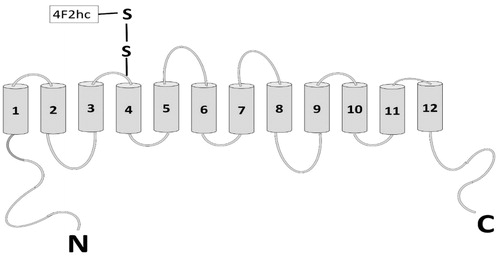
In contrast to other known endogenous amino acid transporters studied until now, system (Kilberg & Hausingger, Citation1992), which belongs to the family heterodimeric amino acid transporters, comprises of two subunits: a light chain and a heavy chain () (Verrey et al., Citation2004; Wagner et al., Citation2001). This heterodimer consists of the 4F2 heavy chain (4F2hc), covalently linked via a disulphide bond to one of the several hydrophobic, non-glycosylated ‘light chain xCT’ (Bridges et al., Citation2001).
4F2 heavy chain
The human heavy chain of 4F2 is made up of 529 amino acids (AAs) (94 kDa, glycosylated; 72 kDa, unglycosylated) (Mosckovitz et al., Citation1993; Wells et al., Citation1992). The amino acid sequence indicates that the 4F2hc have only one trans-membrane helix where intracellular domain holds N-terminus while C-terminus is located extracellularly (Mosckovitz et al., Citation1993). This chain has been classified as type II membrane proteins. Until now, there is dearth of complete information that clearly elucidates the structure of 4F2hc. Nevertheless, there is information about the glycosylation pattern in 4F2hc which evidently shows that a bulky portion of the C-terminus is positioned extracellularly. Besides this, a part of 4F2 is structurally related to glycosidases; however, no glycosidase activity has been demonstrated (Wells & Hediger, Citation1992).
xCT light chain
The xCT light chain (502 Aas, ∼40 kDa) belongs to the family of solute carrier family 7 (SLC7), which consists of two subgroups. One subgroup comprises cationic amino acid transporters (CAT) while other subgroup the light chains of HATs (Verrey et al., Citation2004). These subunits are hydrophobic in nature and constitute non-glycosylated 12 transmembrane (TM) domains where both N- and C-termini are located intracellularly (Gasol et al., Citation2004) (). For surface expression of these subunits, they require one of the two disulphide-linked heavy chain type-II N glycoprotein. Even though the main transporting activity is carried out through xCT subunit, the presence of 4F2hc subunit is mandatory for functional characteristics and for trafficking of xCT to the plasma membrane (Nakamura et al., Citation1999). Interestingly, in one of the experiments, Sato et al. (Citation1999) showed that l-Cys/l-Glu exchange is only functional when both the xCT and 4F2hc proteins are expressed together in Xenopus oocytes.
Figure 1. Structure of the functional chain-xCT with 12 TM domains and both the N-and C-termini are located intracellularly. Light chain is attached at TM4 by forming disulphide bond. There is a re-entrant loop in between TM2 and 3. On addition to this the binding and transporter activity is associated to TM8 (adapted from Gasol et al., Citation2004).
Localization
Although there have been many reports stating the presence of system at different sites, its functional character is still unclear (Lim et al., Citation2007). Sato et al. (Citation2002) used in situ hybridization to localize the expression of xCT and 4F2hc in the brain. A relatively high expression of system
was found in choroid plexus, leptomeninges, ependymal cells, vascular endothelial cells and on blood brain barrier (Burdo et al., Citation2006). It is also located in the astrocytes, microglia, retinal muller cells, and immature cortical neurons (Allen et al., Citation2001; Cho & Bannai, Citation1990; Gochenauer & Robinson, Citation2001; Piani & Fontana, Citation1994). Besides the CNS, it is also expressed on non-CNS cells like fibroblasts, macrophages, hepatocytes, endothelial cells and pancreatic cells (Bannai et al., Citation1986; Hosoya et al., Citation2002; Sato et al., Citation1999; Sasaki et al., Citation2002). Furthermore, the expression of subunits xCT and 4F2hc was detected immunohistochemically at borderline membrane of kidney and duodenum.
Regulation
The expression of system antiporter is regulated by a number of mechanisms including oxidative stress, xenobiotic exposure, amino acid deprivation, and pathological conditions.
Oxidative stress
In astrocytes glutathione synthesis depends on cysteine derived from cystine imported via sytem (Shih et al., Citation2006). This transportation of cystine through system
is a rate-limiting step in the synthesis of GSH after its entry inside the cultured cells (Sagara et al., Citation1993). More precisely, astrocytes present in the CNS may play a role in the intracellular transport of cystine which gets converted to cysteine and ultimately to glutathione as shown in vitro (Dringen et al., Citation1999).
Free radicals produced through mitochondrial electron transport, paroxysmal fatty acid, cytochrome P450 and phagocytic cells have deleterious effects on cell components and connective tissues and thus it is major cause of oxidative stress (Gemma et al., Citation2007). Treatment of ethanol to mouse hepatic stellate cells was found to significantly increase system mRNA and protein levels in vitro (Lin et al., Citation2013). During oxidative stress, there is an increased demand of GSH which might be one of the noticeable reasons for the up-regulation (through activation of Nrf-2: ARE signalling pathway () of the system
antiporter. However, De Bundel et al. (Citation2011) reported that there is no need of system
for basic GSH production in mouse brains indicating other alternative pathways for GSH production in vivo. In few studies it is reported that system
or the high-affinity glutamate transporters in cultured astrocytes are also capable of importing cysteine (Kranich et al., Citation1998; Shanker & Aschner, Citation2001) which might be one of the alternatives for GSH production.
Figure 2. Nrf-2/ARE activation pathway. Under normal conditions Nrf-2 dimerizes with KEAP1 followed by its degradation due to ubiquitination by Cul3. Stimuli like free radicals and oxidizing agents can interrupt with dimerization of Nrf2 and KEAP by altering cysteine residues in KEAP1 or by phosphorylation of Nrf2 at Ser40 with the help of protein kinases. This phosphorylated Nrf2 is then translocated to the nucleus followed by its binding to the adapter proteins which increase ARE-driven transcription (adapted from Bridges et al., Citation2012b).
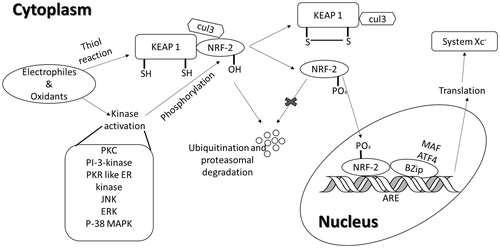
Figure 3. Production of glutathione in astrocytes and neurons via system transporter. SXc, System
, ASC, alanine-serinecysteine; MRP, multi-drug resistance proteins; GCL, glutamate cysteine ligase; GS, glutathione synthase; GSSG-GSH disulphide; TRR1, thioredoxin reductase; GGL, γ-glutamyl transferase (adapted from Lewerenz et al., Citation2013).
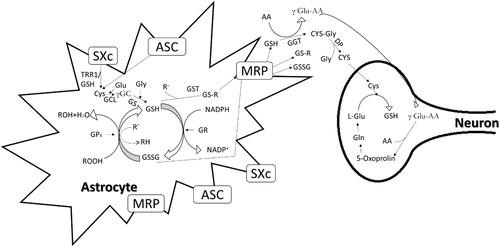
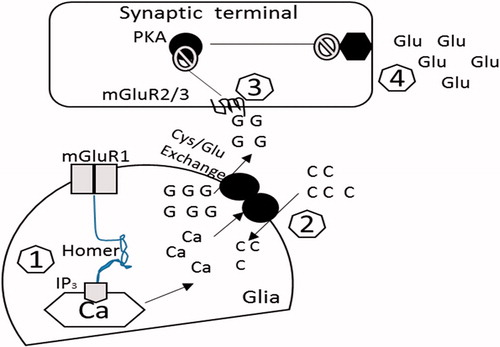
Figure 4. The possible pathways of glutamatergic transmission in the nucleus accumbens that may be responsible for drug-seeking behavior. Cocaine treatment increases the extrasynaptic release of glutamate which promotes the synaptic release of glutamate and ultimately drug-seeking behavior (adapted from Kalivas, Citation2004). G, intracellular glutamate; C, cystine; Glu, released glutamate; Ca, calcium; PKA, protein kinase A. (1) There is a reduction of homer protein in the nucleus accumbens, creating a decrease in signalling through mGluR1 receptors via inositol trisphosphate (IP3) receptor regulation of inner calcium (Ca2+) stocks. (2) The glutamate released through system activates mGlutR1. However, down-regulation of this receptor in addiction may be due to the altered activity of cystine/glutamate transporter. (3) The altered activity of system
results in the decreased release of glutamate and thus reduced activation of the presynaptic mGlu2/3 autoreceptors. (4) This decreased activation of mGlu2/3 results in a prevention of its inhibitory regulation for the glutamate release.
Xenobiotic exposure
In an in vivo experiment, Ramos-Chávez et al. (Citation2015) reported that inorganic arsenic when orally administered to female rats during gestation period resulted in increased expression of system in brain tissues of the offspring. Ceftriaxone, a third-generation cephalosporin, was also found to induce system
expression and increases its mRNA expression in vitro (Lewerenz et al., Citation2009). In addition, there is an evidence of the up-regulation of the system
antiporter in protoplasmic astrocytes when treated with electrophile, tert-butylhydroquinone (tBHQ). However, the same phenomenon was not observed in differentiated culture signifying a distinct regulatory mechanism. Many experiments have failed to mimic this up-regulation by adding H2O2 to the astrocytes and thus, it could be interpreted that it is not the xenobiotics but GSH concentration that triggers the induction (Bridges et al., Citation2012a). Contrary to these above observations, self-administration of nicotine in rats showed decreased expression of system
(Knackstedt et al., Citation2009).
Amino acid deprivation
Recent studies have confirmed that depletion of the AAs results in the stimulation of several genes through the induction of genomic cis-element termed amino acid response element (AARE) (Sato et al., Citation2004). System is very specific for cystine and glutamate, and its activity is known to be induced by cystine deprivation. In one of the studies conducted using mouse NIH3T3 cells, up-regulation of system
was observed in vitro. However, this induction was not only because of cystine deprivation but also due to some other amino acids (Sato et al., Citation2004).
Pathophysiological conditions
System is known to maintain the cell redox system by releasing one molecule of glutamate and simultaneous uptake of one molecule of cystine to maintain neuronal plasticity. Thus, any pathological changes may alter behaviour of system
resulting in increased or decreased expression (Bridges et al., Citation2012a; Lewerenz et al., Citation2013).
Cystine, a vital component of GSH biosynthetic pathway, is required by cells to protect themselves from endogenously-produced reactive oxygen species (ROS). Glioma cells depend upon system to fulfil their GSH requirement. Compromised system
function may cause a rapid depletion of GSH, and the resulting loss of ROS defence causes caspase-mediated apoptosis. Thus, to address the deficit in the glutathione concentration, system
is overexpressed in glioma cells (Chung et al., Citation2005). System
up-regulation has also been demonstrated in vitro at the mRNA or protein level in lymphomas (Gout et al., Citation1997) and pancreatic cancers (Lo et al., Citation2008). Moreover, Qin et al. (Citation2006) demonstrated significantly increased expression of xCT gene in the activated microglia in the cerebral cortex of Thy1-APP751 transgenic mice and adult mouse hippocampus after treatment with amyloid-β protein. Additionally, the injection of 6-hydroxydopamine showed increase in striatal xCT protein levels in vivo after three weeks of injection (Massie et al., Citation2008). However, this study did not include functional significance, the follow-up studies using system
-deficient mice reported no deleterious effects of 6-hydroxydopamine in vivo. (Massie et al., Citation2011). Altered expression of xCT was also found in CNS of patients suffering from MS as well in acute and chronic animal models of MS (Pampliega et al., Citation2011). Furthermore, alterations in xCT expression levels in the hippocampus were also observed in animal models of epilepsy (Takaki et al., Citation2008). Few reports mention the altered xCT expression in the nucleus accumbens in addiction in vivo (Knackstedt et al., Citation2009).
Pathway of regulation
xCT is a functional unit of system antiporter and therefore its regulation has been focused mainly by several studies (Sato et al., Citation1999). The leading pathways for xCT regulation mainly includes: (i) Nuclear factor erythroid 2-related factor 2 (Nrf2)-antioxidant responsive element (ARE), and (ii) eukaryotic initiation factor-2 (eIF2)-activating transcription factor (ATF) 4-amino acid response element (AARE). Along with these two key regulation pathways, xCT expression is also regulated through activation of NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells), activator protein (AP-1), hypoxia-inducible factor (HIF), cAMP, and growth factors.
Nuclear factor erythroid 2-related factor 2 (Nrf2)-antioxidant responsive element (ARE) pathway (). The 5′-flanking region of the mouse xCT gene possesses four ARE-like sequences (also known as electrophile response element) (Sasaki et al., Citation2002) and amongst these four sequences two are completely conserved in 5′-flanking region of the human xCT gene (Sasaki et al., Citation2002; Sato et al., Citation2000). As proposed by Venugopal and Jaiswal (Citation1996), Nrf2 is present in cytosol in bound form with keap-1 (Itoh et al., Citation1999). Oxidative burden releases the Nrf2b from keap-1 through PKC-δ-mediated phosphorylation of Nrf2 which translocates to the nucleus and binds to ARE to initialize specific protein transcription (Niture et al., Citation2009). ARE, a cis-acting regulatory element found in the promoter regions of multiple genes encodes antioxidant proteins including heme oxygenase, γ-glutamylcyteine synthatase, glutamate-cystine ligase, glutathione synthatase, glutathione S-transferase, glutathione reductase, multidrug resistance protein and xCT as well (Bannai, Citation1984; Erickson et al., Citation2002; Lee & Johnson, Citation2004; Sasaki et al., Citation2002).
Eukaryotic initiation factor-2 (eIF2)-activating transcription factor (ATF) 4-amino acid response element (AARE) pathway. The 5′-flanking regions of xCT also contains two amino acids response element (AARE)-like sequence (Sato et al., Citation2004; Lewerenz & Maher, Citation2009). Compromised amino acids levels initiate the signalling cascade followed by the increased translation of activating transcription factor (ATF) 4 and initiation of translational and transcription pathways. Ultimately, levels of proteins like membrane transporters, growth factors and metabolizing enzymes are increased (Kilberg et al., Citation2005). While there are compromised levels of AA in cells, the general control non-repressible proteins 2 (GCN2) kinase is activated through free excessive tRNAs. Activation of GCN2 phosphorylates the eIF2 (Zhang et al., Citation2002). This phosphorylated eIF2 inhibits eIF2B (factor responsible for formation of initiation complex for translation) (Kimball, Citation1999; Rowlands et al., Citation1988).
Another mechanism associated with the activation of AARE regulated gene transcription is phosphorylation of eIF2B (Proud, Citation2005) by Glycogen synthase 3β (GSK3β). Due to eIF2B phosphorylation, it loses its guanine nucleotide exchange factor property and ultimately prevention of formation of initiation complex for translation (Welsh & Proud, Citation1993). Phosphoinositide-3 kinase (PI3K) (Welsh et al., Citation1998) and MEK/ERK (Kleijn & Proud, Citation2000; Quevedo et al., Citation2000) activated by hormones, mitogens, and growth factors, inactivates GSK3β by its phosphorylations and thus protein translation is up-regulated. However, it is has been recently reported that PI3K signalling by inhibiting GSK3beta and thereby activating GCN2 induces eIF2alpha phosphorylation, increases ATF4 protein levels and xCT expression (Lewerenz et al., Citation2014). Thus, phosphorylation of eIF2 and eIF2B reduces total protein translation but at the same time there is increased the translation of certain mRNAs including ATF4. ATF4 in turn activates the transcription of AARE-regulated genes including xCT, the specific subunit of system . Deficiency of cystine was found to activate ATF4-AARE pathway to induce the uptake of cystine in cultured human fibroblast cells after 24 hours (Bannai, Citation1984) suggesting the role of ATF4-AARE pathway in system
regulation.
Also, the 5′-flanking regions of xCT contains binding sites for both NF-κB and AP 1 (Sato et al., Citation2001). However, there is no direct involvement of NF-κB and AP 1 in regulating system reported until now. Additionally, hypoxic preconditioning increased xCT levels in hippocampus (in vivo) and mouse neuronal stem cells (in vitro), through both transcription and translation (Ogunrinu & Sontheimer, Citation2010; Sims et al., Citation2012). However, direct mechanism of system
regulation by hypoxia is yet to be studied.
Growth factors like insulin-like growth factor 1 (IGF-1), transforming growth factor-β (TGF-β), and fibroblast growth factor-2 (FGF-2) have been reported to up-regulate the system function in vitro (Liu et al., Citation2012; Pauly et al., Citation2011; Yang & Yee, Citation2014). Moreover, cAMP also increases the level of xCT mRNA in cultured cells (Gochenauer & Robinson, Citation2001; Seib et al., Citation2011). However, the exact mechanism for the same is yet to be studied.
Functions
Transporters like system present on glial cells take part in maintaining both intra and extracellular environment within CNS (Sato et al., Citation2002). They do so by up taking amino acid (Cystine) required for production of intracellular compounds like GSH with the release of neurotransmitter (Glutamate) in the synaptic cleft to maintain neuronal plasticity (Bannai & Kitamura, Citation1980; Bannai et al., Citation1986). This exchange occurs in 1:1 molar ratio per carrier with the participation of Cl− component (Waniewski & Martin, Citation1984). However, these processes are independent of Na+ availability (Bannai & Kitamura, Citation1981). Additionally, this phenomenon is electroneutral and pH-dependent where each substrate having a net negative charge is exchanged via the transporter (Bannai et al., Citation1986). Overall, system
plays a vital role on GSH, an antioxidant, production in cells and neurotransmission through glutamate release.
System and glutathione production
The CNS requires the highest amount of energy compared to other body systems to function properly. To satisfy this energy requirement, the brain is supplied with enough blood which maintains sufficient glucose and oxygen levels (Sibson et al., Citation1998). During the process, there is ample generation of ROS, which must be neutralized in order to prevent brain damage. Additionally, metabolisms of xenobiotics as well as oxidative stress equally participate in generating ROS (Halliwell, Citation2006). To deactivate ROS, the body requires a counter mechanism that can protect brain from oxidative damage. With regard to this, system antiporter plays a vital role in generating GSH by providing cystine as a synthetic precursor (Bridges et al., Citation2012a; Shih et al., Citation2006).
As mentioned above, system exchanges glutamate present in the astrocytes with cystine available in nanomolar concentration in CSF (Baker et al., Citation2003). The expression of system
is much more on astrocytes cultures as compared to the mature neuron culture and therefore, neurons are dependent on astrocytes for the production of GSH. After the entry of cystine inside the cells, it is reduced to cysteine and then to GSH (Sagara et al., Citation1993). The synthesized GSH is then released in the extracellular space followed by its enzymatic catabolism by γ-glutamyltranspeptidase to form γ-glutamyl-GSH or l-glutamate and l-cysteine-Gly. The released GSH is also converted non-enzymatically to cystine to form cysteine and cysteine-GSH. Thus, cysteine produced by both the pathways enters into the neurons and generate GSH (Dringen et al., Citation1999; Wang & Cynader, Citation2000) ().
Extrasynaptic excitatory neurotransmission
System is involved not only in the uptake of cystine and subsequent production of the GSH, but also in the release of glutamate which is critically important for signalling pathway. Baker et al. (Citation2002b) reported that blockade of system
in striatum results in 60% reduced glutamate levels in extrasynaptic space in vivo, thus nonvesicular release of glutamate plays an important role in extrasynaptic neurotransmission in striatum. This released glutamate through system
plays a physiological role in the tonal regulation of extrasynaptic group II mGluRs and thereby controls neurotransmitter release throughout the CNS (Baker et al., Citation2002a; Mohan et al., Citation2011; Xi et al., Citation2003). The aptitude of system
to contribute in neuronal signalling by releasing glutamate in extrasynaptic environment is still unclear. Recent studies reported that Km for glutamate uptake is 2- to 5-fold higher in glioma cell lines compared to primary cultures of differentiated astrocytes (Seib et al., Citation2011). In another study, cystine in the concentration range of 0.1–0.3 mM was able to produce system
-mediated release of glutamate. This efflux of glutamate decreased the synaptic release of glutamate as a consequence of presynaptic group II mGluRs activation (Moran et al., Citation2005). Based on these research findings it can be concluded that the ability of glutamate released from system
to activate the EAA receptors follows different pathways. Moreover, glutamate released through vesicular and non-vesicular routes is primarily cleared by diffusion through a family of Na+-dependent excitatory amino acid transporter (EAATs) (Bridges et al., Citation2012a). This high capacity of EAATs for clearing glutamate is attributed to their surface density which is almost equivalent to number of glutamate molecules released during action potential (Lehre & Danbolt, Citation1998). Additionally, EAATs have an uptake capacity around 10 µM.ms−1 which is capable of preventing the excitotoxicity-like effects of glutamate released through respective transporters (Asztely et al., Citation1997; Diamond & Jahr, Citation2000; Zheng et al., Citation2008). While this interaction or binding of glutamate occurs very rapidly, the translocation of glutamate may take bit longer (approx. 70 ms, i.e. time required for 90% of glutamate to diffuse out of the synaptic cleft) (Savtchenko & Rusakov, Citation2004; Wadiche et al., Citation1995).
In contrast, Augustin et al. (Citation2007) in their study on Drosophila melanogaster reported, system as a source of glutamate along with its contribution to the signalling and plasticity. The activity of system
transporter was also confirmed by performing studies on xCT homologue gender blind D. melanogaster (Grosjean et al., Citation2008). The damage of this associated gene decreases extracellular l-glutamate levels by about half. Moreover, alterations in the ambient l-glutamate levels arising from altered system
activity were shown to affect both the function (i.e. desensitization) and localization (i.e. clustering) of the ionotropic glutamate receptors (iGluRs) in the Drosophila CNS (Augustin et al., Citation2007; Featherstone, Citation2011).
Although the study conducted by Augustin et al. (Citation2007) states that system -like activity was seen in insects, the real exchange activity by system
through radiolabelled amino acid uptake assay was not been reported. Additionally, phylogenetic analysis performed by Augustin et al. did not include system L and ASC light chains. These were included in another phylogenetic analysis along with xCT, which demonstrated that gender blind is closely related to system L and ASC but not xCT (Lewerenz et al., Citation2013). Moreover phylogenetic analysis for insect SLC7 light chains using the genome of the mosquito Aedes aegypti also demonstrated the absence of an xCT homologue in insects (Carpenter et al., Citation2012).
System and CNS disorders
Drug addiction
Drug addiction is a disorder where neurotransmission is manipulated by administration of some drugs resulting in increased craving for the respective drug. These drug-induced effects on neuronal activity accounts for the addiction which is followed by chronic changes in functioning of corticostriatal pathways initiated in the prefrontal cortex and angled to the nucleus accumbens (Kalivas, Citation2009; Kalivas et al., Citation2005; Uys & LaLumiere, Citation2008; Wolf, Citation2010).
It has been observed that system -mediated transport of cystine and glutamate is effectively reduced in nucleus accumbens by the administration of cocaine and other drugs of abuse (). During this process, glutamate signalling pathway starting from its release to its binding to its receptors is affected, resulting in drug seeking behaviour or addiction (Baker et al., Citation2003; Kau et al., Citation2008; Knackstedt et al., Citation2009; Madayag et al., Citation2007). More precisely, repeated cocaine exposure to animals significantly increased Km values for the cystine uptake in tissue slices of nucleus accumbens (Baker et al., Citation2003; Kau et al., Citation2008; Madayag et al., Citation2007). In one of the pre-clinical studies, it was observed that there was significant decrease in glutamate concentration (in non-habituated rats) followed by system
blockade while there was no change in glutamate levels in cocaine withdrawn rats suggesting decreased expression of system
antiporter (Baker et al., Citation2003). Thus, as a result of reduced activity of system
, there is probably decreased extracellular concentration of glutamate and diminished functional activity in cocaine withdrawn rats. Corroborating the above-mentioned findings, Baker et al. (Citation2003) observed that on infusing cystine into nucleus accumbens of cocaine withdrawn rats, the glutamate levels were elevated to the levels almost equivalent to normal levels. Thus, this increased glutamate levels in nucleus accumbens prevents the cocaine-induced synaptic release of glutamate resulting in reduced cravings for cocaine in vivo (Madayag et al., Citation2007). Furthermore, Baker et al. (Citation2003) also reported that acute administration of N-acetylcysteine (Cysteine prodrug) blocked cocaine-induced addictive behavior in vivo. Moreover, repeated administration of N-acetylcysteine was capable of blocking not only drug-induced cocaine-seeking behavior but also effect of drug related stimuli (Madayag et al., Citation2007; Reichel et al., Citation2011). Though N-acetylcysteine helps in managing addiction, it is not directly transported through system
; in fact this translocation of N-acetylcysteine and its metabolite cysteine is achieved by cysteine transporters (Kupchik et al., Citation2012). Confirming the therapeutic efficacy of N-acetylcysteine in preclinical studies, clinical studies had demonstrated the improvement in drug-seeking behavior when treated with N-acetylcysteine (Knackstedt et al., Citation2009; LaRowe et al., Citation2007; Mardikian et al., Citation2007). Consistent with these studies one more clinical study proved the aforementioned observations stating that N-acetylcysteine was able to improve cocaine induced addiction in humans without affecting the euphoric properties of drugs (Amen et al., Citation2011).
Thus, the above-mentioned findings indicate the potential role of system in drug-seeking behavior and therefore increased urge of cocaine administration with reduced system
activity which might be associated with the pathophysiology of addiction (Breiter et al., Citation1997; Pierce et al., Citation1996; Reid & Berger, Citation1996).
Under normal conditions, released glutamate from astrocytes transports to synaptic cleft and binds to glutamate receptors, i.e. group I and II metabotropic and NMDA receptors (Angulo et al., Citation2004; D’Ascenzo et al., Citation2007; Jabaudon et al., Citation1999; Kupchik et al., Citation2012; Moussawi et al., Citation2011). However, reduced activation of group II mGluRs in cocaine withdrawn rats was observed (Xi et al., Citation2002). This diminished activation of group II mGluRs resulted in increased vesicular release of glutamate in nucleus accumbens after repeated administration of cocaine. N-acetylcyteine was found to increase the glutamate levels and thus activates the group II mGluRs helping in better management of addictive conditions in vivo (Madayag et al., Citation2010).
Schizophrenia
Schizophrenia is a CNS disorder characterized by an interruption in thinking and poor emotional response. Various studies demonstrated the involvement of a variety of factors contributing to schizophrenia including but not limited to genetic predisposition and pre- or postnatal environmental stressors leading to abnormal neurodevelopment (Brown, Citation2011; Chua & Murray, Citation1996; Weinberger, Citation1987). Consequently, there is an abnormal pyramidal neuron activation followed by altered network oscillatory activity in the dorsolateral prefrontal cortex (Cho et al., Citation2006; Gonzalez-Burgos & Lewis, Citation2008; Gonzalez-Burgos et al., Citation2010). The central mechanism for such kind of abnormality includes alteration in glutamatergic activity and changes in N-methyl-d-aspartate (NMDA) as well as other glutamate receptor activation (Coyle et al., Citation2003; Marino & Conn, Citation2002; Rung et al., Citation2005; Verma & Moghaddam, Citation1996).
As already discussed, system also takes part in glutamatergic neurotransmission through non-vesicular release of glutamate which interacts with glutamate receptors (particularly mGluR2/3 and NMDA receptors) along with cystine uptake for GSH production (Baker et al., Citation2002a; Mohan et al., Citation2011). The intracellularly synthesized glutathione is transported outside the cell and then facilitate NMDA receptor activation through reducing extracellular redox sites with release of co-agonist glycine (Metabolite of GSH) (Abbott & Meister, Citation1986; Kohr et al., Citation1994; Lipton et al., Citation2002). However, this hypothesis still requires extensive research in terms of preclinical and clinical studies. Thus, altered or diminished activity of system
transporter may possibly cause neuronal network desynchronization either through decrease in extrasynaptic glutamate signalling or decrease in glutathione concentration. As a consequence of this, there is a diminished receptor activation followed by diminished oscillatory activity. Additionally, reduction in intracellular concentration of GSH may cause compromised management of oxidative stress. Moreover, diminished group II mGluR activation cannot negatively modulate vesicular release which eventually results in abnormal pyramidal neuron activation (Conn & Pin, Citation1997; Xi et al., Citation2002). Hypoglutamatergic condition through reduced non-vesicular release of glutamate and hyperglutamatergic condition at synapse due to reduced inhibition of group II mGluRs contributes to schizophrenia symptoms.
In support of the above discussion, some studies have set an evidence of altered GSH metabolism in schizophrenics (Do et al., Citation2000; Raffa et al., Citation2009), possibly associated with altered activity of system as moderate cystine is available for uptake by system
upon altered metabolism of GSH (Lutgen, Citation2012; Kim et al., Citation2001; Shih et al., Citation2006). Another study also reported slightly increased expression of xCT (active subunit of system
), in the post-mortem brain tissues of schizophrenic patients (Baker et al., Citation2008) probably for acute management of oxidative stress. Additionally, increase in group II mGluRs was observed in schizophrenic patients (Gupta et al., Citation2005), possibly due to chronic understimulation of receptor due to compromised non-vesicular release of glutamate.
Recent research work describing involvement of system in schizophrenia has revealed that the cystine prodrug N-acetylcysteine improved learning ability in rats (Lutgen, Citation2012). Additionally, studies conducted by Berk et al. (Citation2008) and Bulut et al. (Citation2009) showed significant improvement in schizophrenic symptoms upon repeated administration of N-acetylcysteine. Thus, it is possible that targeting system
antiporter may help in attenuating the schizophrenic condition.
Alzheimer’s disease
Amyloid-β (Aβ) peptide is one of the major causes associated with the pathogenesis of AD (Hardy & Selkoe, Citation2002). Aβ is known to stimulate microglia that promotes death of neurons by producing toxins such as glutamate, cytokines and free radicals (Giulian et al., Citation1995; Ishii et al., Citation2000; McDonald et al., Citation1997; Meda et al., Citation1995; Weldon et al., Citation1998). As already discussed, system is located on glial cells and transports glutamate to the extrasynaptic space. Therefore, the release of glutamate may be associated with Aβ’s interaction with system
present on glial cells. Using co-cultures of microglia and neurons, Qin et al. (Citation2006) demonstrated that system
facilitated release of glutamate from Aβ-stimulated microglia is neurotoxic by activating NMDA receptors. In the same study, it was shown that in transgenic mice expressing mutant human amyloid precursor protein in vicinity of amyloid plaques or after injecting Aβ into the brain of C57BL/6 mice stimulated microglia express high levels of xCT. Earlier studies have revealed that exposure of the neurons to Aβ augments neuronal responses to glutamate, along with the rise of intracellular Ca2+ and subsequent cell death (Koh et al., Citation1990; Mattson et al., Citation1992). The up-regulation of system
in Alzheimer’s patients is believed to be due to abundant phosphorylation of α subunit of the eukaryotic initiation factor 2 and activating transcriptional factor 4 (Lewerenz & Maher, Citation2009).
Even though such studies suggest the involvement of system in AD, the impact of such changes is not clear. The reason being the dual function of system
wherein it releases glutamate (activation of extrasynaptic NMDA receptors, may contribute to Aβ production and toxicity) with the uptake of cystine required for the production of glutathione to neutralize ROS in AD (Bordji et al., Citation2010; Olivieri et al., Citation2001; Qin et al., Citation2006). Though such findings is not making the clear role of system
, many Alzheimer’s patients treated with N-acetylcysteine have shown significant improvement in some of the cognitive tasks (Adair et al., Citation2001). Improved GSH production may reduce oxidative stress induced through Aβ followed by diminished abnormal protein expression normalizing the system
transporter activity (Begni et al., Citation2004; Olivieri et al., Citation2001; Onyango et al., Citation2005; Tanel & Averill-Bates, Citation2007).
It is very important to meet the antioxidant demand with the reduced release of glutamate for better management of AD. The system inhibitors may serve the purpose with compromised GSH synthesis. In such conditions, compounds having both system
inhibition and ROS-neutralizing capability may be of great advantage in establishing a good drug candidate for treatment of AD.
Parkinson’s disease
Two prime factors involved in pathogenesis of neurodegenerative disease are excitotoxicity and oxidative stress. More particularly, in PD neurodegeneration is a result of increased extracellular striatal glutamate levels along with hyperactivity of the subthalamic nucleus (STN) (Blandini et al., Citation2000). Moreover, GSH is the key antioxidant in the brain, having significant and early role in the pathogenesis of PD. Thus, loss of GSH production may worsen the condition in PD (Schulz et al., Citation2000). The involvement of glutamate and cystine (Precursor for GSH) both at the same time may contribute towards the vital role of system antiporter in the pathogenesis of PD.
In an interesting study, Massie et al. (Citation2011) showed that in xCT-knockout mice, there is a decreased glutamate level in the extrasynaptic space. However, there was no change in the GSH production suggesting a different pathway for the intracellular movement of cystine. Recently, a study conducted on hemi-Parkinson’s rats described over expression of xCT in their striatum. (Massie et al., Citation2008). Taken together, it supports the role of system in the pathophysiology of PD.
In contrast to the above mentioned studies, a number of animal studies exhibited that N-acetylcysteine treatment showed significant improvement in PD-related symptoms indicating the involvement of system -mediated cystine transport. In support, N-acetylcysteine was capable of showing positive outcomes in animals treated with 1-methyl-4-phenyl-1,2,3,6- tetrahydropyridine (Aoyama et al., Citation2008; Park et al., Citation2004), or in α-synuclein overexpression and EAA carrier 1-null mice (Berman et al., Citation2011; Clark et al., Citation2010). A clinical trial of a sulphonamide, i.e. zonisamide showed the reduction in disease severity (Murata et al., Citation2001), and the mechanism responsible for such a therapeutic effect may involve increased system
activity. Moreover, frequent zonisamide treatment produced improved GSH synthesis in hemiparkinsonian mice, as a result of zonisamide mediated overexpression of system
(Asanuma et al., Citation2010).
In PD, there is a cascade of functional changes as a result of degeneration of dopaminergic neurons in substantia nigra pars compacta which affects the whole basal ganglia network. The utmost significant changes affect the output nuclei of the circuit, the medial globus pallidus and substantia nigra pars reticulata, which become hyperactive. Enhanced glutamatergic inputs that the output nuclei receive from the subthalamic nucleus sustains such hyperactivity. However, mechanisms leading to the subthalamic disinhibition are still poorly understood (Blandini et al., Citation2000). It is also known that GSH is decreased in substantia nigra in PD patients (Martin & Teismann, Citation2009). While it is necessary to have enough GSH levels in brain, it is also important to decrease glutamate levels for the management of PD. So while inhibiting system antiporter, glutamate release may be reduced but at the same time GSH production is also probably dampened. However, several studies have quoted the alternatives for GSH production during increased generation of ROS (Massie et al., Citation2011) and thus perfect combination of dopaminergic receptor modulator and system
inhibitor may form the decent treatment for PD.
Multiple sclerosis
Multiple sclerosis (MS) is a chronic inflammatory and degenerative disease of the CNS characterized by infiltration of immune cells, demyelination, oligodendroglial death and axonal degeneration (Barnett & Prineas, Citation2004; Lucchinetti et al., Citation1999; Trapp et al., Citation1998). Amongst the etiological characteristics, oligodendroglial death and demyelination are believed to be due to glutamate excitotoxicity through activation of ionotropic glutamate receptors (iGluRs) (Matute et al., Citation1997). Several observations supporting the excitotoxic hypothesis in MS comprise the evidence of elevated glutamate levels in MS, both at CNS and peripheral blood (Pampliega et al., Citation2011). Glutamate is increased in cerebrospinal fluid (CSF) in acute MS conditions as well (Stover et al., Citation1997). Thus, the role of system in transporting glutamate outside the glial cell may contribute to the pathophysiology of MS.
Recent studies have shown evidence of increased xCT expression in activated macrophages-microglia in the CNS in MS as well as in leukocytes from MS patients (Pampliega et al., Citation2011). There is also evidence that the simultaneous glutamate release from activated microglia by the system antiporter and the inhibition of Na+-dependent glutamate uptake by the activated microglia results in local increase in extracellular glutamate that leads to excitotoxic oligodendrocyte death (Domercq et al., Citation2007).
The inflammatory process in MS is one of the reasons for neurodegeneration. Besides this, glutamate is also believed to be one of the major culprits for neurodegeneration. The activation of glutamate receptors through system -mediated glutamate release may be targeted for better efficacy. Either glutamate receptor modulators or system
inhibitors could help in such conditions. However, management of inflammation (as a result of ROS) still remains unanswered. In such circumstances, a known anti-inflammatory drug sulfasalazine which showed system
inhibition capability may become a worthy candidate for treatment of MS. However, few studies reported adverse effects associated with sulfasalazine in animal models of MS (Correale et al., Citation1991), while in case of clinical trials conducted on MS patients showed no significant outcome with sulfasalazine treatment (Noseworthy et al., Citation1998). These further require specific studies to understand the exact reasons why sulfasalazine is not able to improve the MS condition.
Depression
Depression is a psychological condition where individuals thought, feelings and sense of well-being are affected thus leads to low mood and displeasure to activity. Current studies have suggested the involvement of glutamatergic neurotransmission interconnected with GABAergic and monoaminergic pathways in the pathophysiology of neuropsychiatric disorder like depression (McCarthy et al., Citation2012; Sanacora et al., Citation2012). Moreover, many clinical and preclinical studies verified that maladaptive changes in excitatory/inhibitory circuitry, particularly in glutamate homeostasis and neurotransmission, have a main part in the mood and anxiety disorders (Musazzi et al., Citation2012). Hence, the variation in glutamate release, clearance and metabolism within certain brain region may contribute to mood disorders like depression. System transporter being an important medium in managing glutamate homeostasis, may have critical involvement in the pathophysiology of depression and thus, could become a novel target in treating the same.
Fibroblast growth factor-2 (FGF-2) has a vital role in the neurogenesis and normal brain development (Zechel et al., Citation2010). Additionally, recent in vitro studies have also suggested the contribution of FGF-2 in the pathophysiology of depression through system -mediated glutamate release. In support of this, studies have proved that there is an up-regulation of system
as a result of FGF 2 treatment in vitro (Liu et al., Citation2012). The report obtained from the post-mortem of depressed patients’ brains showed reduced mRNA for FGF-2 (Evans et al., Citation2004) which may be, given the up-regulation of system
by FGF2 in vitro, associated with decreased expression of system
. Thus, disruption of FGF-2 signalling pathway followed by dysfunctioning of system
may be associated with depression.
Furthermore, there are evidences of diminished GSH levels in patients suffering from depression (Kodydková et al., Citation2009). Since GSH diminution has been associated in depression along with disturbance of the FGF-2 system, it is likely that the pathological effects of FGF-2 disruption are likely to be associated to reduced system activity but still we require studies proving the role of system
.
It will be of great interest to evaluate whether the inhibition or activation of system can benefit in management of depression. However, glutamate is required for managing depression-like conditions and thus it might be advantageous to explore the role of system
in depression with extensive research.
Glioma and excitotoxicity
Gliomas are tumors of glial cells that spread into the healthy brain parenchyma with the release of glutamate being one factor that might facilitate this mechanism. Elevated glutamate interacts with postsynaptic receptors and generates hyperexcitation of the neurons which ultimately results in neuronal cell death and thus ease the way for cancer cells to distribute (de Groot & Sontheimer, Citation2011). The elevated concentration of glutamate in the synapse is attributed to the over activity of the system antiporter along with down-regulation of other EAATs (Chung et al., Citation2005; Kim et al., Citation2001; Rothstein, Citation2002; Takano et al., Citation2001; Ye & Sontheimer, Citation1999; Ye et al., Citation1999). Furthermore, due to high activity of system
antiporter in glioma cells, the impact of chemotherapy and radiation reduces (Chung et al., Citation2005). Taken together, the increased expression of system
in glioma releases high levels of glutamate that can lead to seizures and neuronal death. At the same time, system
uptakes cystine to generate glutathione and thus protect cancer cells from ROS.
GSH requirement is mandatory for cancer cells to survive. The inhibition of system will not only decrease GSH production but also compromise glutamate release. Thus, targeting system
could help in finding novel compounds for both glioma as well as epileptic seizures. It has been shown that sulfasalazine is able to inhibit system
effectively (Bridges et al., Citation2012b). Moreover, Chung et al. (Citation2005) reported that inhibition of cystine uptake by sulfasalazine halts the progress of primary brain tumours in vivo. However, due to poor pharmacokinetic properties of sulfasalazine, the bioavailability is very low at the site of action (CNS) when administered orally (Kusuhara et al., Citation2012). Additionally, clinical trial of sulfasalazine in recurrent glioma had to be terminated prematurely due to deleterious effects of the compound (Robe et al., Citation2009). Thus, with suitable chemical modification of sulfasalazine moiety, it is possible to synthesize molecules with inhibitory activity like parent moiety with improved pharmacokinetic characteristics. Other than sulfasalazine, erastin and its analogues also demonstrated their system
inhibition potential in vitro. However, erastin is still under preliminary evaluation (Dixon et al., Citation2014; Singer et al., Citation2015). Moreover, an already approved anti-cancer drug for kidney and liver cancer, Sorafenib is under clinical trials for its efficacy in glioblastoma (NCI, Citation2015).
Hypoglycaemic neuronal death
As already mentioned above, the brain’s energy requirement is highest compared to all body systems thus, if glucose concentration falls below desired level, it may cause neuronal injury. It has been shown that when glucose concentrations in the blood are below 2 mM, brain glucose level touches zero leading to neuronal injury (Choi et al., Citation2001; Langan et al., Citation1991; Ryan et al., Citation1990; Suh et al., Citation2007; Xu et al., Citation2011). Neurons, particularly those present in the hippocampus and cortex, are extremely sensitive to glucose deprivation (GD) (Auer et al., Citation1984; Goldberg & Choi, Citation1993; Monyer & Choi, Citation1988; Monyer et al., Citation1989). To balance glucose deprivation, astrocytes contribute to work energy generation via glycogen metabolism. Additionally, glutamate released from the astrocytes undergoes metabolic pathway (TCA cycle) and gets converted to pyruvate for more energy generation. (Bakken et al., Citation1998; Butterworth, Citation1999; Dennis & Clark, Citation1978). In spite of this, neuronal cell death does not seem to be a straight consequence of the energy failure. Numerous studies exhibited that hypoglycaemic neuronal injury happens subsequently to glutamate excitotoxicity, as insulin-induced hypoglycaemia effects increase glutamate concentration in the rat hippocampus and striatum (Sandberg et al., Citation1986; Silverstein et al., Citation1990) and in the cerebrum of the pig (Ichord et al., Citation1999). Jackman et al. (Citation2012) also confirmed the involvement of high levels of glutamate in the hypoglycaemic neuronal cell death.
As already known, system antiporter located on the astrocytes releases glutamate in the extracellular space. The higher concentration of glutamate in hippocampus and striatum may be responsible for hypoglycaemic cell death. Interestingly, it has also been shown that system
contributes to hypoglycaemic neuronal cell in vitro by glutamate release and subsequent activation of neuronal glutamate receptors (Jackman et al., Citation2012).
System inhibition
The involvement of system in the pathophysiology of a variety of CNS disorders makes it a promising target for treatment. Until now, a lot of work has been done in this area. Almost all the conducted experiments were based on competitive assays that screened structurally similar analogues for their ability to block the system
-mediated exchange of radiolabeled l-glutamate for l-cystine. From the preliminary studies, two structurally dissimilar molecules, sulfasalazine and (S)-4-carboxyphenylglycine 2 (S-4CPG) were found to inhibit system
antiporter (Gout et al., Citation2001; Ye et al., Citation1999). Along with system
inhibition, sulfasalazine is also a well-known NF-kappa B (NF-kB) inhibitor indicating its possible role in ailments like cancer, inflammation, infection and autoimmune diseases (Chávez et al., Citation2012).
Although these molecules exhibited inhibitory properties, further research was limited because of their secondary effects and poor pharmacokinetic profiles (Chung et al., Citation2005; Savaskan et al., Citation2008). Compound 2 also antagonizes group I metabotropic receptors (mGluRs) (Hayashi et al., Citation1994) along with system inhibition while 3 is metabolized by CYP enzymes to produce inactive metabolites (Azad et al., Citation1983).
To overcome such problems, many researchers have modified existing inhibitors like 3 to get metabolically stable and effective analogues (). Apart from the modifications in 3 that exhibited inhibitory activity, a lot of other modifications can be done to increase the pool of inhibitors ().
Table 2. List of system inhibitors along with their IC50 value and potency comparison with sulfasalazine (Dixon et al., Citation2014; Matti et al., Citation2013; Shukla et al., Citation2011). a: [3H]Glu uptake (% control), b: EC50.
Table 3. Possible chemical modifications in sulfasalazine for its improved pharmacokinetic profile.
Recently, an antitumor agent erastin 28 has been found as a selective inhibitor of system . Additionally, analogues for 28 have also been generated to improve potency (Dixon et al., Citation2014; Singer et al., Citation2015). Erastin and its analogues represent a novel scaffold for system
inhibitors. Currently, such novel scaffolds are needed to shift the gears in this field. Other than these two molecules, sorafenib, a protein kinase inhibitor, also inhibits the transporter. Dixon et al. (Citation2014) reported that sorafenib could triggered ER stress and ferroptosis through system
inhibition.
Molecular modelling
The X-ray crystal structure of system transporter is not available in the Protein Data Bank (PDB). Few groups of researchers have tried to build the homology model of the transporter based on a related amino acid, polyamine and organocation transporter (ApcT) from a thermophile Methanocaldococcus jannaschii (PDB ID 3GIA) (Matti et al., Citation2013). The developed and validated homology model was then used for docking studies of the novel system
inhibitors belonging to isoxazole hydrazide class (). The binding mode clearly showed the importance of –COOH group located on the isoxazole ring along with the hydrazide –C=O which were found to interact with Arg135 (electrostatic interaction) and Ser330 (H-bonding), respectively. In addition, the O group of isoxazole ring was found to interact with Tyr251 via H-bonding. Overall, the homology model assisted the authors in rationalizing the observed potency. Due to lack of significant number of inhibitors, ligand-based approaches such as 3D pharmacophore and quantitative structure-activity relationship (QSAR) studies are limited in the literature. Substantial efforts in this direction will help in discovering novel classes of system
inhibitors.
Structure-activity relationship
Sulfasalazine and analogues
Shukla et al. (Citation2011) synthesized sulfasalazine analogues and determined the concentration required by newly synthesized molecules to reduce the cellular cystine uptake by 50% compared to the solvent control in U87 cell line. Sulfasalazine (3, IC50 = 30 µM), a prodrug of sulfapyridine (4, IC50 > 1 mM) and 5-aminosalicylic acid (5, IC50 > 1 mM) is a well-known system inhibitor. Both sulfapyridine (4) and 5-aminosalicylic acid (5) exhibited complete loss of system
activity. Modification of the central planar linker with trans geometry (N = N) in 3, with planar ethyne linker led to conservation of activity (7, IC50 = 30 µM) demonstrating that the azo linkage was not absolute necessity. The corresponding ester of 7 did not show any activity (6, ). On the similar lines, the replacement of azo linkage with ethylene linker (9, ) led to 2-fold reduction in activity. Disruption of the conjugation in 3, resulted in over 10-fold reduction in activity (11, ). All other modifications in the parent structure led to loss of activity ().
Isooxazole analogues
Matti et al. (Citation2013) reported a set of isooxazole derivatives which were evaluated for their system -inhibiting potential using SNB-19 cell lines. Cells were tested for l-[3H]Glu uptake under Cl− conditions in the presence of isoxazole-hydrazide derivatives. A novel series of isooxazole 3-carboxylic acids exhibited less sensitivity to the structured modification ([3H]Glu uptake). The reported series explored effect of substituents on the distant aryl ring (). The aryl ring was found to tolerate a variety of substituents such as –OCH3, –OH, –F, –H etc. without appreciable change in activity.
In a similar attempt, Newell et al. (Citation2014) further explored the SAR of 22 reported earlier to be a competitive inhibitor. Extension of 5-methyl group as (un)substituted arylmethyl group also resulted in potent analogues (22a–22c, ) as non-competitive inhibitors. This demonstrated the tight structural requirements of inhibitors with respect to the kinetics of inhibition (competitive vs. non-competitive). Nonetheless, the isoxazole scaffold is a novel scaffold, quite different from the sulfasalazine and analogs. This may be further explored synthetically or computationally to fill the void in the availability of novel, orally bioavailable system inhibitors.
Erastin and analogues
Lethal effective concentration (EC) for all synthesized erastin analogues was determined in HT-1080 cells in a 10-point, two-fold dilution assay, starting at a high concentration of 20 μM, ±β-ME (18 μM). IC50 values were also determined for the synthesized analogues in CCF-STTG1 cells using glutamate uptake assay by Dixon et al. (Citation2014). Erastin (32, , IC50 = 0.20 µM) a quinolone-4-one, provided another lead structure for the design and development of potent system inhibitors. Systematic structural modifications resulted in quite potent analogues with IC50 as low as 3.5 nM (49, ). The majority of the analogues retained the original structural framework of 32, with modification in the N3- substituent which seem to require other bulky substitutes at C-5 on the aromatic ring such as –Br, –Ph etc. for improved potency. The 2-isopropoxy substituents also seemed necessary for the potency (32 vs. 33, ). Addition of substituents on the core quinolone-4-one nucleus led to >1.5-fold reduction in potency (33 vs. 34, ). Placement of 5-Cl substituent on the quinazoline-4-one nucleus, however increase potency 2-fold (33 vs. 36, ). Conversion of the pendant piperazine ring in 32 to the open chain congener led to 3-fold drop in potency (39, ). Also, replacing ether (–OPh) in distal part of 32 with –NH– led to 2.5-fold decrease in potency (43, ). Other modifications led to reduction in potency.
Conclusions
The prevalence of CNS disorders is increasing day-by-day with a high mortality rate throughout the world. The adequate management of CNS disorders is becoming a tough task because of insufficient knowledge in such areas. Advances in understanding how system contributes to the pathophysiology of CNS disorders might pave the way for future treatment strategies. The dual role of system
transporter, i.e. glutamate release and cysteine uptake, has participation in the pathophysiology of many CNS diseases. Thus, targeting system
could serve the purpose of treatment in CNS complaints. A number of research groups have generated small molecule hits/leads which are potent system
inhibitors but still their clinical efficacy will be a major concern. Hence, further research is needed to explore the system
target as a therapeutic option for CNS disorders.
Future prospects
This review describes various aspects of system in variety of CNS disorders. Until now, only preliminary research or in vitro studies have been performed for modifying system
activity and there is very little documentation about preclinical and clinical studies. With regard to this, extensive research is required to draw a valid conclusion about how system
inhibition or activation will benefit in CNS disorders. Additionally, there are limited numbers of sulfasalazine analogues that have demonstrated therapeutic potential and therefore synthesis of additional active sulfasalazine analogues should also be pursued. Like sulfasalazine and its analogs, other drugs can also be repurposed and evaluated for their system
inhibition activity.
Acknowledgements
The authors acknowledge Achievers League, USA, for their editorial assistance.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Abbott WA, Meister A. 1986. Intrahepatic transport and utilization of biliary glutathione and its metabolites. Proc Natl Acad Sci USA 83:1246–1250
- Adair JC, Knoefel JE, Morgan N. 2001. Controlled trial of N-acetylcysteine for patients with probable Alzheimer’s disease. Neurology 57:1515–1517
- Alazne D, Álvarez A, Hilario E, Suarez-Merino B, Goñi-de-Cerio F. 2013. Central nervous system diseases and the role of the blood-brain barrier in their treatment. Neurosci Discov 1:1–8
- Allen JW, Shanker G, Aschner M. 2001. Methylmercury inhibits the in vitro uptake of the glutathione precursor, cystine, in astrocytes, but not in neurons. Brain Res 894:131–140
- Amen SL, Piacentine LB, Ahmad ME, Li SJ, Mantsch JR, Risinger RC, Baker DA. 2011. Repeated N-acetyl cysteine reduces cocaine seeking in rodents and craving in cocaine-dependent humans. Neuropsychopharmacology 36:871–878
- Angulo MC, Kozlov AS, Charpak S, Audinat E. 2004. Glutamate released from glial cells synchronizes neuronal activity in the hippocampus. J Neurosci 24:6920–6927
- Aoyama K, Matsumura N, Watabe M, Nakaki T. 2008. Oxidative stress on EAAC1 is involved in MPTP-induced glutathione depletion and motor dysfunction. Eur J Neurosci 27:20–30
- Asanuma M, Miyazaki I, Diaz-Corrales FJ, Kimoto N, Kikkawa Y, Takeshima M, et al. 2010. Neuroprotective effects of zonisamide target astrocyte. Ann Neurol 67:239–249
- Asztely F, Erdemli G, Kullmann DM. 1997. Extrasynaptic glutamate spillover in the hippocampus: dependence on temperature and the role of active glutamate uptake. Neuron 18:281–293
- Auer RN, Wieloch T, Olsson Y, Siesjo BK. 1984. The distribution of hypoglycemic brain damage. Acta Neuropathol 64:177–191
- Augustin H, Grosjean Y, Chen K, Sheng Q, Featherstone D. 2007. Nonvesicular release of glutamate by glial xCT transporters suppresses glutamate receptor clustering in vivo. J Neurosci 27:111–123
- Azad Khan AK, Guthrie G, Johnston HH, Truelove SC, Williamson DH. 1983. Tissue and bacterial splitting of sulphasalazine. Clin Sci (Lond) 64:349–354.
- Baker DA, Madayag A, Kristiansen LV, Meador-Woodruff JH, Haroutunian V, Raju I. 2008. Contribution of cystine-glutamate antiporters to the psychotomimetic effects of phencyclidine. Neuropsychopharmacology 33:1760–1772
- Baker DA, McFarland K, Lake RW, Shen H, Tang XC, Toda S, Kalivas PW. 2003. Neuroadaptations in cystine-glutamate exchange underlie cocaine relapse. Nat Neurosci 6:743–749
- Baker DA, Shen H, Kalivas PW. 2002a. Cystine/glutamate exchange serves as the source for extracellular glutamate: modifications by repeated cocaine administration. Amino Acids 23:161–162
- Baker DA, Xi ZX, Shen H, Swanson CJ, Kalivas PW. 2002b. The origin and neuronal function of in vivo nonsynaptic glutamate. J Neurosci 22:9134–9141
- Bakken IJ, White LR, Unsgard G, Aasly J, Sonnewald U. 1998. [U- 13C] glutamate metabolism in astrocytes during hypoglycemia and hypoxia. J Neurosci Res 51:636–645
- Bannai S, Kitamura E. 1981. Role of proton dissociation in the transport of cystine and glutamate in human diploid fibroblasts in culture. J Biol Chem 256:5770–5772
- Bannai S, Kitamura E. 1980. Transport interaction of L-cystine and L-glutamate in human diploid fibroblasts in culture. J Biol Chem 255:2372–2376
- Bannai S. 1984. Induction of cystine and glutamate transport activity in human fibroblasts by diethyl maleate and other electrophilic agents. J Biolog Chem 259:2435–2440
- Bannai S, Takada A, Kasuga H, Tateishi N. 1986. Induction of cystine transport activity in isolated rat hepatocytes by sulfobromophthalein and other electrophilic agents. Hepatology 6(6):1361–1368
- Barger SW. 2004. An unconventional hypothesis of oxidation in Alzheimer’s disease: intersections with excitotoxicity. Front Biosci 1(9):3286–3295
- Barnett MH, Prineas JW. 2004. Relapsing and remitting multiple sclerosis: pathology of the newly forming lesion. Ann Neurol 55:458–468
- Begni B, Brighina L, Sirtori E, Fumagalli L, Andreoni S, Beretta S, et al. 2004. Oxidative stress impairs glutamate uptake in fibroblasts from patients with Alzheimer’s disease. Free Radic Biol Med 37:892–901
- Berk M, Copolov D, Dean O, Lu K, Jeavons S, Schapkaitz I, et al. 2008. N-Acetyl cysteine as a glutathione precursor for schizophrenia – a double-blind, randomized, placebo-controlled trial. Biol Psychiatry 64:361–368
- Berman AE, Chan WY, Brennan AM, Reyes RC, Adler BL, Suh SW, et al. 2011. N-acetylcysteine prevents loss of dopaminergic neurons in the EAAC1-/- mouse. Ann Neurol 69:509–520
- Blandini F, Nappi G, Tassorelli C, Martignoni E. 2000. Functional changes in the basal ganglia circuitry in Parkinson’s disease. Prog Neurobiol 62:63–88
- Bordji K, Becerril-Ortega J, Nicole O, Buisson A. 2010. Activation of extrasynaptic, but not synaptic, NMDA receptors modifies amyloid precursor protein expression pattern and increases amyloid-β production. J Neurosci 30:15927–15942
- Breiter HC, Gollub RL, Weisskoff RM, Kennedy DN, Makris N, Berke JD, et al. 1997. Acute effects of cocaine on human brain activity and emotion. Neuron 19:591–611
- Bridges CC, Kekuda R, Wang H, Prasad PD, Mehta P, Huang W, et al. 2001. Structure, function, and regulation of human cystine/glutamate transporter in retinal pigment epithelial cells. Invest Ophthalmol Vis Sci 42(1):47–54
- Bridges R, Lutgen V, Lobner D, Baker DA. 2012a. Thinking outside the cleft to understand synaptic activity: contribution of the cystine-glutamate antiporter (System ) to normal and pathological glutamatergic signaling. Pharmacol Rev 64(3):780–802
- Bridges RJ, Natale NR, Patel SA. 2012b. System xc cystine/glutamate antiporter: an update on molecular pharmacology and roles within the CNS. Br J Pharmacol 165:20–34
- Brown AS. 2011. The environment and susceptibility to schizophrenia. Prog Neurobiol 93:23–58
- Bulut M, Savas HA, Altindag A, Virit O, Dalkilic A. 2009. Beneficial effects of N-acetylcysteine in treatment resistant schizophrenia. World J Biol Psychiatry 10:626–628
- Burdo J, Dargusch R, Schubert D. 2006. Distribution of the cystine/glutamate antiporter system x-c in the brain, kidney, and duodenum. J Histochem Cytochem 54:549–557
- Butterworth RF. 1999. Hypoglycemic encephalopathy. In: Siegel GJ, Agranoff BW, Albers RW, Fisher SK, Uler MD, eds. Basic neurochemistry: Molecular, cellular and medical aspects. 6th ed. Philadelphia: Lippincott-Raven
- Carpenter VK, Drake LL, Aguirre SE, Price DP, Rodriguez SD, Hansen IA. 2012. SLC7 amino acid transporters of the yellow fever mosquito Aedes aegypti and their role in fat body TOR signaling and reproduction. J Insect Physiol 58(4):513–522
- Chávez E, Castro-Sánchez L, Shibayama M, Tsutsumi V, Moreno MG, Muriel P. 2012. Sulfasalazine prevents the increase in TGF-β, COX-2, nuclear NFκB translocation and fibrosis in CCl4-induced liver cirrhosis in the rat. Hum Exp Toxicol 31(9):913–920
- Cho RY, Konecky RO, Carter CS. 2006. Impairments in frontal cortical gamma synchrony and cognitive control in schizophrenia. Proc Natl Acad Sci USA 103:19878–19883
- Cho Y, Bannai S. 1990. Uptake of glutamate and cystine in C-6 glioma cells and in cultured astrocytes. J Neurochem 55:2091–2097
- Choi IY, Lee SP, Kim SG, Gruetter R. 2001. In vivo measurements of brain glucose transport using the reversible Michaelis-Menten model and simultaneous measurements of cerebral blood flow changes during hypoglycemia. J Cereb Blood Flow Metab 21:653–663
- Chua SE, Murray RM. 1996. The neurodevelopmental theory of schizophrenia: evidence concerning structure and neuropsychology. Ann Med 28:547–555
- Chung WJ, Lyons SA, Nelson GM, Hamza H, Gladson CL, Gillespie GY, Sontheimer H. 2005. Inhibition of cystine uptake disrupts the growth of primary brain tumors. J Neurosci 25(31):7101–7110
- Clark J, Clore EL, Zheng K, Adame A, Masliah E, Simon DK. 2010. Oral N-acetyl-cysteine attenuates loss of dopaminergic terminals in alpha-synuclein overexpressing mice. PLoS One 5:12333
- Conn PJ, Pin JP. 1997. Pharmacology and functions of metabotropic glutamate receptors. Annu Rev Pharmacol Toxicol 37:205–237
- Correale J, Olsson T, Björk J, Smedegård G, Höjeberg B, Link H. 1991. Sulfasalazine aggravates experimental autoimmune encephalomyelitis and causes an increase in the number of autoreactive T cells. J Neuroimmunol 34:109–120
- Coyle JT, Tsai G, Goff D. 2003. Converging evidence of NMDA receptor hypofunction in the pathophysiology of schizophrenia. Ann NY Acad Sci 1003:318–327
- D’Ascenzo M, Fellin T, Terunuma M, Revilla-Sanchez R, Meaney DF, Auberson YP, et al. 2007. mGluR5 stimulates gliotransmission in the nucleus accumbens. Proc Natl Acad Sci USA 104:1995–2000
- Diamond JS, Jahr CE. 2000. Synaptically released glutamate does not overwhelm transporters on hippocampal astrocytes during high-frequency stimulation. J Neurophysiol 83:2835–2843
- De Bundel D, Schallier A, Loyens E, Fernando R, Miyashita H, Van Liefferinge J, Vermoesen K, Bannai S, Sato H, Michotte Y, Smolders I, Massie A. 2011. Loss of system does not induce oxidative stress but decreases extracellular glutamate in hippocampus and influences spatial working memory and limbic seizure susceptibility. J Neurosci 13:31(15):5792–5803
- de Groot J, Sontheimer H. 2011. Glutamate and the biology of gliomas. Glia 59:1181–1189
- Dennis SC, Clark JB. 1978. The regulation and glutamate metabolism by tricarboxylic acid-cycle activity in rat brain mitochondria. Biochem J 172:155–162
- DiNunzio JC, Williams RO III. 2008. CNS disorders – current treatment options and the prospects for advanced therapies. Drug Dev Ind Pharm 34(11):1141–1167
- Dixon SJ, Patel DN, Welsch M, Skouta R, Lee ED, Hayano M, et al. 2014. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife 20(3):2523
- Do KQ, Trabesinger AH, Kirsten-Kruger M, Lauer CJ, Dydak U, Hell D, et al. 2000. Schizophrenia: glutathione deficit in cerebrospinal fluid and prefrontal cortex in vivo. Eur J Neurosci 12:3721–3728
- Domercq M, Sánchez-Gómez MV, Sherwin C, Etxebarria E, Fern R, Matute C. 2007. System and glutamate transporter inhibition mediates microglial toxicity to oligodendrocytes. J Immunol 178(10):6549–6556
- Dringen R, Pfeiffer B, Hamprecht B. 1999. Synthesis of the antioxidant glutathione in neurons: supply by astrocytes of CysGly as precursor for neuronal glutathione. J Neurosci 19:562–569
- Erickson AM, Nevarea Z, Gipp JJ, Mulcahy RT. 2002. Identification of a variant antioxidant response element in the promoter of the human glutamate-cysteine ligase modifier subunit gene. Revision of the ARE consensus sequence. J Biolog Chem 277:30730–30737
- Evans SJ, Choudary PV, Neal CR, Li JZ, Vawter MP, Tomita H, et al. 2004. Dysregulation of the fibroblast growth factor system in major depression. Proc Natl Acad Sci USA 101:15506–15511
- Featherstone DE. 2011. Glial solute carrier transporters in Drosophila and mice. Glia 59(9):1351–1363
- Gasol E, Jiménez-Vidal M, Chillarón J, Zorzano A, Palacín M. 2004. Membrane topology of system light subunit reveals a re-entrant loop with substrate-restricted accessibility. J Biol Chem 279(30):31228–31236
- Gemma C, Vila J, Bachstetter A, Bickford PC. Oxidative stress and the aging brain: From theory to prevention. In: Riddle DR, editor. Brain aging: Models, methods, and mechanisms. Boca Raton, FL: CRC Press; 2007. Chapter 15
- Giulian D, Haverkamp LJ, Li J, Karshin WL, Yu J, Tom D, et al. 1995. Senile plaques stimulate microglia to release a neurotoxin found in Alzheimer brain. Neurochem Int 27:119–137
- Gochenauer GE, Robinson MB. 2001. Dibutyryl-cAMP (dbcAMP) upregulates astrocytic chloride-dependent L-[3H] transport and expression of both system subunits. J Neurochem 78:276–286
- Goldberg MP, Choi DW. 1993. Combined oxygen and glucose deprivation in cortical cell culture: calcium-dependent and calcium-independent mechanisms of neuronal injury. J Neurosci 13:3510–3524
- Gonzalez-Burgos G, Hashimoto T, Lewis DA. 2010. Alterations of cortical GABA neurons and network oscillations in schizophrenia. Curr Psychiatry Rep 12:335–344
- Gonzalez-Burgos G, Lewis DA. 2008. GABA neurons and the mechanisms of network oscillations: implications for understanding cortical dysfunction in schizophrenia. Schizophr Bull 34:944–961
- Gout PW, Buckley AR, Simms CR, Bruchovsky N. 2001. Sulfasalazine, a potent suppressor of lymphoma growth by inhibition of the x(c)- cystine transporter: a new action for an old drug. Leukemia 15(10):1633–1640
- Gout PW, Kang YJ, Buckley DJ, Bruchovsky N, Buckley AR. 1997. Increased cystine uptake capability associated with malignant progression of Nb2 lymphoma cells. Leukemia 11:1329–1337
- Grosjean Y, Grillet M, Augustin H, Ferveur JF, Featherstone DE. 2008. A glial amino-acid transporter controls synapse strength and courtship in Drosophila. Nat Neurosci 11:54–61
- Gupta DS, McCullumsmith RE, Beneyto M, Haroutunian V, Davis KL, Meador-Woodruff JH. 2005. Metabotropic glutamate receptor protein expression in the prefrontal cortex and striatum in schizophrenia. Synapse 57:123–131
- Halliwell B. 2006. Oxidative stress and neurodegeneration: where are we now? J Neurochem 97:1634–1658
- Hardy J, Selkoe DJ. 2002. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297:353–356
- Hayashi Y, Sekiyama N, Nakanishi S, Jane DE, Sunter DC, Birse EF, et al. 1994. Analysis of agonist and antagonist activities of phenylglycine derivatives for different cloned metabotropic glutamate receptor subtypes. J Neurosci 5(2):3370–3377
- Hosoya K, Tomi M, Ohtsuki S, Takanaga H, Saeki S, Kanai Y, et al. 2002. Enhancement of L-cystine transport activity and its relation to xCT gene induction at the blood-brain barrier by diethyl maleate treatment. J Pharmacol Exp Ther 302:225–231
- Ichord RN, Northington FJ, van Wylen D, Johnston MV, Kwon C, Traystman RJ. 1999. Brain O2 consumption and glutamate release during hypoglycemic coma in piglets are temperature sensitive. Am J Physiol Heart Circ Physiol 276:2053–2062
- Ishii K, Muelhauser F, Liebl U, Picard M, Kühl S, Penke B, et al. 2000. Sub-acute NO generation induced by Alzheimer’s beta-amyloid in the living brain: reversal by inhibition of the inducible NO synthase. FASEB J 14:1485–1489
- Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD, Yamamoto M. 1999. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev 13:76–86
- Jabaudon D, Shimamoto K, Yasuda-Kamatani Y, Scanziani M, Gähwiler BH, Gerber U. 1999. Inhibition of uptake unmasks rapid extracellular turnover of gluamate of nonvesicular origin. Proc Natl Acad Sci USA 96:8733–8738
- Jackman NA, Melchior SE, Hewett JA, Hewett SJ. 2012. Non-cell autonomous influence of the astrocyte system on hypoglycaemic neuronal cell death. ASN Neuro 4(1):e00074
- Kalivas PW. 2004. Glutamate systems in cocaine addiction. Curr Opin Pharmacol 4(1):23–29
- Kalivas PW. 2009. The glutamate homeostasis hypothesis of addiction. Nat Rev Neurosci 10:561–572
- Kalivas PW, Volkow N, Seamans J. 2005. Unmanageable motivation in addiction: a pathology in prefrontal-accumbens glutamate transmission. Neuron 45:647–650
- Kau KS, Madayag A, Mantsch JR, Grier MD, Abdul hameed O, Baker DA. 2008. Blunted cystine-glutamate antiporter function in the nucleus accumbens promotes cocaine-induced drug seeking. Neuroscience 155:530–537
- Kilberg MS, Pan Y-X, Chen H, Leung-Pineda V. 2005. Nutritional control of gene expression: how mammalian cells respond to amino acid limitation. Annu Rev Nutr 25:59–85
- Kilberg MS, Hausingger D. 1992. Mammalian amino acid transport mechanism and control. Cambridge (MA): Cell Press
- Kim JY, Kanai Y, Chairoungdua A, Cha SH, Matsuo H, Kim DK, et al. 2001. Human cystine/glutamate transporter: cDNA cloning and upregulation by oxidative stress in glioma cells. Biochim Biophys Acta 1512:335–344
- Kimball SR. 1999. Eukaryotic initiation factor eIF2. Int J Biochem Cell Biol 31:25–29
- Kleijn M, Proud CG. 2000. The activation of eukaryotic initiation factor (eIF)2B by growth factors in PC12 cells requires MEK/ERK signaling. FEBS Lett 476:262–265
- Knackstedt LA, LaRowe S, Mardikian P, Malcolm R, Upadhyaya H, Hedden S, et al. 2009. The role of cystine-glutamate exchange in nicotine dependence in rats and humans. Biol Psychiatry 65:841–845
- Kodydková J, Vávrová L, Zeman M, Jirák R, Macásek J, Stanková B, et al. 2009. Antioxidative enzymes and increased oxidative stress in depressive women. Clin Biochem 42:1368–1374
- Koh JY, Yang LL, Cotman CW. 1990. Beta-amyloid protein increases the vulnerability of cultured cortical neurons to excitotoxic damage. Brain Res 533:315–320
- Kohr G, Eckardt S, Luddens H, Monyer H, Seeburg PH. 1994. NMDA receptor channels: subunit-specific potentiation by reducing agents. Neuron 12:1031–1040
- Kranich O, Dringen R, Sandberg M, Hamprecht B. 1998. Utilization of cysteine and cysteine precursors for the synthesis of glutathione in astroglial cultures: preference for cystine. Glia 22:11–18
- Kupchik YM, Moussawi K, Tang XC, Wang X, Kalivas BC, Kolokithas R, et al. 2012. The effect of N-acetylcysteine in the nucleus accumbens on neurotransmission and relapse to cocaine. Biol Psychiatry 71:978–986
- Kusuhara H, Furuie H, Inano A, Sunagawa A, Yamada S, Wu C, et al. 2012. Pharmacokinetic interaction study of sulphasalazine in healthy subjects and the impact of curcumin as an in vivo inhibitor of BCRP. Br J Pharmacol 166:1793–1803
- Langan SJ, Deary IJ, Hepburn DA, Frier BM. 1991. Cumulative cognitive impairment following recurrent severe hypoglycaemia in adult patients with insulin-treated diabetes mellitus. Diabetologia 34:337–344
- LaRowe SD, Myrick H, Hedden S, Mardikian P, Saladin M, McRae A, et al. 2007. Is cocaine desire reduced by N-acetylcysteine? Am J Psychiatry 164:1115–1117
- Lee JM, Johnson JA. 2004. An important role of Nrf2-ARE pathway in the cellular defense mechanism. J Biochem Mol Biol 37:139–143
- Lehre KP, Danbolt NC. 1998. The number of glutamate transporter subtype molecules at glutamatergic synapses: chemical and stereological quantification in young adult rat brain. J Neurosci 18:8751–8757
- Lewerenz J, Baxter P, Kassubek R, Albrecht P, Van Liefferinge J, Westhoff MA, et al. 2014. Phosphoinositide 3-kinases upregulate system xc(-) via eukaryotic initiation factor 2α and activating transcription factor 4. A pathway active in glioblastomas and epilepsy. Antioxid Redox Signal 20(18):2907–2922
- Lewerenz J, Hewett SJ, Huang Y, Lambros M, Gout PW, Kalivas PW, et al. 2013. The cystine/glutamate antiporter system x(c)(-) in health and disease: from molecular mechanisms to novel therapeutic opportunities. Antioxid Redox Signal 18(5):522–555
- Lewerenz J, Albrecht P, Tien ML, Henke N, Karumbayaram S, Kornblum HI, et al. 2009. Induction of Nrf2 and xCT are involved in the action of the neuroprotective antibiotic ceftriaxone in vitro. J Neurochem 111:332–343
- Lewerenz J, Maher P. 2009. Basal levels of eIF2alpha phosphorylation determine cellular antioxidant status by regulating ATF4 and xCT expression. J Biol Chem 284:1106–1115
- Lim J, Li L, Kistler J, Donaldson PJ. 2007. Mapping of glutathione and its precursor amino acids reveals a role for GLYT2 in glycine uptake in the lens core. Invest Ophth Vis Sci 48:5142–5151
- Lin X, Yang H, Zhang H, Zhou L, Guo Z. 2013. A novel transcription mechanism activated by ethanol: induction of Slc7a11 gene expression via inhibition of the DNA-binding activity of transcriptional repressor octamer-binding transcription factor 1 (OCT-1). J Biol Chem 288:14815–14823
- Lipton SA, Choi YB, Takahashi H, Zhang D, Li W, Godzik A, Bankston LA. 2002. Cysteine regulation of protein function – as exemplified by NMDA-receptor modulation. Trends Neurosci 25:474–480
- Liu X, Resch J, Rush T, Lobner D. 2012. Functional upregulation of system by fibroblast growth factor-2. Neuropharmacology 62(2):901–906
- Lo M, Wang YZ, Gout PW. 2008. The x(c)- cystine/glutamate antiporter: a potential target for therapy of cancer and other diseases. J Cell Physiol 215:593–602
- Lucchinetti C, Bruck W, Parisi J, Scheithauer B, Rodriguez M, Lassmann H. 1999. A quantitative analysis of oligodendrocytes in multiple sclerosis lesions. A study of 113 cases. Brain 122:2279–2295
- Lutgen V. 2012. The contribution of system to prefrontal cortical mediated behaviors associated with schizophrenia. Dissertations, Marquette University, Milwaukee, Wisconsin, USA
- Madayag A, Kau KS, Lobner D, Mantsch JR, Wisniewski S, Baker DA. 2010. Drug-induced plasticity contributing to heightened relapse susceptibility: neurochemical changes and augmented reinstatement in high-intake rats. J Neurosci 30:210–217
- Madayag A, Lobner D, Kau KS, Mantsch JR, Abdulhameed O, Hearing M, et al. 2007. Repeated N-acetylcysteine administration alters plasticity dependent effects of cocaine. J Neurosci 27:13968–13976
- Mardikian PN, LaRowe SD, Hedden S, Kalivas PW, Malcolm RJ. 2007. An open-label trial of N-acetylcysteine for the treatment of cocaine dependence: a pilot study. Prog Neuropsychopharmacol Biol Psychiatry 31:389–394
- Marino MJ, Conn PJ. 2002. Direct and indirect modulation of the N-methyl D-aspartate receptor. Curr Drug Targets CNS Neurol Disord 1:1–16
- Martin HL, Teismann P. 2009. Glutathione – a review on its role and significance in Parkinson’s disease. FASEB J 23(10):3263–3272
- Massie A, Schallier A, Kim SW, Fernando R, Kobayashi S, Beck H, et al. 2011. Dopaminergic neurons of system x(c)−-deficient mice are highly protected against 6-hydroxydopamine-induced toxicity. FASEB J 25(4):1359–1369
- Massie A, Schallier A, Mertens B, Vermoesen K, Bannai S, Sato H, et al. 2008. Time-dependent changes in striatal xCT protein expression in hemi-Parkinson rats. NeuroReport 19:1589–1592
- Matti A, Mirzaei J, Rudolph J, Smith SA, Newell JL, Patel SA, et al. 2013. Microwave accelerated synthesis of isoxazole hydrazide inhibitors of the system transporter: initial homology model. Bioorg Med Chem Lett 23:5931–5935
- Mattson MP, Cheng B, Davis D, Bryant K, Lieberburg I, Rydel RE. 1992. β-Amyloid peptides destabilize calcium homeostasis and render human cortical neurons vulnerable to excitotoxicity. J Neurosci 12:376–389
- Matute C, Sanchez-Gomez MV, Martinez-Millan L, Miledi R. 1997. Glutamate receptor-mediated toxicity in optic nerve oligodendrocytes. Proc Natl Acad Sci USA 94:8830–8835
- McCarthy DJ, Alexander R, Smith MA, Pathak S, Kanes S, Lee CM, et al. 2012. Glutamate-based depression GBD. Med Hypotheses 78(5):675–681
- McDonald DR, Brunden KR, Landreth GE. 1997. Amyloid fibrils activate tyrosine kinase-dependent signaling and superoxide production in microglia. J Neurosci 17:2284–2294
- Meda L, Cassatella MA, Szendrei GI, Otvos L Jr, Baron P, Villalba M, et al. 1995. Activation of microglial cells by beta-amyloid protein and interferon-gamma. Nature 374:647–650
- Mohan A, Pendyam S, Kalivas PW, Nair SS. 2011. Molecular diffusion model of neurotransmitter homeostasis around synapses supporting gradients. Neural Comput 23:984–1014
- Monyer H, Choi DW. 1988. Morphinans attenuate cortical neuronal injury induced by glucose deprivation in vitro. Brain Res 446:144–148
- Monyer H, Goldberg MP, Choi DW. 1989. Glucose deprivation neuronal injury in cortical culture. Brain Res 483:347–354
- Moran MM, McFarland K, Melendez RI, Kalivas PW, Seamans JK. 2005. Cystine/glutamate exchange regulates metabotropic glutamate receptor presynaptic inhibition of excitatory transmission and vulnerability to cocaine seeking. J Neurosci 25:6389–6393
- Mosckovitz R, Yan N, Heimer E, Felix A, Tate SS, Udenfriend S. 1993. Characterization of the rat neutral and basic amino acid transporter utilizing anti-peptide antibodies. Proc Natl Acad Sci USA 90:4022–4026
- Moussawi K, Zhou W, Shen H, Reichel CM, See RE, Carr DB, Kalivas PW. 2011. Reversing cocaine-induced synaptic potentiation provides enduring protection from relapse. Proc Natl Acad Sci USA 108:385–390
- Murata M, Horiuchi E, Kanazawa I. 2001. Zonisamide has beneficial effects on Parkinson’s disease patients. Neurosci Res 41:397–399
- Musazzi L, Treccani G, Popoli M. 2012. Glutamate hypothesis of depression and its consequences for antidepressant treatments. Expert Rev Neurother 12(10):1169–1172
- Nakamura E, Sato M, Yang H, Miyagawa F, Harasaki M, Tomita K, et al. 1999. 4F2 (CD98) heavy chain is associated covalently with an amino acid transporter and controls intracellular trafficking and membrane topology of 4F2 heterodimer. J Biol Chem 274(5):3009–3016
- National Cancer Institute (NCI). 2015. Sorafenib and temsirolimus in treating patients with recurrent glioblastoma. Available from: ClinicalTrials.gov [Internet]. Bethesda, MD: National Library of Medicine (USA). 2000- [accessed 2 April 2015]
- Newell JL, Keyari CM, McDaniel SW, Diaz PJ, Natale NR, Patel SA, Bridges RJ. 2014. Novel di-aryl-substituted isoxazoles act as noncompetitive inhibitors of the system Xc(-) cystine/glutamate exchanger. Neurochem Int 73:132–138
- Niture SK, Jain AK, Jaiswal AK. 2009. Antioxidant-induced modification of INrf2 cysteine 151 and PKC-delta-mediated phosphorylation of Nrf2 serine 40 are both required for stabilization and nuclear translocation of Nrf2 and increased drug resistance. J Cell Sci 15(122):4452–4464
- Noseworthy JH, O’Brien P, Erickson BJ, Lee D, Sneve D, Ebers GC, et al. 1998. The Mayo Clinic-Canadian Cooperative trial of sulfasalazine in active multiple sclerosis. Neurology 151:1342–1352
- Olivieri G, Baysang G, Meier F, Müller-Spahn F, Stähelin HB, Brockhaus M, Brack C. 2001. N-Acetyl-L-cysteine protects SHSY5Y neuroblastoma cells from oxidative stress and cell cytotoxicity: effects on beta-amyloid secretion and tau phosphorylation. J Neurochem 76:224–233
- Ogunrinu TA, Sontheimer H. 2010. Hypoxia increases the dependence of glioma cells on glutathione. J Biolog Chem 285:37716–37724
- Onyango IG, Bennett JP Jr, Tuttle JB. 2005. Endogenous oxidative stress in sporadic Alzheimer’s disease neuronal cybrids reduces viability by increasing apoptosis through pro-death signaling pathways and is mimicked by oxidant exposure of control cybrids. Neurobiol Dis 19:312–322
- Pampliega O, Domercq M, Soria FN, Villoslada P, Rodríguez-Antigüedad A, Matute C. 2011. Increased expression of cystine/glutamate antiporter in multiple sclerosis. J Neuroinflammation 3(8):63
- Park SW, Kim SH, Park KH, Kim SD, Kim JY, Baek SY, et al. 2004. Preventive effect of antioxidants in MPTP-induced mouse model of Parkinson’s disease. Neurosci Lett 363:243–246
- Pauly K, Fritz K, Furey A, Lobner D. 2011. Insulin-like growth factor 1 and transforming growth factor-β stimulate cystine/glutamate exchange activity in dental pulp cells. J Endod 37:943–947
- Piani D, Fontana A. 1994. Involvement of the cystine transport system in the macrophage-induced glutamate-dependent cytotoxicity to neurons. J Immunol 152:3578–3585
- Pierce RC, Bell K, Duffy P, Kalivas PW. 1996. Repeated cocaine augments excitatory amino acid transmission in the nucleus accumbens only in rats having developed behavioral sensitization. J Neurosci 16:1550–1560
- Pow DV. 2001. Visualizing the activity of the cystine-glutamate antiporter in glial cells using antibodies to aminoadipic acid, a selectively transported substrate. Glia 34:27–38
- Proud CG. 2005. eIF2 and the control of cell physiology. Semin Cell Dev Biol 16:3–12
- Qin S, Colin C, Hinners I, Gervais A, Cheret C, Mallat M. 2006. System and apolipoprotein E expressed by microglia have opposite effects on the neurotoxicity of amyloid-beta peptide 1–40. J Neurosci 26:3345–3356
- Quevedo C, Alcázar A, Salinas M. 2000. Two different signal transduction pathways are implicated in the regulation of initiation factor 2B activity in insulin-like growth factor-1-stimulated neuronal cells. J Biolog Chem 275:19192–19197
- Raffa M, Mechri A, Othman LB, Fendri C, Gaha L, Kerkeni A. 2009. Decreased glutathione levels and antioxidant enzyme activities in untreated and treated schizophrenic patients. Prog Neuropsychopharmacol Biol Psychiatry 33:1178–1183
- Ramos-Chávez LA, Rendón-López CRR, Zepeda A, Silva-Adaya D, Del Razo LM, Gonsebatt ME. 2015. Neurological effects of inorganic arsenic exposure: altered cysteine/glutamate transport, NMDA expression and spatial memory impairment. Front Cell Neurosci 9:21
- Reichel CM, Moussawi K, Do PH, Kalivas PW, See RE. 2011. Chronic N-acetylcysteine during abstinence or extinction after cocaine self-administration produces enduring reductions in drug seeking. J Pharmacol Exp Ther 337:487–493
- Reid MS, Berger SP. 1996. Evidence for sensitization of cocaine induced nucleus accumbens glutamate release. Neuroreport 7:1325–1329
- Robe PA, Martin DH, Nguyen-Khac MT, Artesi M, Deprez M, Albert A, et al. 2009. Early termination of ISRCTN45828668, a phase 1/2 prospective, randomized study of Sulfasalazine for the treatment of progressing malignant gliomas in adults. BMC Cancer 9:372
- Rothstein JD. 2002. Paving new pathways. Nat Med 8:938–940
- Rowlands AG, Panniers R, Henshaw EC. 1988. The catalytic mechanism of guanine nucleotide exchange factor action and competitive inhibition by phosphorylated eukaryotic initiation factor 2. J Biolog Chem 263:5526–5533
- Rung JP, Carlsson A, Ryden Markinhuhta K, Carlsson ML. 2005. (+)-MK-801 induced social withdrawal in rats; a model for negative symptoms of schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry 29:827–832
- Ryan CM, Atchison J, Puczynski S, Puczynski M, Arslanian S, Becker D. 1990. Mild hypoglycemia associated with deterioration of mental efficiency in children with insulin-dependent diabetes mellitus. J Pediatr 117:32–38
- Sagara JI, Miura K, Bannai S. 1993. Maintenance of neuronal glutathione by glial cells. J Neurochem 61:1672–1676
- Sanacora G, Treccani G, Popoli M. 2012. Towards a glutamate hypothesis of depression: an emerging frontier of neuropsychopharmacology for mood disorders. Neuropharmacology 62(1):63–77
- Sandberg M, Butcher SP, Hagberg H. 1986. Extracellular overflow of neuroactive amino acids during severe insulin-induced hypoglycemia: in vivo dialysis of the rat hippocampus. J Neurochem 47:178–184
- Sasaki H, Sato H, Kuriyama-Matsumura K, Sato K, Maebara K, Wang H, et al. 2002. Electrophilic response element-mediated induction of the cystine/gluamate exchange transporter gene expression. J Biol Chem 274:44765–44771
- Sato H, Nomura S, Maebara K, Sato K, Tamba M, Bannai S. 2004. Transcriptional control of cystine/glutamate transporter gene by amino acid deprivation. Biochem Biophys Res Commun 325(1):109–116
- Sato H, Tamba M, Okuno S, Sato K, Keino-Masu K, Masu M, Bannai S. 2002. Distribution of cystine/glutamate exchange transporter, system x(c)-, in the mouse brain. J Neurosci 22(18):8028–8033
- Sato H, Kuriyama-Matsumura K, Hashimoto T, Sasaki H, Wang H, Ishii T, et al. 2001. Effect of oxygen on induction of the cystine transporter by bacterial lipopolysaccharide in mouse peritoneal macrophages. J Biolog Chem 276:10407–10412
- Sato H, Tamba M, Kuriyama-Matsumura K, Okuno S, Bannai S. 2000. Molecular cloning and expression of human xCT, the light chain of amino acid transport system . Antioxid Redox Signal 2:665–671
- Sato H, Tamba M, Ishii T, Bannai S. 1999. Cloning and expression of a plasma membrane cystine/glutamate exchange transporter composed of two distinct proteins. J Biol Chem 274(17):11455–11458
- Savaskan NE, Heckel A, Hahnen E, Engelhorn T, Doerfler A, Ganslandt O, et al. 2008. Small interfering RNA-mediated xCT silencing in gliomas inhibits neurodegeneration and alleviates brain edema. Nat Med 14(6):629–632
- Savtchenko LP, Rusakov DA. 2004. Glutamate escape from a tortuous synaptic cleft of the hippocampal mossy fibre synapse. Neurochem Int 45:479–484
- Schallier A, Smolders I, Van Dam D, Loyens E, De Deyn PP, Michotte A, et al. 2011. Region- and age-specific changes in glutamate transport in the AβPP23 mouse model for Alzheimer’s disease. J Alzheimers Dis 24(2):287–300
- Schulz JB, Lindenau J, Seyfried J, Dichgans J. 2000. Glutathione, oxidative stress and neurodegeneration. Eur J Biochem 267:4904–4911
- Seib TM, Patel SA, Bridges RJ. 2011. Regulation of the system x(C)- cystine/glutamate exchanger by intracellular glutathione levels in rat astrocyte primary cultures. Glia 59(10):1387–1401
- Shanker G, Aschner M. 2001. Identification and characterization of uptake systems for cystine and cysteine in cultured astrocytes and neurons: evidence for methylmercury-targeted disruption of astrocyte transport. J Neurosci Res 66:998–1002
- Shih AY, Erb H, Sun X, Toda S, Kalivas PW, Murphy TH. 2006. Cystine/glutamate exchange modulates glutathione supply for neuroprotection from oxidative stress and cell proliferation. J Neurosci 26(41):10514–10523
- Shukla K, Thomas AG, Ferraris DV, Hin N, Sattler R, Alt J, et al. 2011. Inhibition of transporter-mediated cystine uptake by sulfasalazine analogs. Bioorg Med Chem Lett 21(20):6184–6187
- Sibson NR, Dhankhar A, Mason GF, Rothman DL, Behar KL, Shulman RG. 1998. Stoichiometric coupling of brain glucose metabolism and glutamatergic neuronal activity. Proc Natl Acad Sci USA 95(1):316–321
- Silverstein FS, Simpson J, Gordon KE. 1990. Hypoglycemia alters striatal amino acid efflux in perinatal rats: an in vivo microdialysis study. Ann Neurol 28:516–521
- Sims B, Clarke M, Francillion L, Kindred E, Hopkins ES, Sontheimer H. 2012. Hypoxic preconditioning involves system Xc- regulation in mouse neural stem cells. Stem Cell Res 8:285–291
- Singer E, Judkins J, Salomonis N, Matlaf L, Soteropoulos P, McAllister S, Soroceanu L. 2015. Reactive oxygen species-mediated therapeutic response and resistance in glioblastoma. Cell Death Dis 6:e1601–e1611
- Stover JF, Pleines UE, Morganti-Kossmann MC, Kossmann T, Lowitzsch K, Kempski OS. 1997. Neurotransmitters in cerebrospinal fluid reflect pathological activity. Eur J Clin Invest 27:1038–1043
- Suh SW, Hamby AM, Swanson RA. 2007. Hypoglycemia, brain energetics, and hypoglycemic neuronal death. Glia 55:1280–1286
- Takaki M, Ueda Y, Doi T, Nagatomo K, Murashima YL, Kannan H. 2008. Molecular regulation of antioxidant ability in the hippocampus of EL mice. Brain Res 1228:1–5
- Takano T, Lin JH, Arcuino G, Gao Q, Yang J, Nedergaard M. 2001. Glutamate release promotes growth of malignant gliomas. Nat Med 7:1010–1015
- Takeuchi S, Wada K, Toyooka T, Shinomiya N, Shimazaki H, Nakanishi K, et al. 2013. Increased xCT expression correlates with tumor invasion and outcome in patients with glioblastomas. Neurosurgery 72(1):33–41
- Tanel A, Averill-Bates DA. 2007. Inhibition of acrolein-induced apoptosis by the antioxidant N-acetylcysteine. J Pharmacol Exp Ther 321:73–83
- Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mork S, Bo L. 1998. Axonal transection in the lesions of multiple sclerosis. N Engl J Med 338:278–285
- Uys JD, LaLumiere RT. 2008. Glutamate: the new frontier in pharmacotherapy for cocaine addiction. CNS Neurol Disord Drug Targets 7:482–491
- Venugopal R, Jaiswal AK. 1996. Nrf1 and Nrf2 positively and c-Fos and Fra1 negatively regulate the human antioxidant response element-mediated expression of NAD(P)H:quinone oxidoreductase1 gene. Proc Natl Acad Sci USA 93:14960–14965
- Verma A, Moghaddam B. 1996. NMDA receptor antagonists impair prefrontal cortex function as assessed via spatial delayed alternation performance in rats: modulation by dopamine. J Neurosci 16:373–379
- Verrey F, Closs EI, Wagner CA, Palacin M, Endou H, Kanai Y. 2004. CATs and HATs: the SLC7 family of amino acid transporters. Pflugers Arch 447(5):532–542
- Wadiche JI, Arriza JL, Amara SG, Kavanaugh MP. 1995. Kinetics of a human glutamate transporter. Neuron 14:1019–1027
- Wagner CA, Lang F, Bröer S. 2001. Function and structure of heterodimeric amino acid transporters. Am J Physiol Cell Physiol 1(4):C1077–1093
- Wang XF, Cynader MS. 2000. Astrocytes provide cysteine to neurons by releasing glutathione. J Neurochem 74:1434–1442
- Waniewski RA, Martin DL. 1984. Characterization of L-glutamic acid transport by glioma cells in culture: evidence for sodium-independent, chloride-dependent high affinity influx. J Neurosci 4(9):2237–2246
- Weinberger DR. 1987. Implications of normal brain development for the pathogenesis of schizophrenia. Arch Gen Psychiatry 44:660–669
- Weldon DT, Rogers SD, Ghilardi JR, Finke MP, Cleary JP, O’Hare E, et al. 1998. Fibrillar β-amyloid induces microglial phagocytosis, expression of inducible nitric oxide synthase, and loss of a select population of neurons in the rat CNS in vivo. J Neurosci 18:2161–2173
- Wells RG, Hediger MA. 1992. Cloning of a rat kidney cDNA that stimulates dibasic and neutral amino acid transport and has sequence similarity to glucosidases. Proc Natl Acad Sci USA 89:5596–5600
- Wells RG, Lee WS, Kanai Y, Leiden JM, Hediger MA. 1992. The 4F2 antigen heavy chain induces uptake of neutral and dibasic amino acids in Xenopus oocytes. J Biol Chem 267:15285–15288
- Welsh GI, Miller CM, Loughlin AJ, Price NT, Proud CG. 1998. Regulation of eukaryotic initiation factor eIF2B: glycogen synthase kinase-3 phosphorylates a conserved serine which undergoes dephosphorylation in response to insulin. FEBS Lett 421:125–130
- Welsh GI, Proud CG. 1993. Glycogen synthase kinase-3 is rapidly inactivated in response to insulin and phosphorylates eukaryotic initiation factor eIF-2B. Biochem J 294:625–629
- Wolf ME. 2010. The Bermuda Triangle of cocaine-induced neuroadaptations. Trends Neurosci 33:391–398
- Xi ZX, Ramamoorthy S, Baker DA, Shen H, Samuvel DJ, Kalivas PW. 2002. Modulation of group II metabotropic glutamate receptor signaling by chronic cocaine. J Pharmacol Exp Ther 303:608–615
- Xi ZX, Shen H, Baker DA, Kalivas PW. 2003. Inhibition of non-vesicular glutamate release by group III metabotropic glutamate receptors in the nucleus accumbens. J Neurochem 87:1204–1212
- Xu C, Yogaratnam J, Lua R, Naik S, Khoo CL, Pillai SS, Sim K. 2011. Persistent, severe hypoglycemia-induced organic brain syndrome with neurological sequelae: a case report. Gen Hosp Psychiatry 33:9–11
- Ye ZC, Rothstein JD, Sontheimer H. 1999. Compromised glutamate transport in human glioma cells: reduction-mislocalization of sodium-dependent glutamate transporters and enhanced activity of cystine-glutamate exchange. J Neurosci 19:10767–10777
- Ye ZC, Sontheimer H. 1999. Glioma cells release excitotoxic concentrations of glutamate. Cancer Res 59:4383–4391
- Yang Y, Yee D. 2014. IGF-I regulates redox status in breast cancer cells by activating the amino acid transport molecule . Cancer Res 74(8):2295–2305
- Zechel S, Werner S, Unsicker K, von Bohlen und Halbach O, . 2010. Expression and functions of fibroblast growth factor 2 (FGF-2) in hippocampal formation. Neuroscientist 16:357–373
- Zhang P, McGrath BC, Reinert J, Olsen DS, Lei L, Gill S, et al. 2002. The GCN2 eIF2alpha kinase is required for adaptation to amino acid deprivation in mice. Mol Cell Biol 22:6681–6688
- Zheng K, Scimemi A, Rusakov DA. 2008. Receptor actions of synaptically released glutamate: the role of transporters on the scale from nanometers to microns. Biophys J 95:4584–4596