Abstract
Reconsolidation is the process by which previously consolidated memories are stabilized after retrieval. Several lines of evidence indicate that glucocorticoids modulate distinct phases of learning and memory. These effects are considered to be mediated by mineralocorticoid receptors and glucocorticoid receptors (GRs), which display a high concentration and distinct distribution in the hippocampus. The role of glucocorticoid system in fear memory reconsolidation is the subject of some controversy. Moreover, we found no studies that assessed the role of hippocampal GRs in fear memory reconsolidation. Here, we investigated the effect of GR blockade on fear memory reconsolidation in rats. Rats were trained and tested in an inhibitory avoidance task. Intrahippocampal or systemic administration of the GR antagonist RU38486 immediately following memory reactivation produced a deficit in post-retrieval long-term memory that persisted over test sessions, and memory did not re-emerge following a footshock reminder. These results indicate that hippocampal GRs are required for reconsolidation of fear-based memory.
Introduction
During memory consolidation, labile short-term memories are converted into long-term memory. This process is time dependent and requires gene expression and protein synthesis (McGaugh Citation2000). Once consolidated, memory is sustained over long periods of inactivity, but it returns to a labile and vulnerable state in which it can be reinforced or altered. For instance, administration of protein synthesis inhibitors after the retrieval of a previously consolidated memory results in amnesia in subsequent retention tests (Debiec et al. Citation2002; Duvarci and Nader Citation2004), indicating that when recalled, consolidated memory returns to a labile state which initiates another time-dependent memory similar to that seen after novel learning. This phenomenon is now referred to as reconsolidation (Nader and Einarsson Citation2010). Molecular processes that underlie long-term behavioral changes during consolidation and reconsolidation share some common mechanisms but also display different characteristics (Alberini Citation2005).
Stressful stimuli activate the hypothalamic–pituitary–adrenocortical (HPA) axis. Glucocorticoids (corticosterone in rodents and cortisol in humans) are the final products of the HPA axis and are released by the adrenal glands. Glucocorticoid actions are mediated through two distinct intracellular receptor types: mineralocorticoid receptors (MRs) and glucocorticoid receptors (GRs). GRs have a low affinity for corticosterone and become occupied only during stress and at the circadian peak, when circulating levels of glucocorticoids are high. In contrast, MRs have a 10-fold higher affinity for corticosterone and are almost saturated under basal conditions (De Kloet et al. Citation1998). These two types of receptors also have distinctly different distributions in the brain. GRs are widely expressed throughout the brain, although the hippocampus has particularly high amounts. MRs are much more localized than GRs. Again the hippocampus has a high concentration, whereas expression is sparse elsewhere (De Kloet et al. Citation1998).
Glucocorticoids regulate a variety of biological functions including learning and memory. Animal and human studies indicate that adrenal glucocorticoids enhance memory consolidation while impairing memory retrieval (Roozendaal Citation2000; Pakdel and Rashidy-Pour Citation2006; Sajadi et al. Citation2006). GRs play differential roles in cognitive behavior. GRs are involved in the consolidation of recently acquired information, whereas MRs are essential for interpretation of environmental stimuli and selection of the appropriate behavioral response (Oitzl and de Kloet Citation1992). Recently, we have shown that MRs play an essential role in retrieval of spatial information (Khaksari et al. Citation2007).
Recent studies have shown that post-retrieval administration of glucocorticoids can influence subsequent expression of fear memory. Administration of corticosterone immediately after reactivation of a contextual fear memory disrupts subsequent recall in rats (Abrari et al. Citation2008). Systemic as well as intra-amygdala injections of the GR antagonist RU38486 impaired reconsolidation of fear memory (Tronel and Alberini Citation2007; Taubenfeld et al. Citation2009). We have found no studies assessed the role of GRs in the hippocampus in fear memory reconsolidation. This area contains a high density of GRs (De Kloet et al. Citation1998) and plays a crucial role in fear memory reconsolidation (Debiec et al. Citation2002). Thus, this study was designed to examine the hypothesis that hippocampal GRs have a role in the reconsolidation of an inhibitory avoidance, a form of contextual fear conditioning currently used as a model of a traumatic memory (Tronel and Alberini Citation2007; Taubenfeld et al. Citation2009). The learning of this task is known to require hippocampal protein synthesis (Quevedo et al. Citation1999).
Materials and methods
Animals
Adult male Wistar rats (230–260 g) were housed four per cage (60 × 40 × 20 cm) in a room with a natural light cycle and controlled temperature (24 ± 2°C). Food and water were available ad libitum. Behavioral procedures were conducted during the light phase of the cycle between 10:00 and 13:00 h. All procedures were conducted in agreement with the National Institutes of Health Guide for care and use of laboratory animals and were approved by the Ethics Committee of Semnan University of Medical Sciences, Iran. Every effort was made to minimize the number of animals used and their suffering.
Surgery and histology procedures
Approximately, 1 week prior to the initiation of the behavioral experiments, the rats were anesthetized with ketamine hydrochloride (70 mg/kg, i.p.) plus xylazine (14 mg/kg, i.p.). The rat's head was fixed in a stereotaxic apparatus, and a midline incision of the skin over the skull was made. The skull was dried and cleaned of fascia. Two permanent stainless steel guide cannulae (22 gauge, 10 mm) were aimed at 1 mm above the dorsal hippocampus, using the following coordinates relative to bregma: AP − 3.6 mm; L ± 2.48 mm (midline); DV − 3.4 mm from skull surface; with the nose bar 3.30 mm below the interaural line, implanted bilaterally (Paxinos and Watson Citation2005). The cannulae were affixed to the skull with dental acrylic; stylettes were inserted into the cannulae to keep them patent.
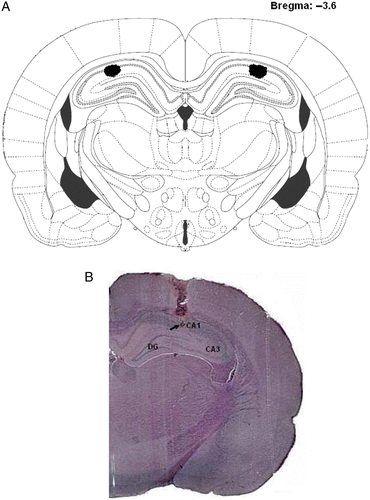
After completion of the behavioral tests, the rats were anesthetized with an overdose of thiopental sodium (100 mg/kg, i.p.). The brains were removed and placed in a 10% formalin solution for approximately 1 week, then sectioned into 40-μm coronal slices with a freezing microtome, and stained with cresyl violet. Cannula location () was determined using a light microscope and atlas plates (Paxinos and Watson Citation1998, Citation2005) by an observer blind to the behavioral results. Only rats with both cannula tips terminating within the dorsal hippocampus were included in the behavioral analysis ().
Figure 1. (A) Diagrammatic representation of a histology section, redrawn from Paxinos and Watson (Citation2005). The dark irregular areas define the range of cannula tip locations within the dorsal hippocampus. (B) A representative photomicrograph of a coronal section, stained with cresyl violet, illustrating placement of cannula and injection needle tip (arrow) in the dorsal hippocampus. CA1, CA3, Ammon's horn; DG, dentate gyrus.
Drugs and injection procedures
RU38486 (purchased from Simgma-Alderich, Stelnheim, Germany), a specific GR antagonist (Mao et al. Citation1992), was dissolved in propylene glycol and subsequently diluted in 0.9% saline to a final concentration of 0.3 or 3 ng/μl for intrahippocampal and 20 mg/kg for systemic injections. The final concentration of propylene glycol was 5%. These doses of RU38486 were sufficient to interfere with behavioral responses (Roozendaal and McGaugh Citation1997).
Intrahippocampal infusions of RU38486 (0.3 or 3 ng in 1 μl vehicle/hemisphere) or vehicle (1 μl/hemisphere) were performed (1 μl/min) through an injection needle (30 gauge, 11 mm) attached to a 10-μl Hamilton syringe via polyethylene tubing. The needle was equipped with a stopper limiting the depth of insertion to 1 mm beyond the tip of the cannula. The injection needle remained in the cannula for 1 min, following the infusion, in order to maximize diffusion away from the needle tip and to minimize dorsal diffusion. The volume and duration of intrahippocampal injections were chosen based on previously published reports (Amaral et al. Citation2007; Luft et al. Citation2008). Systemic injections were intraperitoneal and given in volumes of 2 ml/kg. Rats received RU38486 (20 mg in 2 ml/kg) or vehicle (2 ml/kg).
Inhibitory avoidance training and testing
The experimental apparatus was a shuttle box (Ugo Basile, Gemonio, Italy) divided into dark and light compartments. Both compartments had a grid floor (3-mm stainless steel rods spaced at 9 mm) connected to a shock generator. An automated apparatus registered the latency of passage from the light to the dark side of the box (step through latency). The apparatus was located in a sound attenuated room.
For the study of an inhibitory avoidance memory reconsolidation, we used a protocol similar to that of Tronel and Alberini (Citation2007). All experimental rats were first habituated to the apparatus. The rat was placed in an illuminated compartment and the guillotine door was raised 7 s later. Upon entering the dark compartment, the door was closed and the rat was taken from the dark compartment into the home cage. The acquisition trial was done 30 min later during which the door was closed and a 50 Hz, 1 mA constant current shock was applied for 3 s immediately after the rat had entered the dark compartment. The rat was removed from the dark compartment about 10 s after receiving the shock and returned to its home cage.
Forty-eight hours after training, memory reactivation was induced (Test 1; ). The rat was again placed in the illuminated compartment and the guillotine door was opened. Rats that entered the dark compartment were returned to their home cages immediately after entering. For rats that did not enter the dark side, the test was terminated at 540 s. Foot shock was not delivered during the retention test.
Figure 2. Effects of RU38486 administration following memory reactivation on fear memory reconsolidation. (A) Passive avoidance training/testing and drug administration schedule; intervals are 2 or 7 days (d). (B) Median ( ± interquartile ranges) step-through retention latencies of groups of rats systemically injected with 20 mg/kg of RU38486 (n = 8) or vehicle (VEH; n = 8) immediately after Test 1 (T1) and re-tested 2 days (Test 2), 7 days (Test 3), and after a reminder shock (Test 4). (C) Median ( ± interquartile ranges) step-through latencies of groups of rats received intrahippocampal injections of RU38486 (0.3 or 3 ng) (n = 8 in each group) or VEH (n = 8) immediately after Test 1 and re-tested 2 days (Test 2) and 7 days (Test 3) later and after a reminder shock (Test 4). Latencies were significantly different between groups in all retention test sessions. (Mann–Whitney U-tests, two-tailed, *P = 0.0005 compared with the vehicle (VEH) group).
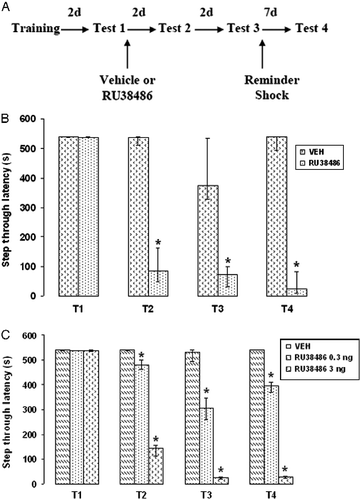
Two days (Test 2) and four days (Test 3) after memory reactivation (Test 1), rats were retested for fear memory retention. To determine whether memory could re-emerge, immediately after Test 3, rats were exposed to a reminder shock (0.5 mA, 1.5 s) in a different box and retested 7 days later (Test 4). All retention tests (Tests 1–4) were done as described for Test 1.
Experimental groups
To determine the role of GRs in fear memory reconsolidation, rats were randomly divided into five groups (n = 8 in each group) and given different treatment immediately after Test 1. Two groups of rats were systemically injected with vehicle or RU38486 (20 mg/kg). Three groups received bilateral injections of vehicle or RU38486 (0.3 or 3 ng/μl). Control groups for systemic or intrahippocampal injections of RU38486 received the same volume of vehicle.
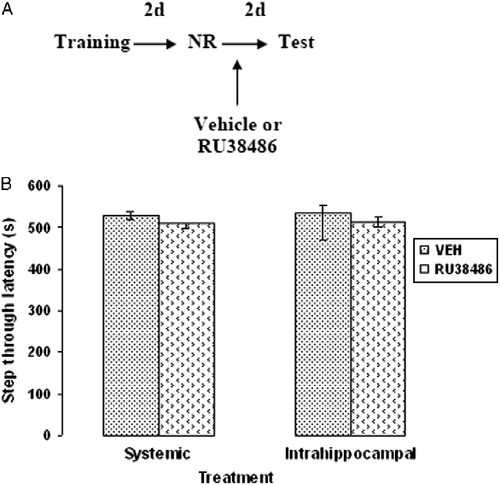
Furthermore, to determine whether the effect of GR blockade on fear memory reconsolidation was reactivation dependent, four additional groups of rats were given one of the following treatments 2 days after training in the absence of memory reactivation (): systemic vehicle (2 ml/kg) and intrahippocampal vehicle (1 μl per side, bilateral), systemic RU38486 (20 mg/kg), or intrahippocampal RU38486 (3 ng/μl, bilateral).
Figure 3. Effects of RU38486 administration in the absence of memory reactivation on fear memory reconsolidation. (A) Passive avoidance training/testing and drug administration schedule; intervals are 2 days (d); NR, no memory reactivation. (B) Median (interquartile ranges) step-through latencies of groups of rats given systemic (20 mg/kg) or intrahippocampal (3 ng in 1 μl per side) injection of RU38486 or vehicle (VEH; n = 8) in the absence of memory reactivation and tested 2 days; VEH, vehicle.
Statistical analysis
Because the distribution of the retention scores in the inhibitory avoidance task was artificially truncated at 540 s, nonparametric statistics were used to analyze the data. Data were analyzed, when appropriate, with the Kruskal–Wallis analysis of variance, and the differences between groups were evaluated by the two-tailed Mann–Whitney U-tests. Values of P < 0.05 were considered significant.
Results
When systemic administration of RU38486 (20 mg/kg) was done following memory reactivation (Test 1), a significant memory impairment was found in subsequent tests (). A Kruskal–Wallis one-way ANOVA on retention latencies of the vehicle-injected and the RU38486-injected groups at Tests 1–3 revealed significant differences between the groups (H = 35.76, P < 0.001). The U-tests indicated that there was a significant difference between the vehicle group and RU38486 groups in Tests 2 and 3 (both, P = 0.0005), but not Test 1. Also, the retention levels of the rats that received RU38486 injections in Tests 2 and 3 were significantly lower than those of Test 1 (both, P = 0.0005). Application of a weak reminder shock did not recover the original memory ().
As shown in , injection of RU38486 in the hippocampus after memory reactivation impaired subsequent memory retention in a dose-dependent manner. A Kruskal–Wallis one-way ANOVA on retention latencies of the vehicle-injected and the RU38486-injected groups at Tests 1–3 revealed significant differences between the groups (H = 59.93, P < 0.001). The U-tests indicated that there were significant differences between the vehicle group and RU38486 (0.3 and 3 ng) groups in Tests 2 and 3 (all, P = 0.005). Application of a weak reminder shock did not strengthen memory retention in rats that received 0.3 or 3 ng RU38486. However, differences between the vehicle and RU38486 (both 0.3 and 3 ng) treated rats were significant in Test 4 (both, P = 0.0005). This finding indicates that RU38486-induced amnesia is long lasting, and memory does not recover following a shock reminder ().
Also, the retention levels of the rats injected with the vehicle or RU38486, 48 h after training in the absence of memory reactivation (Test 1), did not differ during the retention test that was done 2 days later () (P = 0.4 and P = 0.09 for systemic and intrahippocampal comparisons, respectively).
Discussion
The main purpose of this study was to investigate the effects of blockade of GRs on memory reconsolidation. Our results indicate that systemic and intrahippocampal administrations of the GR antagonist RU38486 after memory reactivation produced a long-lasting deficit in subsequent expression of memory. This impairment is only seen after reactivation of memory and not in the absence of memory reactivation, indicating that adequate memory reactivation must occur for RU38486 to alter post-reactivation memory processes. These findings demonstrate that hippocampal GRs play an important role in the regulation of fear memory reconsolidation.
Does RU38486 impair memory reconsolidation or enhance extinction? Memory retrieval by re-exposure to the conditioned stimulus in the absence of an unconditioned stimulus may trigger two opposite processes: reconsolidation and extinction. Memory reconsolidation is a category of processes that serves to maintain, strengthen, and modify the original memory that is already stored in long-term memory (Debiec et al. Citation2002; Alberini Citation2005). Conversely, the extinction process that also requires protein synthesis tends to weaken the original memory, resulting in long-term changes in behavior (Eisenberg et al. Citation2003). Recent studies have shown that the duration of exposure may be an important determinant of subsequent memory processing: short exposure to the original learning context results in reconsolidation, whereas longer exposure to the context leads to extinction (Suzuki et al. Citation2004). Two important characteristics of extinction are spontaneous recovery (the return of the original memory in the absence of explicit retraining) and re-establishment of the original memory with a reminder shock (Nader and Einarsson Citation2010). In the present experiments, we have not found restoration of the memory following the presentation of a weak reminder shock or spontaneous recovery. Thus, we can conclude that post-retrieval administration of RU38486 disrupted memory reconsolidation. Another possibility is that the memory impairment induced by RU38486 might be due to a retrieval failure rather than an interference with the memory reconsolidation process. Indeed, some recent studies have shown that apparent impairments in an inhibitory avoidance reconsolidation induced by the pharmacological inhibition of the hippocampus can undergo spontaneous recovery with time in the subsequent test sessions, suggesting a transient blockade of memory retrieval (Amaral et al. Citation2007; Luft et al. Citation2008). However, the observed impairment of memory in this study is not due to a transient retrieval impairment because the memory impairment was obvious at the testing times that were remote from the time of treatment, when the direct effect of RU38486 was eliminated.
The finding of this study that the GR antagonist impaired memory reconsolidation when administered into the hippocampus following memory reactivation provides evidence that hippocampal GRs are required for fear memory reconsolidation. As mentioned before, hippocampus has a high density of GRs (de Kloet Citation1998) and plays an important role in mediating the effects of glucocorticoids on consolidation and retrieval processes (Roozendaal Citation2000; Sajadi et al. Citation2006). The mechanism by which hippocampal GRs are involved in fear memory reconsolidation is not known. Considering that reconsolidation of fear memory requires protein synthesis in the hippocampus (Debiec et al. Citation2002), it is possible that activation of GRs affects cellular and molecular reconsolidation that occurs within the hippocampus via genomic (Yudt and Cidlowski Citation2002) and non-genomic mechanisms (Joels and de Kloet Citation1992; Sajadi et al. Citation2006).
Our findings support and extend recent results, indicating an essential role of GRs in the basolateral amygdala (BLA) in fear memory reconsolidation. It has been shown that systemic and intra-amygdala administrations of RU38486 impaired fear memory reconsolidation (Tronel and Alberini Citation2007; Taubenfeld et al. Citation2009), indicating a critical role for GRs located in the BLA in this process. Findings that GRs in both the hippocampus and the BLA are required for fear memory reconsolidation, indicates that glucocorticoids influence memory reconsolidation by acting at many different brain structures. This view is consistent with evidence that GRs are widely distributed throughout the brain (de Kloet Citation1998). Previous studies have shown that systemic and intrahippocampal injections of GR agonists enhance the consolidation of an inhibitory avoidance memory, and that lesion of the BLA blocks these effects (Roozendaal Citation2000), showing a critical role of the BLA in regulating glucocorticoid effects on memory storage. Presently, it is not clear whether the effects of glucocorticoids on fear memory reconsolidation in the hippocampus depend critically on the BLA. Further studies are needed to examine this possibility.
Current evidence indicates that the post-retrieval process of an inhibitory avoidance memory can be disrupted, transiently or permanently, by administration of a variety of pharmacological agents after memory reactivation (Amaral et al. Citation2007; Tronel and Alberini Citation2007; Luft et al. Citation2008; Taubenfeld et al. Citation2009). The mechanism of the post-retrieval retention deficit has not been resolved. According to the reconsolidation hypothesis, memory reactivation triggers a new phase of memory lability, and that a blockade of memory storage underlies the deficits (Debiec et al. Citation2002). If the impairment reflects memory storage, then the disruption should be permanent; memory should not return at subsequent tests (Duvarci and Nader Citation2004). Consistent with previous studies (Tronel and Alberini Citation2007; Taubenfeld et al. Citation2009), we found that blockade of GRs induced a permanent retention deficit. These findings are in agreement with the reconsolidation hypothesis but are in conflict with the alternative interpretations that similar treatments may temporarily disrupt memory retrieval, and apparent impairments in memory can undergo spontaneous recovery (Amaral et al. Citation2007; Luft et al. Citation2008). The reasons for the reported discrepancies are not entirely understood, but they are probably related to the behavioral tasks and reactivation conditions used, memory age, predictability of the reactivation stimulus, and training intensity (Nader and Einarsson Citation2010).
Pharmacological manipulations and intervention in memory reconsolidation open up new avenues for providing long-term cure for patients with traumatic memories such as post-traumatic stress disorder (PTSD; Kindt et al. Citation2009). Our findings indicate that administration of the GR antagonist may be beneficial for patients with PTSD by reducing excessive retrieval of aversive memories. This idea contrasts with human studies indicating that administration of glucocorticoids has beneficial effects in patients with PTSD (Schelling et al. Citation2004).
In conclusion, our findings demonstrate that systemic and intrahippocampal administrations of RU38486 disrupts reconsolidation of fear memory. Thus, we propose that GR antagonists administered following a traumatic memory reactivation could, therefore, potentially represent a novel treatment for pathogenic memories such as those seen in patients with PTSD and phobia as suggested by others (Tronel and Alberini Citation2007; Taubenfeld et al. Citation2009).
Acknowledgements
This work was supported by a grant (251) from Semnan University of Medical Sciences to Dr Ali Rashidy-Pour and Dr Abbas Ali Vafaei.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Abrari K, Rashidy-Pour A, Semnanian S, Fathollahi Y. 2008. Administration of corticosterone after memory reactivation disrupts subsequent retrieval of a contextual conditioned fear memory: Dependence upon training intensity. Neurobiol Learn Mem. 89:178–184.
- Alberini CM. 2005. Mechanisms of memory stabilization: Are consolidation and reconsolidation similar or distinct processes?. Trends Neurosci. 28:51–56.
- Amaral OB, Luft T, Cammarota M, Izquierdo I, Roesler R. 2007. Temporary inactivation of the dorsal hippocampus induces a transient impairment in retrieval of aversive memory. Behav Brain Res. 180:113–118.
- Debiec J, LeDoux JE, Nader K. 2002. Cellular and systems reconsolidation in the Hippocampus. Neuron. 36:527–538.
- De Kloet ER, Vreugdenhil E, Oitzl MS, Joëls M. 1998. Brain corticosteroid receptor balance in health and disease. Endocr Rev. 19:269–301.
- Duvarci S, Nader K. 2004. Characterization of fear memory reconsolidation. J Neurosci. 24:9269–9275.
- Eisenberg M, Kobilo T, Berman DE, Dudai Y. 2003. Stability of retrieved memory: Inverse correlation with trace dominance. Science. 301:1102–1104.
- Joels M, de Kloet ER. 1992. Control of neuronal excitability by corticosteroid hormones. Trends Neurosci. 15:25–30.
- Khaksari M, Rashidy-Pour A, Vafaei AA. 2007. Central mineralocorticoid receptors are indispensable for corticosterone-induced impairment of memory retrieval in rats. Neuroscience. 149:729–738.
- Kindt M, Soeter M, Vervliet B. 2009. Beyond extinction: Erasing human fear responses and preventing the return of fear. Nat Neurosci. 12:256–258.
- Luft T, Amaral OB, Schwartsmann G, Roesler R. 2008. Transient disruption of fear-related memory by post-retrieval inactivation of gastrin-releasing peptide or N-methyl-d-aspartate receptors in the hippocampus. Curr Neurovasc Res. 5:21–27.
- Mao J, Regelson W, Kalimi M. 1992. Molecular mechanisms of RU 486 action: A review. Mol Cell Biol. 109:1–8.
- McGaugh JL. 2000. Memory-a century of consolidation. Science. 287:248–251.
- Nader K, Einarsson EO. 2010. Memory reconsolidation: An update. Ann NY Acad Sci. 1191:27–41.
- Oitzl MS, de Kloet ER. 1992. Selective corticosteroid antagonists modulates specific aspects of spatial orientation learning. Behav Neurosci. 106:62–71.
- Pakdel R, Rashidy-Pour A. 2006. Glucocorticoid-induced impairment of long-term memory retrieval in rats: An interaction with dopamine D2 receptors. Neurobiol Learn Mem. 85:300–306.
- Paxinos G, Watson C. 1998. The rat brain in stereotaxic coordinates . 4th ed. Orlando, FL: Academic Press.
- Paxinos G, Watson C. 2005. The rat brain in stereotaxic coordinates . 5th ed. Orlando, FL: Academic Press.
- Quevedo J, Vianna MRM, Roesler R, de Paris F, Izquierdo I, Rose SPR. 1999. Two time windows of anisomycin-induced amnesia for inhibitory avoidance training in rats: Protection from amnesia by pretraining but not pre-exposure to the task apparatus. Learn Mem. 6:600–607.
- Roozendaal B. 2000. Glucocorticoids and regulation of memory consolidation. Psychoneuroendocrinology. 25:213–238.
- Roozendaal B, McGaugh JL. 1997. Glucocorticoid receptor agonist and antagonist administration into the basolateral but not central amygdala modulates memory storage. Neurobiol Learn Mem. 67:176–179.
- Sajadi AA, Samaei SA, Rashidy-Pour A. 2006. Intra-hippocampal microinjections of anisomycin did not block glucocorticoid-induced impairment of memory retrieval in rats: An evidence for non-genomic effects of glucocorticoids. Behav Brain Res. 173:158–162.
- Schelling G, Roozendaal B, de Quervain DJ. 2004. Can posttraumatic stress disorder prevented with glucocorticoids?. Ann NY Acad Sci. 1032:158–166.
- Suzuki A, Josselyn SA, Frankland P, Masushige S, Silva AJ, Kida S. 2004. Memory reconsolidation and extinction have distinct temporal and biochemical signatures. J Neurosci. 24:4787–4795.
- Taubenfeld SM, Riceberg JS, New AS, Alberini CM. 2009. Preclinical assessment for selectively disrupting a traumatic memory via postretrieval inhibition of glucocorticoid receptors. Biol Psychiatry. 65:249–257.
- Tronel S, Alberini CM. 2007. Persistent disruption of a traumatic memory by post-retrieval inactivation of glucocorticoid receptors in the amygdala. Biol Psychiatry. 62:33–39.
- Yudt MR, Cidlowski JA. 2002. The glucocorticoid receptor: Coding a diversity of proteins and responses through a single gene. Mol Endocrinol. 16:1719–1726.