Abstract
Previous studies showed the protective effects of oxytocin (OT) on myocardial injury in ischemic and reperfused rat heart. Moreover, exposure to various stressors not only evokes sudden cardiovascular effects but also triggers the release of OT in the rat. The present study was aimed to evaluate the possible cardioprotective effects of endogenous OT released in response to stress (St), and effects of administration of exogenous OT on the ischemic–reperfused isolated heart of rats previously exposed to St. Wistar rats were divided into six groups: ischemia/reperfusion (IR); St: rats exposed to swim St for 10 min before anesthesia; St+atosiban (ATO): an OT receptor antagonist, was administered (1.5 mg/kg i.p.) prior to St; St+OT: OT was administered (0.03 mg/kg i.p.) prior to St; OT: OT was administrated prior to anesthesia; ATO was given prior to anesthesia. Isolated hearts were perfused with Krebs buffer solution by the Langendorff method and subjected to 30 min of regional ischemia followed by 60 min of reperfusion. The infarct size (IS) and creatine kinase MB isoenzyme (CK-MB) and lactate dehydrogenase (LDH) in coronary effluent were measured. Hemodynamic parameters were recorded throughout the experiment. The plasma concentrations of OT and corticosterone were significantly increased by St. Unexpectedly St decreased IR injury compared with the IR alone group. OT administration significantly inhibited myocardial injury, and administration of ATO with St abolished recovery of the rate pressure product, and increased IS and levels of CK-MB and LDH. These findings indicate that activation of cardiac OT receptors by OT released in response to St may participate in cardioprotection and inhibition of myocardial IR injury.
Introduction
Coronary heart disease is the leading cause of death worldwide (Sans et al. Citation1997), arising mainly from ischemic heart disease. In the heart, transient ischemia followed by reperfusion (ischemia/reperfusion, IR) induces necrosis and apoptosis, leading to myocardial dysfunction (Eefting et al. Citation2004). Currently, the most effective method for reducing mortality in patients suffering from a coronary occlusion is rapid reperfusion of the ischemic myocardium. Paradoxically, reperfusion can at the same time activate a cascade of reactions leading to increased myocardial damage (Braunwald and Kloner Citation1985), and as the reperfusion is initiated with the treatment of myocardial infarction, it is important to limit the extent of this injury (Rao et al. Citation2005).
Preconditioning (PC) is a defensive adaptive cellular phenomenon and ischemic PC is a protective mechanism produced by short periods of ischemic stress (St) rendering the heart more protected against another similar or greater St (Das and Das Citation2008). Cardiac PC represents the most potent and consistently reproducible method of rescuing heart tissue from undergoing irreversible ischemic damage. Although several pharmacological agents that appear to limit reperfusion injury have been identified (Smits et al. Citation1998; Baxter et al. Citation2001; Jonassen et al. Citation2001; Xu et al. Citation2001; Liao et al. Citation2002; Yang et al. Citation2004a), none of these is available for clinical use (Yang et al. Citation2004b).
Several recent clinical studies have documented a significant role of St in evoking sudden cardiovascular dysfunction (Szczepanska-Sadowska Citation2008). Anatomical studies of oxytocin (OT) pathways in the brain have revealed extensive innervation of the brain stem structures regulating the cardiovascular, behavioral, and neuroendocrine responses to St by OT fibers projecting from the paraventricular nucleus (PVN; Sawchenko and Swanson Citation1982). It is possible that OT released during St is involved in modulating hypothalamic–pituitary–adrenocortical axis and selective sympathetic functions (Ondrejcakova et al. Citation2010).
Some investigators have provided evidence that OT may be involved in regulation of the cardiovascular system by means of direct peripheral and indirect central actions (Petersson et al. Citation1996). OT is produced and released by the heart and acts on its cardiac receptors to decrease heart rate (HR) and force of contraction (Gutkowska et al. Citation2000). Ondrejcakova et al. (Citation2009) reported that OT has protective effects on myocardial injury in isolated rat heart induced by IR. In addition, our previous studies confirmed the protective effects of OT on myocardial injury of the ischemic–reperfused heart in the anesthetized rat (Houshmand et al. Citation2009; Alizadeh et al. Citation2010). Also, exposure to various stressor such as swim St (10 min, 19–21°C) as a combined emotional and physical stressor triggers the release of OT within both the supraoptic nucleus (SON) and PVN, in male and female rats (Wotjak et al. Citation1998; Wigger and Neumann Citation2002; Neumann Citation2007), which paralleled OT secretion into blood (Lang et al. Citation1983; Wotjak et al. Citation1998; Szczepanska-Sadowska Citation2008).
Engagement of OT in the control of neuroendocrine responses to St, its putative contribution to the regulation of cardiovascular parameters, and its protective effect on myocardial injury induced by IR raise the question of whether St-induced release of OT may also be involved in modulation of the cardiovascular responses and ischemia-induced heart injury to St.
The present study was designed to evaluate the hypothesis that secretion of OT induced by St has cardioprotective effects. This was investigated by examining effects of administration of exogenous OT and of an OT antagonist on the ischemic–reperfused isolated heart of rats previously exposed to St.
Methods
Animals
Male Wistar rats (200–250 g) were obtained from Tehran University of Medical Sciences and were housed in an air-conditioned colony room on a light/dark (12 h/12 h) cycle (lights on at 07:00 h) at 21–23°C with free access to food and water. The rats were housed individually in stainless steel cages. All experiments were conducted in accordance with the institutional guidelines of Tehran University of Medical Sciences (Tehran, Iran) and the National Institutes of Health guidelines for the care and use of laboratory animals.
Preparation of isolated hearts
The rats were anesthetized with sodium pentobarbital (60 mg/kg, 15 mg/0.5 ml i.p.) and given heparin sodium (500 IU/0.5 ml i.p.). Hearts were rapidly excised and placed in ice-cold buffer, and mounted on a constant pressure (80 mmHg) Langendorff perfusion apparatus.
Hearts were perfused retrogradely with modified Krebs–Henseleit bicarbonate buffer containing (in mmol/l): NaHCO3 25; KCl 4.7; NaCl 118.5; MgSO4 1.2; KH2PO4 1.2; glucose 11; CaCl2 2.5 gassed with 95%O2/5% CO2 (pH 7.35–7.45 at 37°C). A latex, fluid-filled isovolemic balloon was inserted into the left ventricle through the left atrial appendage and inflated to give a preload of 8–10 mmHg and connected to a pressure transducer (Harvard). For HR monitoring, electrocardiogram recording was made by fixation of thin stainless steel electrodes on the ventricular apex and right atrium. A surgical needle (6–0 silk suture) was passed under the origin of the left anterior descending coronary artery, and the two ends of the suture were passed through two plastic pipette tips to form a snare. Regional ischemia was induced by tightening the snare, and reperfusion was performed by releasing the ends of the suture. The perfusion apparatus was enclosed in a water jacket to maintain a constant perfusion temperature of 37°C. Hearts were allowed to beat spontaneously throughout the experiments. Hemodynamic parameters [left ventricular developed pressure (LVDP, the difference between left ventricular systolic and diastolic pressure) and HR] were monitored with a homemade program (Ossilo Graph Monitor, Biomed, Tehran, Iran). Left ventricular function was assessed by the rate pressure product (RPP; HR × LVDP). In addition, coronary effluent was collected at the end of reperfusion to measure enzymes, including creatine kinase MB (CK-MB) and lactate dehydrogenase (LDH) as biochemical markers of myocyte necrosis.
Experimental protocol
All of the hearts were subjected to 30 min ischemia and 60 min reperfusion ().
Figure 1. Illustration of the experimental protocols. Hearts perfused in vitro in all groups were subjected to 30 min of ischemia followed by 60 min reperfusion. IR, in vitro and vehicle treatment in vivo; in vivo treatments: St, swim stress; OT, i.p. oxytocin injection; ATO, i.p. atosiban injection; NS, normal saline.
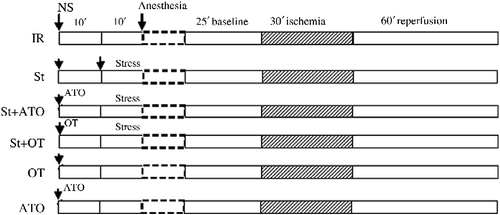
Rats were divided into six groups: (1) IR: hearts were subjected to 30 min ischemia and 60 min reperfusion; (2) St: rats exposed to swim St for 10 min before anesthesia; (3) St+ atosiban (ATO): ATO was used as an OT receptor antagonist (1.5 mg/kg i.p.) 10 min prior to St; (4) St+OT: OT was administered [0.03 mg/kg i.p.; effective dose of OT on IR injury (Houshmand et al. Citation2009)] 10 min prior to St; (5) OT: OT was administered (0.03 mg/kg i.p.) 20 min prior to anesthesia; (6) ATO was administered (1.5 mg/kg i.p.) 20 min prior to anesthesia.
Forced swimming
For St induction, the rats were forced to swim at 09:00 h for 10 min in deep water at 19–20°C in a plexiglass cylinder that was 50 cm high and 30 cm in diameter, filled with tap water to a depth of 35 cm. Rats were transferred to the cylinder from their home cages, returned to their home cages after the forced swim (Wotjak et al. Citation2001; Jorgensen et al. Citation2002). Blood samples for OT and steroid analysis were taken from a tail cut under anesthesia in the IR group, and 15 min after onset of the swim St in the St group. Blood samples were centrifuged (1677 g, 4°C for 5 min) in tubes containing Ethylenediaminetetraacetic acid (EDTA) (10% solution, 10 μl/100 μl blood), aprotinin (a protease inhibitor, 10 μl/tube), and phenylmethylsulfonyl fluoride (5 μl/tube); plasma was removed and aliquots were frozen at − 70°C until assay.
Infarct size measurement
After completion of the reperfusion period, the left coronary artery was re-occluded, and Evans blue dye was infused via the aorta to differentiate the ischemic zone (area at risk, AAR) from the non-ischemic zone. Hearts were frozen overnight and then sliced, using a stainless steel rat heart slicer matrix with 2.0 mm coronal section slice intervals, into 2.0 mm transverse sections from apex to base. Slices were then incubated with 1% triphenyl tetrazolium chloride (TTC in 0.1 M phosphate buffer, pH 7.4) for a period of 20 min at 37°C. TTC reacts with viable tissue, producing a red formazan derivative, which is distinct from the white necrotic tissue once it is fixed in 10% formalin for 24 h. The areas of the left ventricle AAR and infarcted tissue were measured by planimetery from the scanned hearts by using the Photoshop program. AAR was expressed as a percentage of left ventricular size for each heart, and the infarct size (IS) was expressed as a percentage of AAR (Imani et al. Citation2009; Naderi et al. Citation2010a,Citationb).
Enzyme activities
The levels of CK-MB and LDH activity were assayed in coronary effluent samples at 60 min of reperfusion with a specific CK-MB and LDH kit (Pars Azmoon, Tehran, Iran), using an autoanalyzer (Roche Hitachi Modular DP Systems, Mannheim, Germany). The sensitivities of the assays for CK-MB and LDH were 2-1000 IU/l and 0-1000 IU/l, respectively.
Hormone assays
Oxytocin
OT was analyzed in extracted plasma using an enzyme immunoassay (ELISA, Phoenix Pharmaceuticals Inc., Burlingame, CA, USA) kit. The manufacturer's instructions for the extraction procedure were followed. The intra-assay coefficient of variation was 6.8%.
Corticosterone
Plasma corticosterone concentration as a St marker was measured using an enzyme immunoassay (ELISA, DRG, Marburg, Sigma-Aldrich, Steinheim, Germany) kit. The sensitivity of the assay was 1.63 nmol/l. The intra-assay coefficient of variation was 6.4%.
Chemicals
ATO, OT acetate salt hydrate, TTC, and sodium pentobarbital were obtained from Sigma-Aldrich, Steinheim, Germany and heparin sodium was acquired from Caspian Tamin Pharmaceutical Co., Rasht, Iran.
Statistical analysis
Repeated measures ANOVA was used for comparison of hemodynamic parameters within groups. One-way ANOVA and post hoc Dunnet's test was used for comparison of hemodynamic parameters, IS, CK-MB, and LDH between different groups. Statistical analysis of hormone concentrations was determined by unpaired t-test. All data are expressed as mean ± SEM. Statistical significance was defined as P < 0.05.
Results
Hemodynamic parameters
The results in show HR, LVDP, coronary flow (CF), and RPP (as a percentage of an individual baseline). Since HR and LVDP may recover to different degrees, RPP was calculated by multiplying HR by LVDP and presented as a reliable left ventricular function parameter for the isolated heart. There were significant differences between RPP under baseline, end of ischemia, and end of reperfusion (repeated measures ANOVA, F(2,72) = 151.71, P < 0.001). A significant main effect was found for the treatment on RPP (F(2,36) = 12.86, P < 0.001). The main effect of St was not significant (F(1,36) = 0.09). The results showed a significant interaction between treatment and time on RPP (F(4,72) = 7.69, P < 0.001) and significant interaction between treatment and St (F(2,36) = 3.49, P < 0.05). After combining treatment and St groups, Tukey's post hoc test revealed pretreatment with OT (OT group) which increased the recovery of RPP at the end of reperfusion in comparison with the IR group (P < 0.05). Administration of ATO prior to stress (St+ATO group) significantly decreased RPP compared with St alone (St group; P < 0.05).
Table I. Hemodynamic parameters.
Area at risk and IS
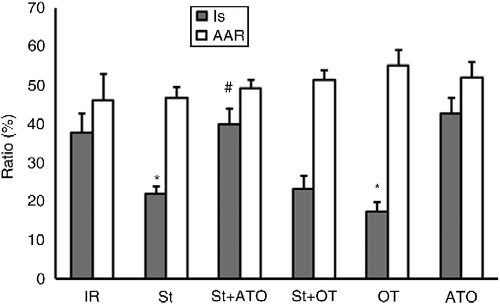
No significant treatment by St interaction and no main effect of treatment and St were revealed on AAR. A significant main effect was found for treatment in IS (two-way ANOVA, F(2,34) = 15.66, P < 0.001). The main effect of St was not significant (F(1,34) = 1.99). Result showed a significant interaction between treatment and St for IS (F(2,34) = 4.35, P < 0.05). For combined treatment and St groups, all pairs of means were compared by Tukey's post hoc test adjusting for multiple comparisons. St significantly decreased IS in the St group compared with the IR group (P < 0.05). IS was decreased significantly in the OT group compared with the IR group (P < 0.01). Administration of ATO (St+ATO group) increased IS in comparison with the St group (P < 0.001). Pretreatment with OT prior to stress (St+OT) did not change IS compared to the St group. ATO alone had no significant effect on IS compared with the IR group ().
Figure 2. Myocardial area at risk (AAR/V%) and infarct size (IS/AAR%) in IR (n = 6), St (n = 8), St+ATO (n = 9), St+OT (n = 7), OT (n = 5), and ATO (n = 5) groups. St, stress; OT, oxytocin; ATO, atosiban; IR, ischemia–reperfusion. Data are presented as mean ± SEM. Two-way ANOVA: *P < 0.05 vs. IR group. #P < 0.001 vs. St group.
Enzyme activities
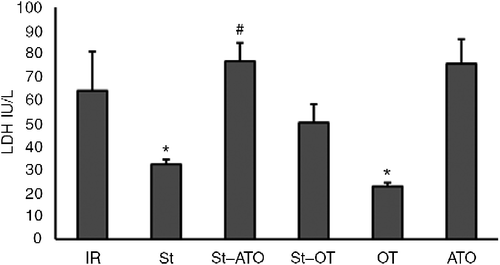
A significant main effect was found for the treatment on CK-MB and LDH (two-way ANOVA; F(2,34) = 14.3, P < 0.001, and F(2,37) = 9.15, P < 0.001, respectively). The main effect of St was not significant on CK-MB and LDH (F(1,34) = 1.8 and F(1,37) = 0.019, respectively). There was a significant interaction between treatment and St on CK-MB and LDH levels (F(2,34) = 6.22, P < 0.001 and F(2,37) = 5.9, P < 0.001, respectively). All pairs of means were compared by Tukey's post hoc test adjusting for multiple comparisons. CK-MB and LDH levels in coronary effluent were significantly decreased by OT-induced PC compared with the IR group at the end of the reperfusion period (P < 0.05). St administration prior to IR significantly decreased CK-MB and LDH levels (P < 0.05). Administration of ATO prior to St in the St+ATO group significantly increased CK-MB and LDH levels compared with the St group (P < 0.05 and P < 0.01, respectively). OT treatment prior to stress (St+OT group) had no significant effect on CK-MB and LDH levels compared with the St alone group. Administration of ATO alone prior to IR, significantly increased CK-MB, but had no significant effect on LDH level in comparison with the IR group (P < 0.05) ().
Figure 3. Activity of creatine kinase MB (CK-MB) in coronary effluent at the end of reperfusion in IR (n = 6), St (n = 8), St+ATO (n = 9), St+OT (n = 7), OT (n = 5), and ATO (n = 5) groups. IR, ischemia–reperfusion; St, stress; OT, oxytocin; ATO, atosiban. Data are presented as mean ± SEM. Two-way ANOVA: *P < 0.05 vs. IR group. #P < 0.05 vs. St group.
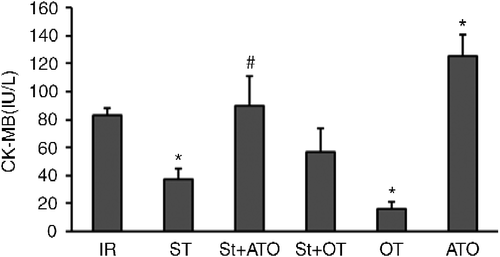
Figure 4. Activity of LDH in coronary effluent at the end of reperfusion in IR (n = 6), St (n = 8), St+ATO (n = 9), St+OT (n = 7), OT (n = 5), and ATO (n = 5) groups. IR, ischemia–reperfusion; St, stress; OT, oxytocin; ATO, atosiban. Data are presented as mean ± SEM. Two-way ANOVA: *P < 0.05 vs. IR group. #P < 0.01 vs. St group.
Plasma hormone concentrations
The plasma concentrations of OT and corticosterone were 7.03 ± 0.87 pg/ml and 26.80 ± 4.04 nmol/l, respectively, in IR group; however following the induction of St, these concentrations significantly increased to mean concentrations of 16.9 ± 5.22 pg/ml and 57 ± 5.31 nmol/l, respectively (unpaired t-test, P < 0.05).
Discussion
The results of the present study show that, acute St increased plasma concentrations of OT and corticosterone and unexpectedly decreased the consequences of IR, such as IS, and CK-MB and LDH levels. Protection induced by St was abolished by the administration of ATO (a non-selective OT receptor antagonist) prior to the St. OT administration decreased IR injury, and OT administration prior to St did not further alter the cardioprotective effects of St.
It is well recognized that St can be harmful to the cardiovascular system (Ramachandruni et al. Citation2004). In this regard, many clinical studies have reported that exposure to St correlates with increased morbidity and mortality from cardiovascular diseases, including myocardial ischemia (Ketterer Citation1993; Tennant et al. Citation1994; Krittayaphong et al. Citation1995; Krantz et al. Citation1996). Acute St accelerates HR, cardiac contractility, and increases total peripheral resistance (Zhang and Leenen Citation1999). Consequently, cardiac work and oxygen consumption markedly increase (Clarke et al. Citation2000; Kario et al. Citation2003). Forced exercise could induce maladaptative changes in both brain and heart tissues (Yancey and Overton Citation1993; Pedersen et al. Citation1997; Koller et al. Citation2001; Scopel et al. Citation2006). Mancardi et al. (Citation2009) observed a worsening of IR outcomes in the heart of rats forced to run, and Scheuer and Mifflin (Citation1998) showed that chronic St increased the size of infarction. These observations conflict with other studies that showed heat St significantly reduced IS in the isolated rat heart subjected to an IR sequence (Joyeux et al. Citation1998). Acute exercise training (Locke et al. Citation1995; Taylor et al. Citation1999) and repeated physiological St provide myocardial protection against IR injury (Hoshida et al. Citation2002). Moreover, there is evidence for an effect of cold-restraint St in cardioprotection (Wu et al. Citation2004).
In our study, an acute episode of St experienced just before IR provided myocardial protection against IR injury by decreasing IS as a distinct endpoint of ischemic heart injury. The effects of St may be of two main types: local and remote. It has been documented that St evokes several protective responses that could prevent the development of IR injury (Scheuer and Mifflin Citation1998). The St response is likely due to interaction between the neuroendocrine system, the sympathetic nervous system, and the target organs, with resulting release of specific hormones (Bohus et al. Citation1987). Previous studies have shown that a 10-min forced swimming session triggers the release of OT within the hypothalamic SON and the PVN (Wotjak et al. Citation1998; Wotjak et al. Citation2001) and into the blood (Wotjak et al. Citation1998; Wigger and Neumann Citation2002). By contrast, a single swimming episode causes a strong increase in plasma OT, whereas plasma levels of arginine vasopressin (AVP) remained essentially unchanged (Wotjak et al. Citation1998). Hence, in the rat, vasopressin secretion appears to be unresponsive to several stimuli known to induce adrenocorticotropic hormone (ACTH) and catecholamine release, such as swimming (Jezova et al. Citation1995).
Our study showed that plasma OT concentration was increased by swim St, confirming the results of previous studies.
Importantly, from our results, it may be concluded that St-induced endogenous OT may protect the heart by preventing worsening or development of IR injury. Forced swimming has a cooling effect on the rat, and involves exercise, which may contribute to the effects reported here. Exercise increases synthesis of OT in the heart (Gutkowska et al. Citation2007) and may cause PC (Domenech et al. Citation2002).
Previously it has been shown that administration of OT has a cardioprotective effect on IR injury in both isolated rat heart (Ondrejcakova et al. Citation2009) and in anesthetized rats (Houshmand et al. Citation2009). We have previously reported that OT induces its protective effect dose dependently, and OT treatment revealed a biphasic pattern, i.e. increasing doses correlated with the increasing efficacy of injury inhibition until an optimal dose was attained beyond which higher doses showed less activity. Optimal effect in the reduction of IS, biochemical changes, and hemodynamic improvement was induced by i.p. bolus injection of 0.03 mg/kg OT. For all indices of protection, higher and lower doses of OT showed a less protective effect (Houshmand et al. Citation2009). Interestingly, in the present experiment, OT administration had a protective effect, but administration of OT prior to St did not increase the protective effect of St. There are two possible explanations for these findings. First, swimming could stimulate release of OT in multiple ways, and the release could be sufficient enough to saturate OT receptors in the heart. Second, as OT has a biphasic dose-dependent effect against IR injury, combination of exogenous OT and OT released in response to St provided a higher level which showed less activity.
In the present experiment, ATO inhibited the protective effect of St by increasing the IS. This implies a possible contribution of OT released in response to St acting via its cardiac OT receptors. In support of the current data, Cicutti et al. (Citation1999) confirmed the presence of a specific OT-binding site in left ventricle of rat and in human atrium. It is generally accepted that OT activates two types of receptors: OT and V1 vasopressin receptors (Costa et al. Citation2005), and both OT (Gutkowska et al. Citation2000) and V1 receptors (Hupf et al. Citation1999) are present in the heart. Therefore, the effect of OT may be caused by the activation of OT and V1 receptors (Jankowski et al. Citation2000).
Our previous studies have indicated that possible mechanisms include release of acetylcholine, nitric oxide (NO), and atrial natriuretic peptide (ANP; unpublished), opening of mitochondrial ATP-sensitive K+ channels (Alizadeh et al. Citation2010), closing of mitochondrial permeability transition pore, activation of protein kinase C, and reactive oxygen species (unpublished) are involved in the signal transduction of the early phase protection of PC by OT in the anesthetized rat. OT may release ANP (Gutkowska et al. Citation1997), and as ANP is a mediator of ischemic PC in the rat (Sangawa et al. Citation2004), there is the possibility that the cardioprotective effects of OT PC may be mediated through the release of ANP. Recent reports suggest that the protective effects of OT pretreatment on IR damage may be caused by the release of NO (Nakamura et al. Citation2006; Tugtepe et al. Citation2007; Dusunceli et al. Citation2008).
In our experiment, St reduced the in vitro release of enzyme markers of myocardial damage. Elevated levels of CK-MB have been regarded as a specific biochemical marker of myocyte necrosis (Yilmaz et al. Citation2006), and St exposure significantly reduced the level of this enzyme. Increase in plasma LDH level, which plays an important role in systemic tissue damage (Devi et al. Citation2005), was attenuated by St treatment. The effects of St exposure on the release of CK-MB and LDH were abolished by ATO. Administration of OT also decreased CK-MB and LDH release. These effects were in the same direction as the changes in IS. In addition, we showed that ATO increased CK-MB level as compared with the IR alone group, indicating a probable protective role for endogenous OT in IR injury under experimental conditions (Houshmand et al. Citation2009).
These findings indicate that activation of cardiac OT receptors (and/or V1 receptors) due to OT released in response to St may participate in cardioprotection and inhibition of myocardial IR injury.
Acknowledgements
This study was supported by the grant of Tehran University of Medical Science.
Decleration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Alizadeh AM, Faghihi M, Sadeghipour HR, Mohammadghasemi F, Imani A, Houshmand F, Khori V. 2010. Oxytocin protects rat heart against ischemia–reperfusion injury via pathway involving mitochondrial ATP-dependent potassium channel. Peptides. 31:1341–1345.
- Baxter GF, Mocanu MM, Brar BK, Latchman DS, Yellon DM. 2001. Cardioprotective effects of transforming growth factor-beta1 during early reoxygenation or reperfusion are mediated by p42/p44 MAPK. J Cardiovasc Pharmacol. 38:930–939.
- Bohus B, Benus RF, Fokkema DS, Koolhaas JM, Nyakas C, va nOortmerssen GA, Prins AJ, de Ruiter AJ, Scheurink AJ, Steffens AB. 1987. Neuroendocrine states and behavioral and physiological stress responses. Prog Brain Res. 72:57–70.
- Braunwald E, Kloner RA. 1985. Myocardial reperfusion: A double-edged sword?. J Clin Invest. 76:1713–1719.
- Cicutti NJ, Smyth CE, Rosaeg OP, Wilkinson M. 1999. Oxytocin receptor binding in rat and human heart. Can J Cardiol. 15:1267–1273.
- Clarke SP, Frasure-Smith N, Lesperance F, Bourassa MG. 2000. Psychosocial factors as predictors of functional status at 1 year in patients with left ventricular dysfunction. Res Nurs Health. 23:290–300.
- Costa ESRH, Pereira-Junior PP, Oliveira PF, Olivares EL, Werneck-de-Castro JP, Mello DB, Nascimento JH, Campos-de-Carvalho AC. 2005. Cardiac effects of oxytocin: Is there a role for this peptide in cardiovascular homeostasis?. Regul Pept. 132:107–112.
- Das M, Das DK. 2008. Molecular mechanism of preconditioning. IUBMB Life. 60:199–203.
- Devi R, Banerjee SK, Sood S, Dinda AK, Maulik SK. 2005. Extract from Clerodendron colebrookianum Walp protects rat heart against oxidative stress induced by ischemic-reperfusion injury (IRI). Life Sci. 24:2999–3009.
- Domenech R, Macho P, Schwarze H, Sanchez G. 2002. Exercise induces early and late myocardial preconditioning in dogs. Cardiovasc Res. 55:561–566.
- Dusunceli F, Iseri SO, Ercan F, Gedik N, Yegen C, Yegen BC. 2008. Oxytocin alleviates hepatic ischemia–reperfusion injury in rats. Peptides. 29:1216–1222.
- Eefting F, Rensing B, Wigman J, Pannekoek WJ, Liu WM, Cramer MJ, Lips DJ, Doevendans PA. 2004. Role of apoptosis in reperfusion injury. Cardiovasc Res. 61:414–426.
- Gutkowska J, Jankowski M, Lambert C, Mukaddam-Daher S, Zingg HH, McCann SM. 1997. Oxytocin releases atrial natriuretic peptide by combining with oxytocin receptors in the heart. Proc Natl Acad Sci. 94:11704–11709.
- Gutkowska J, Jankowski M, Mukaddam-Daher S, McCann SM. 2000. Oxytocin is a cardiovascular hormone. Braz J Med Biol Res. 33:625–633.
- Gutkowska J, Paquette A, Wang D, Lavoie JM, Jankowski M. 2007. Effect of exercise training on cardiac oxytocin and natriuretic peptide systems in ovariectomized rats. Am J Physiol Regul Integr Comp Physiol. 293:267–275.
- Hoshida S, Yamashita N, Otsu K, Hori M. 2002. Repeated physiologic stress provide persistent cardioprotection against ischemia–reperfusion injury in rats. J Am Coll Cardiol. 40:826–831.
- Houshmand F, Faghihi M, Zahediasl S. 2009. Biphasic protective effect of oxytocin on cardiac ischemia/reperfusion injury in anaesthetized rats. Peptide. 30:2301–2308.
- Hupf H, Grimm D, Riegger GA, Schunkert H. 1999. Evidence for a vasopressin system in the rat heart. Circ Res. 84:365–370.
- Imani A, Faghihi M, Keshavarz M, Karimian SM, Niaraki SS. 2009. Effect of different doses of noradrenaline against ischemia-induced ventricular arrhythmias in rat heart in vivo. Indian Pacing Electrophysiol J. 9:35–44.
- Jankowski M, Wang D, Hajjar F, Mukaddam-Daher S, McCann SM, Gutkowska J. 2000. Oxytocin and its receptors are synthesized in the rat vasculature. Proc Natl Acad Sci USA. 97:6207–6211.
- Jezova D, Skultetyova I, Tokarev DI, Bakos P, Vigas M. 1995. Vasopressin and oxytocin in stress. Ann NY Acad Sci. 771:192–203.
- Jonassen AK, Sack MN, Mjos OD, Yellon DM. 2001. Myocardial protection by insulin at reperfusion requires early administration and is mediated via Akt and p70s6 kinase cell-survival signaling. Circ Res. 89:1191–1198.
- Jorgensen H, Knigge U, Kjaer A, Warberg J. 2002. Serotonergic involvement in stress-induced vasopressin and oxytocin secretion. Eur J Endocrinol. 147:815–824.
- Joyeux M, Godin-Ribuot D, Patel A, Demenge P, Yellon DM, Ribuot C. 1998. Infarct size-reducing effect of heat stress and alpha1 adrenoceptors in rats. BrJ Pharmacol. 125:645–650.
- Kario K, McEwen BS, Pickering TG. 2003. Disasters and the heart: A review of the effects of earthquake-induced stress on cardiovascular disease. Hypertens Res. 26:355–367.
- Ketterer MW. 1993. Secondary prevention of ischemic heart disease. The case for aggressive behavioral monitoring and intervention. Psychosomatics. 34:478–484.
- Koller A, Mertelseder S, Moser H. 2001. Is exercise-induced myocardial injury self-abating?. Med Sci Sports Exerc. 33:850–851.
- Krantz DS, Kop WJ, Santiago HT, Gottdiener JS. 1996. Mental stress as a trigger of myocardial ischemia and infarction. Cardiol Clin. 14:271–287.
- Krittayaphong R, Light KC, Biles PL, Ballenger MN, Sheps DS. 1995. Increased heart rate response to laboratory-induced mental stress predicts frequency and duration of daily life ambulatory myocardial ischemia in patients with coronary artery disease. Am J Cardiol. 76:657–660.
- Lang RE, Heil JW, Ganten D, Hermann K, Unger T, Rascher W. 1983. Oxytocin unlike vasopressin is a stress hormone in the rat. Neuroendocrinology. 37:314–316.
- Liao Z, Brar BK, Cai Q, Stephanou A, O'Leary RM, Pennica D, Yellon DM, Latchman DS. 2002. Cardiotrophin-1 (CT-1) can protect the adult heart from injury when added both prior to ischaemia and at reperfusion. Cardiovasc Res. 53:902–910.
- Locke M, Tanguay RM, Klabunde RE, Ianuzzo CD. 1995. Enhanced postischemic myocardial recovery following exercise induction of HSP 72. Am J Physiol. 269:320–325.
- Mancardi D, Tullio F, Crisafulli A, Rastaldo R, Folino A, Penna C. 2009. Omega 3 has a beneficial effect on ischemia/reperfusion injury, but cannot reverse the effect of stressful forced exercise. Nutr Metab Cardiovasc Dis. 19:20–26.
- Naderi R, Imani A, Faghihi M. Phenylephrine produces late pharmacological preconditioning in the isolated rat heart. Eur J Pharmacol. 2010a; 627:203–208.
- Naderi R, Imani A, Faghihi M, Moghimian M. Phenylephrine induces early and late cardioprotection through mitochondrial permeability transition pore in the isolated rat heart. J Surg Res. 2010b; 164:37–42.
- Nakamura K, Al-Ruzzeh S, Gray C, Yacoub M, Amrani M. 2006. Effect of myocardial reperfusion on the release of nitric oxide after regional ischemia: An experimental model of beating-heart surgery. Tex Heart Inst J. 33:35–39.
- Neumann ID. 2007. Stimuli and consequences of dendritic release of oxytocin within the brain. Biochem Soc Trans. 35:1252–1257.
- Ondrejcakova M, Bakos J, Garafova A, Kovacs L, Kvetnansky R, Jezova D. 2010. Neuroendocrine and cardiovascular parameters during simulation of stress-induced rise in circulating oxytocin in the rat. Stress. 13:314–322.
- Ondrejcakova M, Ravingerova T, Bakos J, Pancza D, Jezova D. 2009. Oxytocin exerts protective effects on in vitro myocardial injury induced by ischemia and reperfusion. Can J Physiol Pharmacol. 87:137–142.
- Pedersen BK, Bruunsgaard H, Klokker M, Kappel M, MacLean DA, Nielsen HB, Rohde T, Ullum H, Zacho M. 1997. Exercise-induced immunomodulation–possible roles of neuroendocrine and metabolic factors. Int J Sports Med. 18:52–57.
- Petersson MAP, Lundeburg T, Uvna¨s-Moberg K. 1996. Oxytocin causes a long time decrease of blood pressure in female and male rats. Physiol Behav. 60:1311–1315.
- Ramachandruni S, Handberg E, Sheps DS. 2004. Acute and chronic psychological stress in coronary disease. Curr Opin Cardiol. 19:494–499.
- Rao PR, Kumar VK, Viswanath RK, Subbaraju GV. 2005. Cardioprotective activity of alcoholic extract of Tinospora cordifolia in ischemia–reperfusion induced myocardial infarction in rats. Biol Pharm Bull. 28:2319–2322.
- Sangawa K, Nakanishi K, Ishino K, Inoue M, Kawada M, Sano S. 2004. Atrial natriuretic peptide protects against ischemia–reperfusion injury in the isolated rat heart. Ann Thorac Surg. 77:233–237.
- Sans S, Kesteloot H, Kromhout D. 1997. The burden of cardiovascular diseases mortality in Europe. Task force of the european society of cardiology on cardiovascular mortality and morbidity statistics in Europe. Eur Heart J. 18:1231–1248.
- Sawchenko PE, Swanson LW. 1982. Immunohistochemical identification of neurons in the paraventricular nucleus of the hypothalamus that project to the medulla or to the spinal cord in the rat. J Comp Neurol. 205:260–272.
- Scheuer DA, Mifflin SW. 1998. Repeated intermittent stress exacerbates myocardial ischemia–reperfusion injury. Am J Physiol. 274:470–475.
- Scopel D, Fochesatto C, Cimarosti H, Rabbo M, Bello-Klein A, Salbego C, Netto CA, Siqueira IR. 2006. Exercise intensity influences cell injury in rat hippocampal slices exposed to oxygen and glucose deprivation. Brain Res Bull. 71:155–159.
- Smits GJ, McVey M, Cox BF, Perrone MH, Clark KL. 1998. Cardioprotective effects of the novel adenosine A1/A2 receptor agonist AMP 579 in a porcine model of myocardial infarction. J Pharmacol Exp Ther. 286:611–618.
- Szczepanska-Sadowska E. 2008. Role of neuropeptides in central control of cardiovascular responses to stress. J Physiol Pharmacol. 59:61–89.
- Taylor RP, Harris MB, Starnes JW. 1999. Acute exercise can improve cardioprotection without increasing heat shock protein content. Am J Physiol. 276:1098–1102.
- Tennant CC, Palmer KJ, Langeluddecke PM, Jones MP, Nelson G. 1994. Life event stress and myocardial reinfarction: A prospective study. Eur Heart J. 15:472–478.
- Tugtepe H, Sener G, Biyikli NK, Yuksel M, Cetinel S, Gedik N, Yegen BC. 2007. The protective effect of oxytocin on renal ischemia/reperfusion injury in rats. Regul Pept. 140:101–108.
- Wigger A, Neumann ID. 2002. Endogenous opioid regulation of stress-induced oxytocin release within the hypothalamic paraventricular nucleus is reversed in late pregnancy: A microdialysis study. Neuroscience. 112:121–129.
- Wotjak CT, Ganster J, Kohl G, Holsboer F, Landgraf R, Engelmann M. 1998. Dissociated central and peripheral release of vasopressin, but not oxytocin, in response to repeated swim stress: New insights into the secretory capacities of peptidergic neurons. Neuroscience. 85:1209–1222.
- Wotjak CT, Naruo T, Muraoka S, Simchen R, Landgraf R, Engelmann M. 2001. Forced swimming stimulates the expression of vasopressin and oxytocin in magnocellular neurons of the rat hypothalamic paraventricular nucleus. Eur J Neurosci. 13:2273–2281.
- Wu S, Wong MC, Chen M, Cho CH, Wong TM. 2004. Role of opioid receptors in cardioprotection of cold-restraint stress and morphine. J Biomed Sci. 11:726–731.
- Xu Z, Downey JM, Cohen MV. 2001. Amp 579 reduces contracture and limits infarction in rabbit heart by activating adenosine A2 receptors. J Cardiovasc Pharmacol. 38:474–481.
- Yancey SL, Overton JM. 1993. Cardiovascular responses to voluntary and treadmill exercise in rats. J Appl Physiol. 75:1334–1340.
- Yang XM, Krieg T, Cui L, Downey JM, Cohen MV. NECA and bradykinin at reperfusion reduce infarction in rabbit hearts by signaling through PI3K, ERK, and NO. J Mol Cell Cardiol. 2004a; 36:411–421.
- Yang XM, Proctor JB, Cui L, Krieg T, Downey JM, Cohen MV. Multiple, brief coronary occlusions during early reperfusion protect rabbit hearts by targeting cell signaling pathways. J Am Coll Cardiol. 2004b; 44:1103–1110.
- Yilmaz A, Yalta K, Turgut OO, Yilmaz MB, Ozyol A, Kendirlioglu O, Karadas F, Tandogan I. 2006. Clinical importance of elevated CK-MB and troponin I levels in congestive heart failure. Adv Ther. 23:1060–1067.
- Zhang WHB, Leenen FH. 1999. Brain renin-angiotensin system and sympathetic hyperactivity in rats after myocardial infarction. Am J Physiol Heart Circ Physiol. 276:1608–1615.