Abstract
Patients with inflammatory bowel diseases (IBDs) have a higher risk of developing colorectal cancer (CRC) than the general population. Furthermore, chronic psychosocial stress increases the likelihood of developing IBD and multiple types of malignant neoplasms, including CRC. Here, for the first time, we investigate the effects of chronic psychosocial stress in male mice on an artificially induced CRC, by employing the chronic subordinate colony (CSC) housing paradigm in combination with the reliable azoxymethane (AOM)/dextran sodium sulfate (DSS) CRC model. Colonoscopy revealed that CSC mice showed accelerated macroscopic suspect lesions. In addition, more CSC mice developed low-grade dysplasia (LGD) and/or high-grade dysplasia (HGD) in the colonic tissue compared to the single-housed control mice (SHC). CSC mice showed an increased number of Ki67+ and a decreased number of terminal deoxynucleotidyl transferase dUTP nick end labeling epithelial cells in colonic tissue. Colonic liver receptor homolog-1 (LRH-1), cyclooxygenase II (COXII), tumor necrosis factor, forkhead box P3 (FoxP3) mRNA as well as colonic ß-catenin, COXII, and LRH-1 protein expression were also increased in CSC compared with SHC mice. Although the number of CD4+ Th cells was increased, a tendency toward a decreased colonic interferon-γ (IFN-γ) mRNA expression was observed. Furthermore, despite an increased percentage of CD3+ cells and CD3+/FoxP3+ double-positive cells within mesenteric lymph node cells of CSC mice, IFN-γ secretion from these cells was unaffected. Altogether, our results suggest that chronic psychosocial stress increases the risk for AOM/DSS-induced and, thus, inflammation-related CRC. Finally, assessment of additional time points may test whether the shift from tumor-protective Th1 cell to regulatory T-cell immunity represents a consequence of increased carcinogenesis or a causal factor involved in its development.
Introduction
Chronic stress, in particular chronic psychosocial stress, is a general burden of modern societies and an acknowledged risk factor for numerous disorders, including inflammatory bowel disease (IBD; Bernstein et al. Citation2010) and cancer (Levav et al. Citation2000; Reiche et al. Citation2004).
The concept of stress-promoted carcinogenesis and the hypothetical link between stress-induced suppression of cellular immunity and tumor progression is also suggested by animal studies. For example, a reduction in blood CD4+ and CD8+T cells and in T-cell and natural killer cell activities following prolonged social (7–48 h) stress was paralleled by a significantly lower clearance of injected MADB 106 tumor cells (Stefanski and Ben-Eliyahu Citation1996; Stefanski Citation2001). In addition to these studies, there is also evidence indicating that stress exposure can promote spontaneous cancer development. Thus, repeated immobilization (6 h/day; 3 weeks) increased the susceptibility to UV radiation-induced development of squamous cell carcinoma (Saul et al. Citation2005). Importantly, this was again paralleled by a decrease in the number of protective CD4+ Th1 cells and Th1 cytokine expression, both measured in dorsal skin tissue, as well as an increase in the number of blood regulatory T cells.
Colorectal cancer (CRC) represents one of the most common types of cancer worldwide (Tanaka Citation2009). As no less than one-third of all human cancer is associated with inflammation, it is not surprising that the overall risk of CRC is 10-fold higher in IBD patients, a figure which rises to a 38-fold increase if IBD is diagnosed before the age of 30 (Yang et al. Citation2009). This is also supported by indirect evidence gathered from human (Ekbom et al. Citation1990; Choi and Zelig Citation1994; Rutter et al. Citation2004) and mouse studies (Chulada et al. Citation2000; Laurent et al. Citation2010), showing that dampening colonic inflammation ultimately reduces tumor incidence. CRC, in most cases, is only diagnosed at an advanced stage (O'Shaughnessy et al. Citation2002) and poses one of the most serious complications in patients with IBD (Eaden and Mayberry Citation2000; Eaden et al. Citation2001; van Hogezand et al. Citation2002). These observations are most likely due to a lack of appropriate biomarkers and insight into CRC pathogenesis (O'Shaughnessy et al. Citation2002).
Using a novel colitis-related mouse model (Tanaka et al. Citation2003), in which CRC is initiated with azoxymethane (AOM) and promoted by repeated cycles of dextran sodium sulfate (DSS) administration, initial steps toward a better understanding of the pathogenesis of IBD-related CRC have been taken (Tanaka Citation2009). CRC is initiated by abnormal growth patterns, called low-grade dysplasia (LGD) or high-grade dysplasia (HGD), and/or uncontrolled growth of the initiated (either naturally occurring or AOM-induced) cryptal cells, followed by the formation of adenomatous polyps (neoplasms). After a prolonged time period, these eventually evolve into cancer (malignant neoplasm; Xie and Itzkowitz Citation2008). However, although it is known that IBD increases the risk for CRC and that perceived life stress is involved in the development/progression of both IBD and cancer in general, to the best of our knowledge the influence of environmental factors, such as chronic psychosocial stress, on the development of inflammation-related CRC has not been addressed to date.
Exposure to chronic subordinate colony (CSC) housing a clinically relevant animal model of psychosocial stress (Reber et al. Citation2007, Citation2008, Citation2011; Reber and Neumann Citation2008; Veenema et al. Citation2008; Singewald et al. Citation2009; Schmidt et al. Citation2010) results in spontaneous colitis (Reber et al. Citation2007, Citation2011) and aggravates DSS-induced (Reber et al. Citation2008; Veenema et al. Citation2008) colitis. Moreover, DSS colitis has been shown to promote AOM-induced colon carcinogenesis depending on its severity (Suzuki et al. Citation2005). Therefore, it was the aim of the current study (i) to test the hypothesis that 19 days of CSC increases the risk for inflammation-related CRC and (ii) to dissect the underlying mechanisms. In terms of aim (i), development of HGD and/or low-grade dysplasia (LGD) was assessed, as dysplasia occurring in adenomas is considered to be a selective marker for the increased risk of developing cancer in humans (Konishi and Morson Citation1982). In addition, we quantified epithelial cell proliferation and apoptosis, as an abnormal pattern of cell replication has been detected in several clinical conditions associated with an increased risk for colorectal malignancies (Tanaka Citation2009). With respect to aim (ii), ß-catenin and liver receptor homolog-1 (LRH-1) mRNA and/or protein were assessed, as uncontrolled activation of the ß-catenin signaling pathway causes gastrointestinal tumor development (Oshima et al. Citation1995) and as LRH-1 has been shown to be involved in the control of intestinal cell renewal (Botrugno et al. Citation2004) and in the promotion of CRC (Schoonjans et al. Citation2005). Furthermore, the number of F4/80+ colonic macrophages as well as tumor necrosis factor (TNF) and cyclooxygenase II (COXII) mRNA and/or protein expression were quantified, as macrophage-derived TNF has been shown to promote tumor development in the gastric mucosa (Oguma et al. Citation2008) and as COXII is considered to contribute to tumor development by modulating apoptosis, angiogenesis, and tumor invasiveness (McConnell and Yang Citation2009). As an impaired clearance of abnormal cells by immune competent cells might also be involved in the promotion of AOM/DSS-induced CRC, we assessed the number, and cytokine profile [forkhead box P3 (FoxP3) and interferon-γ (IFN-γ)], of Th cells in colonic and mesenteric lymph node (mesLN) tissue in CSC and single-housed control (SHC) mice.
Methods
Animals
Male C57BL/6 mice (Charles River, Sulzfeld, Germany) weighing 19–22 g (5–6 weeks of age; experimental mice) or 30–35 g (dominant mice) were individually housed in standard polycarbonate mouse cages (16 × 22 × 14 cm) for at least 1 week before the CSC procedure started. All mice were kept under standard laboratory conditions (12-h light/dark cycle, lights on at 06:00 h, 22°C, 60% humidity) and had free access to tap water and standard mouse diet. All experimental protocols were approved by the Committee on Animal Health and Care of the local government, and conformed to the international guidelines on the ethical use of animals. All efforts were made to minimize the number of animals used and their suffering.
Experimental procedure () and AOM/DSS model of colon carcinogenesis (CRC)
Figure 1. Schematic illustration of the experimental procedure. Following injection of AOM (10 mg/kg; i.p.) on day - 6, mice were either exposed to CSC housing (n = 12) or kept as SHCs (n = 11) from day 1 to 20. Subsequently, both SHC and CSC mice were treated with 1% DSS from day 50 to 57, with 0.5% DSS from day 121 to 131, and with 1% DSS from day 163 to 167. Importantly, three CSC mice died during first DSS cycle. Macroscopic changes were monitored by three colonoscopies performed on day 80, 117, and 160 in the same two narcotized mice of each group. On day 186, all mice were killed. Mice that underwent colonoscopy were not included in the analysis of any other readout parameter.
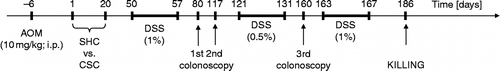
In order to investigate whether chronic psychosocial stress influences the severity of AOM/DSS-induced CRC, mice were split into two weight-matched groups, a control (n = 11) and a chronic stress group (n = 12). Mice of both groups were injected (i.p.) with 10 mg/kg AOM (74.08 kDa, Fa. Sigma-Aldrich, Steinheim, Germany) 6 days prior (day - 6) to the start (day 1) of chronic psychosocial stress induced by 19 days of CSC. On day 20, CSC mice were single housed. Both groups received DSS (36–50 kDa; ICN Biomedicals, cat. no. 160110, Eschwege, Germany) via the drinking water (Obermeier et al. Citation1999): on days 50–57 (1% DSS), 121–131 (0.5% DSS), and 163–167 (1% DSS). Importantly, three CSC mice died during the first DSS cycle. The progression of colon carcinogenesis was monitored in the same two mice per group, employing colonoscopy on days 80, 117, and 160. These mice were not included in the analysis of any other readout parameter assessed in the current study. On day 186, mice of both groups were killed by decapitation following CO2 anesthesia for assessment of colon carcinogenesis and immunological status at the level of the colonic and draining mesLN tissue. CSC has been shown to cause and to aggravate DSS-induced colitis. As AOM-induced carcinogenesis has been shown to be promoted by subsequent inflammation, CSC and DSS were given subsequent to AOM application.
CSC housing
The CSC paradigm was conducted as described previously (Reber et al. Citation2007, Citation2008; Reber and Neumann Citation2008; Veenema et al. Citation2008; Singewald et al. Citation2009). Briefly, four experimental CSC mice were housed together with a larger dominant male for 19 consecutive days as a chronic stressor. Before the CSC procedure, the future dominant males were tested for their aggressive behavior. Males that started to injure their opponents by harmful bites were not used. To avoid habituation during the chronic stressor exposure, each larger male was replaced by a novel larger male on days 8 and 15. Following prior studies, SHC were considered appropriate controls in this paradigm (Singewald et al. Citation2009). This finding is in agreement with others showing that in the male mouse, single housing is less stressful than group housing (Misslin et al. Citation1982; Bartolomucci et al. Citation2003; Gasparotto et al. Citation2005).
Colonoscopy
The progression of colon carcinogenesis was monitored by colonoscopy employing the Coloview mini-endoscope (STORZ GmbH & Co. KG, Tuttlingen, Germany) on days 80, 117, and 160, performed as previously described (Becker et al. Citation2004). To avoid exposure of all mice to the additional stressor of anesthesia and colonoscopy, the same two mice of each group were narcotized by a ketamin–xylacin (100 mg/kg ketamin and 7 mg/kg xylacin; Betapharm, Augsburg, Germany) injection (i.p.; 120 μl/mouse) 10–15 min prior to the start of each colonoscopy. These mice were randomly selected. During colonoscopy an experienced gastroenterologist blind to treatment group assessed whether macroscopic suspect lesions, i.e. granular mucosa () and/or flat polypoid lesions ()/polyps () were present in none (; indicated by “0”), one (; indicated by “1”), or both (; indicated by “2”) mice per treatment group. Importantly, mice that underwent repeated colonoscopy were not included in analysis of any other readout parameter assessed in the current study.
Table I. Effects of chronic psychosocial stress on the development of macroscopic suspect lesions in the colon.
Immunohistochemistry/immunofluorescence
After killing (by decapitation following CO2 anesthesia), the mesLNs and the colon were removed. The mesLN were separated from fat and one lymph node (LN) per mouse was embedded in histological tissue-freezing medium and snap frozen in liquid nitrogen. The other LNs were used for mesLN cell (mesLNC) stimulation. The colon was mechanically cleaned and rinsed with phosphate-buffered saline (PBS). Then a segment 0.5 cm from the distal colon was cut longitudinally, embedded in histological tissue-freezing medium, and snap frozen in liquid nitrogen. The following day the frozen mesLNs and the colon tissues were cryosectioned (longitudinally for the colon). After further tissue collection for mRNA and western blotting, the rest of the colon was spirally coiled, laid on a filter paper, and fixed in 10% formalin overnight. The next day the fixed tissue was embedded in paraffin and cut longitudinally. Ki67 immunohistochemistry (IHC) was performed in paraffin sections (one 6-μm section) and frozen sections (three 6-μm sections taken 100 μm apart). F4/80 and CD4 IHC were performed only in frozen sections (three 6-μm sections taken 100 μm apart) of the colon. Double-staining immunofluorescence for CD3 and FoxP3 was performed in frozen mesLN tissue only. Paraffin sections (colon) were deparaffinized and demasked by heating in citrate buffer (10 mM, 40 min) in a microwave oven, and frozen sections (colon and mesLN) were fixed with acetone [10 min, room temperature (RT)]. Afterwards, for IHC, deparaffinized and frozen sections were incubated with 0.3% H2O2 (30 min, RT), washed with PBS (10 min, RT; F4/80, CD4) or phosphate buffered saline tween 20 (PBS-T) (10 min, RT; Ki67), and blocked with 5% goat serum for 30 min. For immunofluorescence, acetone-fixed sections were washed with PBS (10 min, RT) and blocked with 20% fetal calf serum (FCS) for 30 min. For IHC, this was followed by incubation with 100 μl of a 5 μg/ml rat anti-mouse F4/80 (NatuTec, Frankfurt am Main, Germany) or rat anti-mouse CD4 (BD Biosciences, Heidelberg, Germany), or 150 μg/ml rat anti-mouse Ki67 (DakoCytomation, Hamburg, Germany) solution (in 1% goat serum) for 1 h at RT. For immunofluorescence, this was followed by incubation with 150 μl of a mixed 5 μg/ml hamster anti-mouse CD3 (BD Biosciences) and 2.5 μg/ml rat anti-mouse FoxP3 (BD Biosciences) solution (in 20% FCS) overnight at 4°C. After washing with PBS (three times, 5 min, RT), IHC slices were incubated with the respective biotinylated goat anti-rat secondary antibodies (1:500 in 5% goat serum; Jackson ImmunoResearch, Suffolk, UK) for 1 h at RT and washed with PBS (three times, 5 min, RT). Immunofluorescence slices were incubated with biotinylated goat anti-hamster secondary antibodies (1:2500 in PBS; Jackson ImmunoResearch). Respective positive cells were visualized either by the use of a Vectastain ABC Kit followed by Vector NovaRed Substrate Kit (Vector Laboratories, Loerrach, Germany) for IHC or by the use of a mixture of streptavidin-coupled Alexa Fluor 594 (1:500 in PBS; Invitrogen GmbH, Darmstadt, Germany) and donkey anti-rat Alexa Flour 488 antibodies (1:200 in PBS; Invitrogen GmbH) for immunofluorescence. Afterwards, immunofluorescence slices were washed again with PBS (three times, 5 min, RT) and mounted using ProLong Gold antifade reagent with 4′,6-Diamidin-2-phenylindol (DAPI) (Invitrogen GmbH). In frozen colon slices the number of Ki67+ (absolute proliferation), CD4+, and F4/80+ cells per slice (averaged from three cross-sectioned slices per mouse) and in paraffin colon slices the number of Ki67+ cells per crypt in LGD/HGD and normal tissue (relative proliferation; averaged from 10 cross-sectioned crypts per mouse) were determined. In frozen mesLN slices, the absolute number of double-stained CD3+/FoxP3+ cells per 25,600 μm2 of the T-cell-rich interfollicular area of each LN was assessed and averaged from three slices per mouse (taken 100 μm apart). Isotype controls were used in each approach to verify specificity.
Terminal deoxynucleotidyl transferase dUTP nick end labeling
Three 6-μm cross sections from frozen colon tissue per mouse (taken 100 μm apart) were used for terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL; Promega, Mannheim, Germany) according to the manufacturer's recommendation. Labeled apoptotic epithelial nuclei were counted per mm2 in three fields of view per cross section (fluorescein filter, 465–495 nm) and averaged per mouse. The mean+SEM per group was calculated and presented in the graphs. Positive controls (DNaseI; Roche Diagnostics GmbH, Mannheim, Germany) and negative controls [no TdT enzyme] were used in each approach to verify specificity.
Quantitative real-time polymerase chain reaction using TaqMan technology
Quantitative real-time polymerase chain reaction (qRT–PCR; Bustin et al. Citation2009) was performed as described previously (Blaas et al. Citation2009). Briefly, after killing (by decapitation following CO2 anesthesia) about 0.5 cm (segment proximal to colon tissue used for IHC and immunofluorescence) of the removed, PBS rinsed, and mechanically cleaned colon was placed in RNA later (Applied Biosystems, Foster City, CA, USA) and stored at − 20°C until assayed. Total RNA was isolated using the RNeasy Mini Kit (Qiagen, Hilden, Germany) and reverse transcribed into first-strand cDNA (Reverse Transcription System; Promega). Expression levels of IFN-γ, TNF, FoxP3, LRH-1, ß-catenin, and COXII were then quantified by TaqMan®-qPCR (ABI PRISM 7900HT Sequence Detection System; Applied Biosystems), as single-tube reactions (20 μl) in 384-well plates. Primers used were as follows. IFN-γ forward: TGCTGATGGGAGGAGATGTCT, IFN-γ reverse: TGCTGTCTGGCCTGCTGTTA; TNF forward: CACAAGATGCTGGGACAGTGA, TNF reverse: TCCTTGATGGTGGTGCATGA; FoxP3 forward: GTGGGCACGAAG GCAAAG, FoxP3 reverse: CCTTGT TTTGCGCTGAGAGTCT; LRH-1 forward: TTGAGTGGGCCAGGAGTAGT, LRH-1 reverse: ATCAAGAGCTCACTCCAGC AGTT; ß-catenin forward: GGTCCGAGCTGCCATGTTCC, ß-catenin reverse: CGTCAAACTGCGTGGATGGG; and COXII forward: GGCAAAGGCCTCCATTGACC, COXII reverse: GAGAAGCTGTTGCGGTACTCA. The probes were labeled 5′ with 6-carboxy-fluorescein (FAM) and 3′ with 6-carboxytetramethyl rhodamine (TAMRA). TaqMan®-qPCR was performed using 1 μl cDNA, 1 μl forward and reverse primer each (18 μM), 1 μl probe (5 μM), 10 μl TaqMan Mastermix (Applied Biosystems), 1 μl glyceraldehyde-3-phosphatedehydrogenase (GAPDH) mix (as reference; Applied Biosystems), and made up to the final volume of 20 μl with sterile H2O. Cycling was as follows: 50°C for 2 min, 95°C for 10 min followed by 40 repeats: 95°C for 15 s, and 60°C for 1 min. mRNA expression for each gene was quantified for each individual mouse in triplets and averaged per mouse.
Western blotting
After killing (by decapitation following CO2 anesthesia), about 0.5 cm (segment proximal to colon tissue used for qRT-PCR) of the removed, PBS rinsed, and mechanically cleaned colon was immediately shock-frozen in liquid nitrogen and stored at − 80°C until assayed. For protein extractions, frozen colonic tissue was homogenized in ethylenediamine tetraacetic acid (EDTA) lysis buffer (50 mM EDTA, 250 mM NaCl, 0.5 mM 2-(4-(2-hydroxyethyl)-1-piperazinyl)-ethanesulfonic acid, 0.5% Igepal, 10% complete mini protease inhibitor; Roche Diagnostics GmbH), and total protein concentration was determined using a commercial kit (Bicinchoninic Acid Protein Assay Kit, Thermo Scientific, Rockford, IL, USA). Western blot analysis was carried out using 60 μg of protein per mouse. Samples were loaded on sodium dodecyl sulfate polyacrilamide gels (7.5%) and transferred onto nitrocellulose membranes. The membranes were then blocked for 2 h in 5% milk (COXII, ß-catenin) or for 1.5 h in 5% bovine serum albumin (LRH-1), both diluted in Tris-buffered saline with 0.05% Tween 20 (TBST, Sigma-Aldrich, Milan, Italy) at 22–24°C, before being probed with primary anti-ß-catenin antibody, anti-COXII antibody (1:400; Santa Cruz Biotechnology, Inc., Heidelberg, Germany), or anti-LRH-1 antibody (1:200; Santa Cruz Biotechnology, Inc.), applied overnight at 4°C. Visualization was performed using horseradish peroxidase-conjugated whole donkey anti-rabbit antibodies (1:1000; GE Healthcare, Freiburg, Germany) followed by ECL Western Blotting Detection Reagents (GE Healthcare). Immunoblots were digitized using Molecular Imager® ChemiDoc™ XRS+system and analyzed using Image Lab™ software (Bio-Rad Laboratories GmbH, München, Germany). Afterwards, each membrane was stripped using Re-Blot Plus Mild Antibody Stripping Solution (Millipore GmbH, Schwalbach, Germany), blocked twice with 5% milk in TBST for 5 min, and probed with primary rabbit anti-ß-tubulin antibodies (1:1000, Cell Signaling Technology, New England Biolabs GmbH, Frankfurt am Main, Germany) for 2 h. Visualization, digitization, and analysis were performed as described above. ß-catenin (92 kDa), COXII (61 kDa), and LRH-1 (70 kDa) protein expression for each mouse was normalized to ß-tubulin (50 kDa) expression and averaged per group.
Determination of morphological changes in the colon
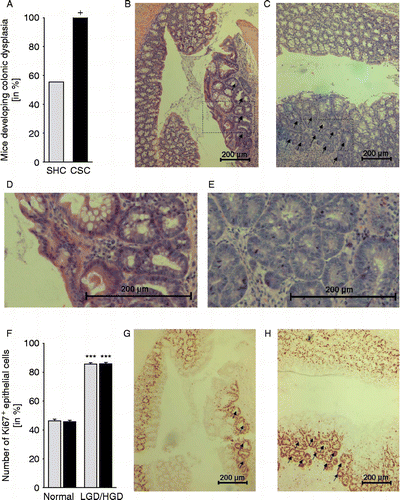
After tissue collection for mRNA and western blotting and IHC, the rest of the colon was spirally coiled, laid on a filter paper, and fixed in 10% formalin overnight. The next day the fixed tissue was embedded in paraffin and cut longitudinally (6 μm sections). Hematoxylin–eosin (HE) stained sections were evaluated by an experienced pathologist with respect to the development of LGD () and HGD (). For statistics, the percentage of mice developing either LGD and/or HGD was compared between the SHC and CSC group.
Figure 2. Effects of chronic psychosocial stress on colonic morphology and relative epithelial cell proliferation. Both SHC (n = 9, gray bars) and CSC housing (n = 7, black bars) mice were killed on day 186 following three cycles of DSS administration. (A) Hematoxylin–eosin stained paraffin sections of the distal colon were evaluated by an experienced pathologist with respect to the development of low-grade dysplasia (LGD; black arrows in B; section surrounded by the rectangle in (B) is shown in higher magnification in (D) and/or high-grade dysplasia (HGD; black arrows in (C); section surrounded by the rectangle in (C) is shown in higher magnification in (E). Data represent percentage of mice developing colonic dysplasia (HGD and/or LGD; +p = 0.069 vs. respective SHC mice; Fisher's exact test). (F) Relative epithelial cell proliferation was assessed in Ki67-stained paraffin sections by determining the percentage of Ki67+ cells per cross-sectioned crypt in non-dysplastic tissue (normal, G&H) and LGD (indicated by arrows in G)/HGD (indicated by arrows in H). Data represent mean+SEM. ***p < 0.001 vs. respective non-dysplastic (normal) tissue mice (two-way ANOVA, factor treatment group and factor dysplasia).
Isolation/incubation of mesLNCs
Isolation/incubation of mesLNCs was performed as previously described (Reber et al. Citation2007, Citation2008; Veenema et al. Citation2008). Briefly, mesLNs from SHC and CSC mice were collected, separated from fat, and after freezing one LN of each mouse in histological tissue-freezing medium for immunofluorescence, the remainder were pooled (three pools per treatment group), respectively, from two to three mice of the same treatment group. From all three pools (per treatment group), cells were isolated and incubated in quadruplicates for 24 h (200,000 cells, 37°C, 5%) in anti-CD3-coated wells in the presence of IL-2 as described previously (Reber et al. Citation2006, Citation2007). After incubation, supernatants of all four wells were harvested; IFN-γ levels were measured by ELISA (Endogene, Woburne, MA, USA); and the values were averaged for each individual pool. Consequently, for each group, the mean+SEM was calculated from these averaged pool data and presented in the graphs.
Flow cytometry
Flow cytometry analysis was performed as described earlier (Obermeier et al. Citation2006) using a Beckman Coulter EPICS® XL-MCL flow cytometer (Coulter Immunotech, Hamburg, Germany). Briefly, isolated and pooled mesLNC (all mice per treatment group) were surface stained with fluorescein isothiocyanate (FITC)-conjugated rat IgG2a anti-CD3 and respective isotype control (rat IgG2a) antibodies (BD Biosciences) to quantify the percentage of CD3+ cells within all gated cells.
Statistics
For statistical comparisons, the software package SPSS (version 12) was used. All parameters depending on just one factor were analyzed using the two-tailed Student's t-test, with the exception of the morphological changes in colonic mucosa (Fisher's exact test) and IFN-γ secretion from mesLNC (Mann–Whitney U test). Epithelial cell proliferation was analyzed using a two-way analysis of variance (ANOVA; factor treatment group and factor dysplasia), followed by a Bonferroni post hoc test if appropriate. Repeated colonoscopy was done in only two mice (always the same) per treatment group, so no statistical analysis of the macroscopic suspect lesions was performed. Data are presented as mean+SEM. Significance was taken at p ≤ 0.05.
Results
Effects of CSC on body weight development
Following AOM treatment, CSC mice (2.09+0.20 g, n = 12) gained significantly less body weight during the 19 days of CSC exposure (p = 0.017; two-tailed Student's t-test) compared with respective SHC control mice (2.91+0.24 g, n = 11). No differences in the body weight change between SHC and CSC mice were observed during any of the three subsequent DSS cycles. However, three CSC mice died during the first cycle of DSS application and have therefore been included only in the analysis of body weight development during CSC.
Effects of CSC on the development of macroscopic suspect lesions in the colon
Data from three colonoscopies repeatedly performed in two mice of each treatment group indicate () that CSC mice develop earlier granular mucosa () and flat polypoid lesions ()/polyps () compared with SHC mice.
Effects of CSC on colonic morphology and relative epithelial cell proliferation
AOM/DSS-treated CSC mice tended to develop more often LGD () and/or HGD () in colonic tissue compared with respective SHC mice (p = 0.069; Fisher's exact test; ). In addition, a significantly increased number of Ki67+ epithelial cells per crypt in LGD ()/HGD () compared with non-dysplastic tissue in both AOM/DSS-treated SHC and CSC (p < 0.001) mice indicated an enhanced relative (per crypt) epithelial cell proliferation in dysplastic compared with normal tissue (factor dysplasia F1,22 = 363.997; p < 0.001; two-way ANOVA, factor treatment group, and factor dysplasia; ).
Effects of CSC on colonic mRNA expression patterns
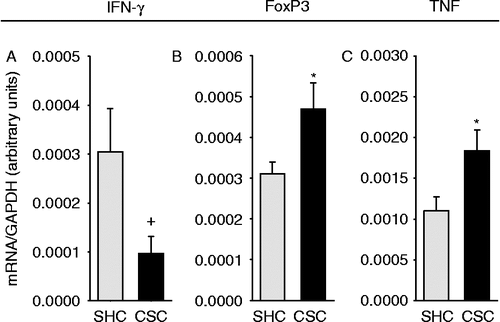
Compared with AOM/DSS-treated SHC mice, colonic mRNA expression for COXII (p = 0.025; two-tailed Student's t-test; ), LRH-1 (p = 0.027; two-tailed Student's t-test; ), FoxP3 (p = 0.038; two-tailed Student's t-test; ), and TNF (p = 0.040; two-tailed Student's t-test; ) were significantly increased in respective CSC mice. Furthermore, IFN-γ mRNA expression in the colonic tissue of AOM/DSS-treated CSC mice tended to be reduced (p = 0.063 vs. SHC; two-tailed Student's t-test; ). Colonic mRNA expression of ß-catenin was not affected by CSC ().
Figure 3. Effects of chronic psychosocial stress on colonic ß-catenin, COXII, and LRH-1 mRNA and protein expression patterns. Both SHC (n = 9) and CSC housing (n = 7) mice were killed on day 186 following three cycles of DSS administration. After killing, mRNA (A–C; tissue was collected proximal to tissue used for IHC/immunofluorescence) and protein (D–I; tissue was collected proximal to tissue used for qRT-PCR) expression levels of ß-catenin (SHC: n = 9, CSC: n = 7; A,D,G), COXII (SHC: n = 8, CSC: n = 6; B,E,H), and LRH-1 (SHC: n = 7, CSC: n = 6; C,F,I) were quantified by TaqMan®-qPCR (relative to the expression of the housekeeping gene GAPDH) or by western blotting (relative to the expression of the housekeeping gene product ß-tubulin). Figures G–I show representative Western blot images of ß-catenin (G, upper bands; 92 kDa), COXII (H, upper bands; 61 kDa), LRH-1 (I, upper bands; 70 kDa), and the corresponding ß-tubulin (G–I, lower bands) protein expression of one SHC and one CSC mouse per treatment group. mRNA expression for each gene was quantified for each individual mouse in triplets and averaged per mouse. Data represent mean+SEM; * p < 0.05 vs. respective SHC mice (two-tailed Student's t-test).
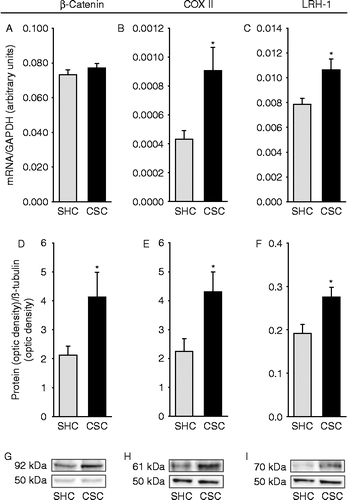
Figure 4. Effects of chronic psychosocial stress on colonic IFN-γ, FoxP3, and TNF mRNA expression patterns. Both SHC (n = 9) and CSC (n = 7) mice were killed on day 186 following three cycles of DSS administration. After killing, the expression levels of IFN-γ (SHC: n = 7, CSC: n = 6; A), FoxP3 (SHC: n = 7, CSC: n = 6; B), TNF (SHC: n = 9, CSC: n = 6; C) were quantified then by TaqMan®-qPCR, relative to the expression of the housekeeping gene (GAPDH). mRNA expression for each gene was quantified for each individual mouse in triplets and averaged per mouse. Data represent mean+SEM; +p = 0.063, *p < 0.05 vs. respective SHC mice (two-tailed Student's t-test).
Effects of CSC on colonic protein expression patterns
Colonic protein expression of ß-catenin (p = 0.028; two-tailed Student's t-test; ), COXII (p = 0.022; two-tailed Student's t-test; ), and LRH-1 (p = 0.016; two-tailed Student's t-test; ) were significantly increased in CSC compared with SHC mice, both treated with AOM/DSS.
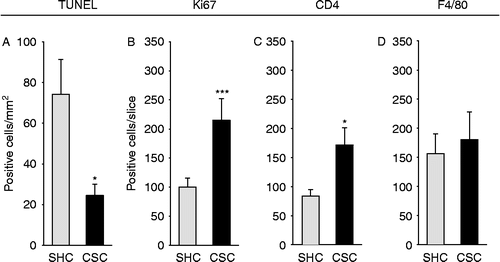
Effects of CSC on epithelial apoptosis, absolute epithelial cell proliferation and the number of CD4+ and F4/80+ cells in the colonic tissue
The number of TUNEL+ (p = 0.050; two-tailed Student's t-test; ) epithelial cells per cross-sectioned slice was significantly decreased, whereas the number of Ki67+ (p = 0.001; two-tailed Student's t-test; ) epithelial cells per cross-sectioned colon slice was significantly increased in AOM/DSS-treated CSC compared with respective SHC mice, indicating decreased epithelial apoptosis and enhanced absolute epithelial cell proliferation in CSC mice. In addition, AOM/DSS-treated CSC mice showed a significantly increased number of CD4+ Th cells (p = 0.038; two-tailed Student's t-test; ) per cross-sectioned slice, whereas the number of colonic F4/80+ macrophages cells was not different between CSC and SHC mice (), both treated with AOM/DSS.
Figure 5. Effects of chronic psychosocial stress on epithelial apoptosis, absolute epithelial cell proliferation, and the number of CD4+ and F4/80+ cells in the colonic tissue. Both SHC (n = 9) and CSC housing (n = 7) mice were killed on day 186 following three cycles of DSS administration. After killing, the colon was removed, mechanically cleaned, rinsed, embedded in histological tissue-freezing medium, snap-frozen, and cut on a cryostat. Three 6 μm cross sections taken from 100 μm apart were acetone fixated and afterwards stained for epithelial apoptosis (TUNEL; SHC: n = 8, CSC: n = 5; A), absolute epithelial proliferation (Ki67; SHC: n = 9, CSC: n = 7; B), and the number of CD4+ (SHC: n = 7, CSC: n = 5; C) or F4/80+ (SHC: n = 8, CSC: n = 6; D) cells. Respective positive cells were counted and averaged from the three cross-sectioned slices per mouse (Ki67, CD4, F4/80) or counted per mm2 of colon tissue in three fields of views in each cross section and averaged per mouse (TUNEL). Data represent mean+SEM; *p < 0.05, ***p ≤ 0.001 vs. respective SHC controls (two-tailed Student's t-test).
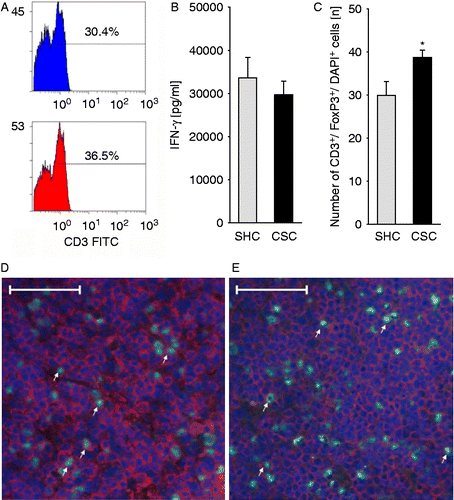
Effects of CSC on the percentage of CD3+ mesLNC, the in vitro IFN-γ secretion from isolated mesLNC, and the number of CD3+/FoxP3+ mesLNC
The percentage of CD3+ cells within all pooled (per treatment group) cells isolated from mesLNs was found to be increased by 20% in AOM/DSS-treated CSC compared with respective SHC mice (; no statistical analysis was done as all mice per group were pooled). However, IFN-γ secretion from isolated and anti-CD3-stimulated mesLNC did not differ between SHC and CSC mice (), both treated with AOM/DSS. Importantly, the number of regulatory CD3+/FoxP3+ mesLNC was significantly higher in AOM/DSS-treated CSC () compared with respective SHC () mice (p = 0.043; two-tailed Student's t-test).
Figure 6. Effects of chronic psychosocial stress on the percentage of CD3+ mesLNC, the in vitro IFN-γ secretion from isolated mesLNC, and the number of CD3+/FoxP3+ mesLNC. Both SHC (n = 9) and CSC housing (n = 7) mice were killed on day 186 following three cycles of DSS administration. After killing, mesLNC were either double-stained for CD3+ (red)/FoxP3+ (green) using immunofluorescence (C; SHC, D; CSC, E; white arrows indicate five representative double-positive cells in each slice) or mesLNC were isolated and pooled (three pools per treatment group containing 2–3 mice each) for assessment of anti-CD3-induced IFN-γ secretion (B). Remaining cells of all three pools were again pooled and surface stained with FITC-conjugated rat IgG2a anti-CD3 and respective isotype control (rat IgG2a) antibodies to quantify the percentage of CD3+ cells within all gated cells (A; no statistical analysis was done as all mice per group were pooled) using flow cytometry. IFN-γ data represent mean+SEM of three pools (2–4 mice per pool; Mann–Whitney U test) per treatment group. Numbers of CD3+/FoxP3+ mesLNC represent mean+SEM of individual mice (*p < 0.05; two-tailed Student's t-test). Scale bars represent 50 μm (SHC, D; CSC, E).
Discussion
In the present study, we demonstrate that chronic psychosocial stress increases the risk of inflammation-related CRC triggered by AOM/DSS. This was indicated by more frequent colonic dysplasia and enhanced expression of colonic ß-catenin, LRH-1, COXII, and TNF. Moreover, absolute epithelial cell proliferation was increased in the colonic tissue of chronically stressed mice. However, assessment of further time points will be essential to test whether the shift from tumor-protective Th1 cell to regulatory T-cell immunity represents a consequence of increased CRC or a causal factor in its development.
In our study, all mice were injected with AOM, a mutagen commonly used to induce carcinogenesis. One week later, mice were either exposed to 19 days of CSC or were kept as SHCs in order to study the effect of chronic psychosocial stress on inflammation-related CRC. CSC has previously been established as a clinically relevant model of chronic psychosocial stress in male mice (Reber et al. Citation2007, Citation2008, Citation2011; Reber and Neumann Citation2008; Veenema et al. Citation2008; Singewald et al. Citation2009; Schmidt et al. Citation2010; Reber Citation2011). In line with previous findings, CSC mice in the present study showed a reduced body weight gain during stressor exposure. As DSS colitis promotes AOM-induced CRC, depending on its severity (Suzuki et al. Citation2005), and as CSC itself only induces a slight colitis but heavily exaggerates DSS colitis (Reber et al. Citation2007, Citation2008; Reber Citation2011), SHC and CSC mice received three cycles of DSS to amplify the hypothesized stress effects on AOM-induced CRC. During the second cycle, DSS concentration was reduced as three CSC mice died during the first DSS cycle recapitulating previous results showing that CSC mice develop a more severe DSS colitis (Reber et al. Citation2008).
Importantly, an exacerbating effect of chronic psychosocial stress on AOM/DSS-induced CRC was suggested by several findings. Repeated colonoscopy (in two mice per treatment group) revealed that macroscopically abnormal lesions, for instance granular mucosa and/or flat polypoid lesions/polyps, developed earlier in CSC mice. Although only a small fraction of these polyps may finally become malignant, there is evidence indicating that a large majority of colorectal carcinomas develop from these adenomatous polyps (Konishi and Morson Citation1982). In line with our data, chronic immobilization (6 h/day for 3 weeks) also resulted in a faster development of UVB exposure-induced skin tumors (Saul et al. Citation2005). Furthermore, assessment of HE-stained colonic slices revealed that CSC mice tended (p = 0.069) to develop more often LGD and/or HGD compared with SHC mice (CSC: 7 out of 7; SHC: 5 out of 9). The lack of a statistically significant difference in dysplasia development between SHC and CSC mice is probably due to the fact that three CSC mice died from severe colitis during the first cycle of DSS treatment, resulting in insufficient statistical power. Humans who develop severe dysplasia in adenomas are considered to be at increased risk of developing cancer (Konishi and Morson Citation1982). Our stressor-independent finding of increased Ki67+ epithelial cells in dysplastic compared with non-dysplastic tissue, indicating increased, possibly uncontrolled, cell proliferation, further strengthens this idea. Although we did not detect differences in relative epithelial cell proliferation (per crypt) between CSC and SHC mice, either in dysplastic- or non-dysplastic-colonic tissue, the absolute number of Ki67+ epithelial cells per colonic cross section was increased in CSC mice. Together with the decreased epithelial apoptosis found in the colon of CSC compared with SHC mice, this clearly indicates abnormal patterns of cell replication, as detected in several clinical conditions associated with an increased risk for colorectal malignancies (Tanaka Citation2009). The recently described reduction in epithelial cell apoptosis following 10 h of CSC (Reber et al. Citation2011) further shows that this effect is of rapid onset and, thus likely to be causally involved in CSC-induced promotion of AOM/DSS-induced CRC. Quantification of proliferating colonic epithelial cells as a common marker for the assessment of CRC severity has also been used in other studies employing the AOM model (Komatsu et al. Citation2001).
At present we can only speculate about the mechanisms underlying the increased epithelial cell proliferation in CSC mice. Under normal conditions degradation of the proliferation-inducing factor ß-catenin is ensured by cytoplasmic proteins belonging to the ß-catenin degradation complex. However, if ß-catenin levels rise, due to either an elevated transcription/translation rate or to reduced degradation, ß-catenin translocates to the nucleus and influences most basic cellular functions including proliferation, cell-fate determination, and survival (Moon et al. Citation2002). Uncontrolled activation of the ß-catenin signaling pathway has further been shown to cause gastrointestinal tumor development (Oshima et al. Citation1995), and the ß-catenin protein is one of the key determinants in the pathogenesis of colon cancer (Kinzler and Vogelstein Citation1996). Therefore, the increased colonic ß-catenin protein expression in CSC mice in the present study is likely to be involved in upregulation of epithelial cell proliferation. However, Western blot results from the current study do not allow any conclusions to be drawn on whether nuclear ß-catenin is also increased in developing cancer lesions. This important information needs to be assessed in future studies employing IHC. In addition to ß-catenin, LRH-1 is also involved in the control of intestinal cell renewal (Botrugno et al. Citation2004) and promotion of CRC (Schoonjans et al. Citation2005). LRH-1 acts as a potent co-activator of ß-catenin via the cyclin D1 promotor (Botrugno et al. Citation2004) or by binding directly to the cyclin E1 promotor, resulting in increased cell proliferation. In the current study, both colonic LRH-1 mRNA and protein expression were significantly elevated in CSC compared with SHC mice indicating also a role for LRH in the enhancement of absolute epithelial cell proliferation in CSC mice.
Besides dysplasia formation and increased epithelial cell proliferation, impaired clearance of abnormal cells might also be involved in the promotion of AOM/DSS-induced CRC in CSC mice. However, an increased number of CD4+ Th cells in the colonic tissue and an increased percentage of CD3+T cells within pooled isolated mesLNC were found in CSC compared with SHC mice, suggesting an upregulation of tumor-protective Th1 cell immunity. This may seem to be contrary to a CSC-induced impairment of tumor cell clearance. However, the tendency to reduced colonic IFN-γ mRNA expression, coupled with the unaltered IFN-γ secretion from mesLNC, in CSC compared with non-stressed control mice, suggests that the increased numbers of intestinal T cells in CSC mice do not represent tumor-eliminating Th1 cells. In contrast, an increased colonic FoxP3 mRNA expression and an increased number of CD3+/FoxP3+ mesLNC indicate a regulatory and, thus, rather immunosuppressive quality of gut-residing T cells, which would promote tumor progression rather than clearance. In support, an increased susceptibility to UV-induced skin cancer by suppressing type-1 cytokines, protective T cells, and increasing regulatory T cell numbers (Saul et al. Citation2005) has been found following repeated immobilization (6 h/day over 3 weeks). Interestingly, several studies have reported that increased regulatory T-cell infiltration in tumor bed predicted reduced survival in cancer-bearing patients (Martin et al. Citation2010).
In a recent study, we showed that Th2, but not Th1, cell subpopulations develop glucocorticoid resistance following 19 days of CSC (Schmidt et al. Citation2010). Moreover, CSC mice overcome stress-induced adrenal insufficiency at least 8 days following termination of CSC, and the inflammatory episodes induced by DSS treatment increase plasma corticosterone levels in both SHC and CSC mice (Reber et al. Citation2008). Therefore, a specific downregulation of tumor-protective Th1 immune responses during repeated cycles of DSS in the current study might have promoted a gradual shift toward regulatory T-cell immunity and, consequently, the development of CRC. Importantly, the latter would also be in line with the observation that CSC mice showed a body weight development comparable with SHC mice during the second and third DSS cycles, indicative of an equally severe colitis, whereas they developed a more severe colitis during the first cycle. The latter is indicated by three CSC mice dying of severe colitis and is in line with an aggravated colonic inflammation in CSC compared with SHC mice when DSS is administered immediately after stressor termination (Reber et al. Citation2008). Together with recent findings showing that the numbers of regulatory T cells were significantly reduced in peripheral LN tissue immediately following termination of CSC (Schmidt et al. Citation2010), these data indicate that regulatory T cell numbers increase gradually following CSC, ameliorating the effects of chronic stress on DSS colitis but promoting those on CRC development.
Most organ-related carcinomas are associated with high levels of local TNF. TNF has been shown to inhibit the activity of tyrosine phosphatases and to diminish class-I major histocompatibility complex (MHC) antigen expression on the cell surface, permitting malignant cells, which enables them to escape immune surveillance. Furthermore, macrophage-derived TNF has been shown to promote signaling via the ß-catenin pathway, thereby contributing to tumor development in the gastric mucosa (Oguma et al. Citation2008). As high levels of perceived life stress are associated with increased expression of TNF, released from the macrophage/monocyte lineage, this might be a general mechanism underlying stress-induced promotion of tumorogenesis (Reiche et al. Citation2004). Although the number of colonic F4/80+ macrophages was not affected by CSC in the present study, an increased colonic TNF mRNA expression indicated an increased activity of these or other TNF-producing cells maybe contributing to the CSC promoting effect on CRC development. Interestingly, recent evidence indicates that TNF is able to activate and expand regulatory T cell number by activating their TNFR2 receptors, especially in the tumor microenvironment (Chen et al. Citation2010; Chen and Oppenheim Citation2011). Therefore, increased TNF mRNA expression in the colonic tissue of CSC mice might be involved in mediating the shift toward regulatory T cell immunity. However, future studies are needed to clarify this hypothesis.
Finally, the increased expression of COXII in CSC mice further supports our stress-promoted carcinogenesis concept. COXII is considered to contribute to tumor development by modulating apoptosis, angiogenesis, and tumor invasiveness (McConnell and Yang Citation2009), and is overexpressed in approximately 80% of CRC and 40% of colorectal adenomas relative to normal mucosa (Eberhart et al. Citation1994). Indeed, as already discussed above, epithelial apoptosis was decreased in CSC compared with SHC mice in the present study.
In conclusion, our findings indicate for the first time that chronic psychosocial stress clearly increases the risk for inflammation-related CRC. Chronically stressed mice more frequently developed colonic LGD and/or HGD, showed an enhanced colonic expression of TNF and COXII, and an increase in epithelial cell proliferation, likely mediated by an enhanced ß-catenin and LRH-1 signaling. However, further time points need to be assessed to clarify in detail whether the detected shift from tumor-protective Th1 cell to regulatory T cell immunity represents cause or consequence of increased CRC found in CSC mice.
Acknowledgements
The authors are grateful to N. Dunger and D. Gaag for their excellent technical help and to Dr D. A. Slattery for his helpful comments regarding the manuscript.
Declaration of interest: This study was not supported by any particular research grant. The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Bartolomucci A, Palanza P, Sacerdote P, Ceresini G, Chirieleison A, Panerai AE, Parmigiani S. 2003. Individual housing induces altered immuno-endocrine responses to psychological stress in male mice. Psychoneuroendocrinology. 28:540–558.
- Becker C, Fantini MC, Schramm C, Lehr HA, Wirtz S, Nikolaev A, Burg J, Strand S, Kiesslich R, Huber S, Ito H, Nishimoto N, Yoshizaki K, Kishimoto T, Galle PR, Blessing M, Rose-John S, Neurath MF. 2004. TGF-[beta] suppresses tumor progression in colon cancer by inhibition of IL-6 trans-signaling. Immunity. 21:491–501.
- Bernstein CN, Singh S, Graff LA, Walker JR, Miller N, Cheang M. 2010. A prospective population-based study of triggers of symptomatic flares in IBD. Am J Gastroenterol. 105:1994–2002.
- Blaas SH, Stieber-Gunckel M, Falk W, Obermeier F, Rogler G. 2009. CpG-oligodeoxynucleotides stimulate immunoglobulin A secretion in intestinal mucosal B cells. Clin Exp Immunol. 155:534–540.
- Botrugno OA, Fayard E, Annicotte JS, Haby C, Brennan T, Wendling O, Tanaka T, Kodama T, Thomas W, Auwerx J, Schoonjans K. 2004. Synergy between LRH-1 and beta-catenin induces G1 cyclin-mediated cell proliferation. Mol Cell. 15:499–509.
- Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL, Vandesompele J, Wittwer CT. 2009. The MIQE guidelines: Minimum information for publication of quantitative real-time PCR experiments. Clin Chem. 55:611–622.
- Chen X, Oppenheim JJ. 2011. The phenotypic and functional consequences of tumour necrosis factor receptor type 2 expression on CD4(+) FoxP3(+) regulatory T cells. Immunology. 133:426–433.
- Chen X, Subleski JJ, Hamano R, Howard OM, Wiltrout RH, Oppenheim JJ. 2010. Co-expression of TNFR2 and CD25 identifies more of the functional CD4+FOXP3+regulatory T cells in human peripheral blood. Eur J Immunol. 40:1099–1106.
- Choi PM, Zelig MP. 1994. Similarity of colorectal cancer in Crohn's disease and ulcerative colitis: Implications for carcinogenesis and prevention. Gut. 35:950–954.
- Chulada PC, Thompson MB, Mahler JF, Doyle CM, Gaul BW, Lee C, Tiano HF, Morham SG, Smithies O, Langenbach R. 2000. Genetic disruption of Ptgs-1, as well as Ptgs-2, reduces intestinal tumorigenesis in Min mice. Cancer Res. 60:4705–4708.
- Eaden JA, Mayberry JF. 2000. Colorectal cancer complicating ulcerative colitis: a review. Am J Gastroenterol. 95:2710–2719.
- Eaden JA, Abrams KR, Mayberry JF. 2001. The risk of colorectal cancer in ulcerative colitis: A meta-analysis. Gut. 48:526–535.
- Eberhart CE, Coffey RJ, Radhika A, Giardiello FM, Ferrenbach S, DuBois RN. 1994. Up-regulation of cyclooxygenase 2 gene expression in human colorectal adenomas and adenocarcinomas. Gastroenterology. 107:1183–1188.
- Ekbom A, Helmick C, Zack M, Adami HO. 1990. Increased risk of large-bowel cancer in Crohn's disease with colonic involvement. Lancet. 336:357–359.
- Gasparotto OC, Lopes DM, Carobrez SG. 2005. Pair housing affects anxiety-like behaviors induced by a social but not by a physiological stressor in male Swiss mice. Physiol Behav. 85:603–612.
- Kinzler KW, Vogelstein B. 1996. Lessons from hereditary colorectal cancer. Cell. 87:159–170.
- Komatsu SI, Watanabe H, Oka T, Tsuge H, Nii H, Kato N. 2001. Vitamin B-6-supplemented diets compared with a low vitamin B-6 diet suppress azoxymethane-induced colon tumorigenesis in mice by reducing cell proliferation. J Nutr. 131:2204–2207.
- Konishi F, Morson BC. 1982. Pathology of colorectal adenomas: A colonoscopic survey. J Clin Pathol. 35:830–841.
- Laurent P, Céline R-B, Aurélie C, Virginie M, Stéphane G, Lionel C, Nathalie I, Agathe R, Paul H, Philippe N, Franck G. 2010. Epithelial vanin-1 controls inflammation-driven carcinogenesis in the colitis-associated colon cancer model. Inflamm Bowel Dis. 16:96–104.
- Levav I, Kohn R, Iscovich J, Abramson JH, Tsai WY, Vigdorovich D. 2000. Cancer incidence and survival following bereavement. Am J Public Health. 90:1601–1607.
- Martin F, Ladoire S, Mignot G, Apetoh L, Ghiringhelli F. 2010. Human FOXP3 and cancer. Oncogene. 29:4121–4129.
- McConnell BB, Yang VW. 2009. The role of inflammation in the pathogenesis of colorectal cancer. Curr Colorectal Cancer Rep. 5:69–74.
- Misslin R, Herzog F, Koch B, Ropartz P. 1982. Effects of isolation, handling and novelty on the pituitary – adrenal response in the mouse. Psychoneuroendocrinology. 7:217–221.
- Moon RT, Bowerman B, Boutros M, Perrimon N. 2002. The promise and perils of Wnt signaling through beta-catenin. Science. 296:1644–1646.
- Obermeier F, Kojouharoff G, Hans W, Scholmerich J, Gross V, Falk W. 1999. Interferon-gamma (IFN-gamma)- and tumour necrosis factor (TNF)-induced nitric oxide as toxic effector molecule in chronic dextran sulphate sodium (DSS)-induced colitis in mice. Clin Exp Immunol. 116:238–245.
- Obermeier F, Hausmann M, Kellermeier S, Kiessling S, Strauch Ulrike G, Duitman E, Bulfone-Paus S, Herfarth H, Bock J, Dunger N, Stoeck M, Schölmerich J, Falk W, Rogler G. 2006. IL-15 protects intestinal epithelial cells. Eur J Immunol. 36:2691–2699.
- Oguma K, Oshima H, Aoki M, Uchio R, Naka K, Nakamura S, Hirao A, Saya H, Taketo MM, Oshima M. 2008. Activated macrophages promote Wnt signalling through tumour necrosis factor-alpha in gastric tumour cells. EMBO J. 27:1671–1681.
- O'Shaughnessy JA, Kelloff GJ, Gordon GB, Dannenberg AJ, Hong WK, Fabian CJ, Sigman CC, Bertagnolli MM, Stratton SP, Lam S, Nelson WG, Meyskens FL, Alberts DS, Follen M, Rustgi AK, Papadimitrakopoulou V, Scardino PT, Gazdar AF, Wattenberg LW, Sporn MB, Sakr WA, Lippman SM, Von Hoff DD. 2002. Treatment and prevention of intraepithelial neoplasia: An important target for accelerated new agent development. Clin Cancer Res. 8:314–346.
- Oshima M, Oshima H, Kitagawa K, Kobayashi M, Itakura C, Taketo M. 1995. Loss of Apc heterozygosity and abnormal tissue building in nascent intestinal polyps in mice carrying a truncated Apc gene. Proc Natl Acad Sci USA. 92:4482–4486.
- Reber SO. Stress and animal models of inflammatory bowel disease – an update on the role of the hypothalamo–pituitary–adrenal axis. Psychoneuroendocrinology. 2011 doi: 10.1016/j.psyneuen.2011.05.014.
- Reber SO, Neumann IO. 2008. Chronic subordinate colony (CSC) housing enhances state anxiety and reduces hypothalamic arginine vasopressin, but not oxytocin, synthesis. Ann NY Acad Sci. 1148:184–195.
- Reber SO, Obermeier F, Straub RH, Falk W, Neumann ID. 2006. Chronic intermittent psychosocial stress (social defeat/overcrowding) in mice increases the severity of an acute DSS-induced colitis and impairs regeneration. Endocrinology. 147:4968–4976.
- Reber SO, Birkeneder L, Veenema AH, Obermeier F, Falk W, Straub RH, Neumann ID. 2007. Adrenal insufficiency and colonic inflammation after a novel chronic psycho-social stress paradigm in mice: Implications and mechanisms. Endocrinology. 148:670–682.
- Reber SO, Obermeier F, Straub RH, Veenema AH, Neumann ID. 2008. Aggravation of DSS-induced colitis after chronic subordinate colony (CSC) housing is partially mediated by adrenal mechanisms. Stress. 11:225–234.
- Reber SO, Peters S, Slattery DA, Hofmann C, Schölmerich J, Neumann ID, Obermeier F. 2011. Mucosal immunosuppression and epithelial barrier defects are key events in murine psychosocial stress-induced colitis. Brain Behav Immun. 25:1153–1161.
- Reiche EM, Nunes SO, Morimoto HK. 2004. Stress, depression, the immune system, and cancer. Lancet Oncol. 5:617–625.
- Rutter M, Saunders B, Wilkinson K, Rumbles S, Schofield G, Kamm M, Williams C, Price A, Talbot I, Forbes A. 2004. Severity of inflammation is a risk factor for colorectal neoplasia in ulcerative colitis. Gastroenterology. 126:451–459.
- Saul AN, Oberyszyn TM, Daugherty C, Kusewitt D, Jones S, Jewell S, Malarkey WB, Lehman A, Lemeshow S, Dhabhar FS. 2005. Chronic stress and susceptibility to skin cancer. J Natl Cancer Inst. 97:1760–1767.
- Schmidt D, Reber SO, Botteron C, Barth T, Peterlik D, Uschold N, Männel DN, Lechner A. 2010. Chronic psychosocial stress promotes systemic immune activation and the development of inflammatory Th cell responses. Brain Behav Immun. 24:1097–1104.
- Schoonjans K, Dubuquoy L, Mebis J, Fayard E, Wendling O, Haby Cl, Geboes K, Auwerx J. 2005. Liver receptor homolog 1 contributes to intestinal tumor formation through effects on cell cycle and inflammation. Proc Natl Acad Sci USA. 102:2058–2062.
- Singewald GM, Nguyen NK, Neumann ID, Singewald N, Reber SO. 2009. Effect of chronic psychosocial stress-induced by subordinate colony (CSC) housing on brain neuronal activity patterns in mice. Stress. 12:58–69.
- Stefanski V. 2001. Social stress in laboratory rats: Behavior, immune function, and tumor metastasis. Physiol Behav. 73:385–391.
- Stefanski V, Ben-Eliyahu S. 1996. Social confrontation and tumor metastasis in rats: defeat and beta-adrenergic mechanisms. Physiol Behav. 60:277–282.
- Suzuki R, Kohno H, Sugie S, Tanaka T. 2005. Dose-dependent promoting effect of dextran sodium sulfate on mouse colon carcinogenesis initiated with azoxymethane. Histol Histopathol. 20:483–492.
- Tanaka T. 2009. Colorectal carcinogenesis: Review of human and experimental animal studies. J Carcinog. 8:5–24.
- Tanaka T, Kohno H, Suzuki R, Yamada Y, Sugie S, Mori H. 2003. A novel inflammation-related mouse colon carcinogenesis model induced by azoxymethane and dextran sodium sulfate. Cancer Sci. 94:965–973.
- van Hogezand RA, Eichhorn RF, Choudry A, Veenendaal RA, Lamers CB. Malignancies in inflammatory bowel disease: Fact or fiction? Scand. J Gastroenterol Suppl. 2002; (236). 200248–58.
- Veenema AH, Reber SO, Selch S, Obermeier F, Neumann ID. 2008. Early life stress enhances the vulnerability to chronic psychosocial stress and experimental colitis in adult mice. Endocrinology. 144:2727–2736.
- Xie J, Itzkowitz SH. 2008. Cancer in inflammatory bowel disease. World J Gastroenterol. 14:378–389.
- Yang GY, Taboada S, Liao J. 2009. Inflammatory bowel disease: A model of chronic inflammation-induced cancer. Methods Mol Biol. 511:193–233.