Abstract
Psychological stress is associated with a systemic inflammatory response. It is unclear, however, whether psychological stress contributes to vascular inflammation. Here, we examined the effects of unpredictable chronic mild stress (UCMS) on vascular inflammation in rabbits. One hundred rabbits were randomly divided into control and stress groups. UCMS was induced by a set of defined adverse conditions applied in a shuffled order for 4, 8, 12, or 16 weeks, and rabbits were killed 24 h after the end of the UCMS protocol. Expression of different inflammatory molecules was analyzed by real-time polymerase chain reaction, immunohistochemistry, or enzyme-linked immunosorbent assay. UCMS resulted in depression-like behaviors, decreased body weight gain, and hypertension with no significant effects on serum lipids. Aortic mRNA and protein expression for tumor necrosis factor-α (TNF-α), C-reactive protein (CRP), monocyte chemoattractant protein-1 (MCP-1), macrophage migration inhibitory factor, and expression of intercellular adhesion molecule-1 (ICAM-1) protein were increased. UCMS increased circulating concentrations of corticosterone, TNF-α, and CRP throughout. Moreover, stress downregulated the expression of endothelial nitric oxide synthase. At 16 weeks of UCMS, macrophage infiltration and lipid accumulation in the subendothelial space were detected in the aorta. In cultured murine vascular smooth muscle cells, treatment with serum from stressed rabbits significantly increased phosphorylation of p38 and c-Jun N-terminal kinase (JNK), and upregulated expression of MCP-1 and ICAM-1 mRNAs, in which the effect was blunted by a TNF-α neutralizing antibody or p38 and JNK inhibitors. Our results indicate that chronic psychological stress induces vascular inflammation via TNF-α and p38/JNK pathways, which may contribute to the development of atherosclerosis.
Introduction
Accumulating epidemiological and experimental evidence has indicated that psychological stress may make significant contributions to the development of cardiovascular diseases like atherosclerosis (Black and Garbutt Citation2002; Strike and Steptoe Citation2004; Lichtman et al. Citation2008). A number of psychosocial factors, including chronic stress, depression, socioeconomic status, personality traits, and social support, have been implicated in the pathogenesis of atherosclerosis (Strike and Steptoe Citation2004). Some of these psychosocial factors have been recognized as independent risk factors for coronary arterial disease (Das and O'Keefe Citation2006; Shen et al. Citation2008), and may account for the major cardiovascular events such as stroke and myocardial infarction in individuals without conventional risk factors (Black and Garbutt Citation2002; Lombard Citation2010). The mechanisms underlying the effects of psychosocial stress in the development of cardiovascular disease are not fully understood, while evidence has suggested that these may include alterations in the neuroendocrine function of the hypothalamic–pituitary–adrenal axis, autonomic regulation, homeostasis of the vascular system, immunity, inflammation, and metabolism (Strike and Steptoe Citation2004). Moreover, the relationship between psychosocial stress and cardiovascular disease is further complicated by the finding that the presence of cardiovascular disease may cause mental abnormalities in humans (Lombard Citation2010). Hence, elucidating the mechanistic link between psychosocial stress and cardiovascular disease depends on the availability of suitable animal models.
The unpredictable chronic mild stress (UCMS) method has been widely used in animal studies and is particularly useful in modeling human depressive disorders because of its sensitivity and reliability in reproducing depression-like behaviors including agitation, anhedonia, and learned helplessness in rodents (Willner Citation1997; Deussing Citation2006). In this model, animals are exposed to a set of defined mild adverse conditions in a random manner. In mice, it has been demonstrated that chronic psychological stress exposure resulted in vascular dysfunction and enhanced atherogenesis (Kumari et al. Citation2003; d'Audiffret et al. Citation2010). However, controversial data have also been reported in mouse models (Bernberg et al. Citation2009). Nonetheless, rabbits have been used as a model to study atherosclerosis for five decades (Constantinides et al. Citation1960). High-cholesterol feeding alone in rabbits can induce foam cell-rich lesions that are similar to those found in human aorta, and concomitant mechanical injury of the aortic wall may substantially accelerate the induction of atherosclerosis (Abela et al. Citation1995; Phinikaridou et al. Citation2009). More importantly, the advanced atherosclerotic plaques formed in the rabbit aorta exhibit features of plaque destabilization and can be pharmacologically triggered to rupture, a process in coronary arteries that is considered to be the direct cause of acute myocardial infarction in humans (Abela et al. Citation1995; Phinikaridou et al. Citation2009). In contrast, plaque destabilization and rupture are difficult to reproduce in mice (Cullen et al. Citation2003). The vascular effects of UCMS in rabbits are currently not clear.
Vascular endothelial dysfunction and inflammation have fundamental roles in the pathogenesis of atherosclerosis. Previous studies have demonstrated that the exposure to an unstable social environment accelerated the progression of atherosclerosis in Watanabe heritable hyperlipidemic rabbits (McCabe et al. Citation2002). Conversely, behavioral interventions tending to reduce psychological stress-retarded atherogenesis (Nerem et al. Citation1980). It has been well documented that psychological stress may contribute to the development of vascular endothelial dysfunction in rodents, primates, and humans (Strawn et al. Citation1991; Williams et al. Citation1993; Sherwood et al. Citation1999; Spieker et al. Citation2002; Neves et al. Citation2009), and this effect may be attributable to decreased nitric oxide availability and increased reactive oxygen production (Kumari et al. Citation2003; d'Audiffret et al. Citation2010). In contrast, the effects of psychological stress on vascular inflammation are not totally clear. Given the pivotal role of vascular inflammation in atherosclerosis, in this study we tested the hypothesis that chronic psychological stress might have direct impacts on vascular inflammatory responses using a rabbit model of UCMS.
Materials and methods
Animals
One hundred adult male white New Zealand rabbits weighing 1.5–2 kg (∼3 months old) were purchased from Shandong Lukang Pharmaceutical Corporation (Jining, Shandong Province, China). The rabbits were maintained on a standard chow and water ad lib, and housed individually in cages 60 × 40 × 50 cm in size, in rooms maintained at 20 ± 2°C with an automated 12-h light–dark cycle (lights on at 07:00 h).
Unpredictable mild stress exposure
All animal treatment procedures and the total number of animals used were approved by the Animal Care Committee of Shandong University (Permit #2008-036). Rabbits were randomly assigned to the control group (n = 50) or psychological stress group (n = 50). Four different stressors were used in this study: exposure to noise (a 110 dB acoustic alarm sounded for 5 s, repeated every 5 min for 3 h); restraint stress (confinement in a wooden box of 16 × 16 × 40 cm, which prevented the rabbit from moving freely and was not exposed to light for 4 h); inversion of the light–dark cycle (lights on at 19:00 h); and cage hanging (by suspending the whole cage with a rope in the middle so that any movement of the rabbit tended to result in loss of balance) for 2 h (Yalcin et al. Citation2005; Kiank et al. Citation2006). Each rabbit was given a single stressor daily for 4, 8, 12, or 16 weeks. To prevent habituation, these conditions were arranged in a shuffled order and applied as a 4-week regime (); for 8-, 12-, and 16-week groups the same regime was repeated. Rabbits without stressor exposure were used as controls. Twenty-four hours after the last stress exposure, the rabbits were killed humanely with 3% pentobarbital sodium injection (30 mg/kg) via the ear vein.
Table I. UCMS protocol.
Behavioral and general physiological parameters
Measurements of behavioral and hemodynamic parameters were performed on the last 3 days before the end of experiment ( and supplementary Figure I). Grooming motivation was assessed by a splash test as an indicator of depression-like behavior. Briefly, a 10% sucrose solution was sprayed on the dorsal coat, and grooming activity (attempts made to clean the fur by licking or scratching) was counted for 5 min (d'Audiffret et al. Citation2010; Isingrini et al. Citation2011). The systolic blood pressure (SBP), the diastolic blood pressure (DBP), and the heart rate (HR) were measured in conscious rabbits using a noninvasive oscillometric device (Softron BP-98E, Softron, Tokyo, Japan) as described previously (Nakamura et al. Citation1998). Briefly, rabbits were acclimatized to the holder supplied and an inflatable cuff was placed around the forearm. The blood pressure and the HR were determined in a fully automated manner. At least five measurements were performed for each rabbit and the readings were averaged. The appetite of the rabbits, another indicator of depression-like behavior, was assessed by monitoring the food intake daily (the weight of food given in the morning minus the remaining next morning) during the last week of treatment, and the number averaged at the end of the week. Body weight was recorded immediately after euthanasia.
Sample collection
Blood samples were collected between 09:00 and 10:00 h on the day of euthanasia by ear vein puncture with gentle manual restraint, into a vacuum blood collection tube without anticoagulant, and left to clot for 10 min at room temperature for clotting. The whole sample was then centrifuged at 1500g for 10 min, and the serum was transferred into a separate vial and stored at − 80°C. Abdominal aorta was dissected and cut into parts. Some of the aorta was snap frozen in liquid nitrogen and stored at − 80°C for RNA extraction; the remaining parts were either embedded in optimum cutting temperature (OCTTM) compound for cryosectioning or fixed in a 10% formalin solution and processed for paraffin embedding and sectioning.
Biochemical measurements
To examine the effects of UCMS on lipid metabolism, total cholesterol, triglyceride, low-density lipoprotein cholesterol, and high-density lipoprotein cholesterol were measured by enzymatic assays using an automated biochemical analyzer (Roche Hitachi 917, Roche Diagnostics, Indianapolis, Indiana, USA). The hypothalamic–pituitary–adrenal axis is a major effector of responses to systemic stress. We therefore measured the serum concentration of corticosterone. To assess the effects of UCMS on circulating inflammatory markers, we measured the serum concentrations of C-reactive protein (CRP) and tumor necrosis factor-α (TNF-α). The concentrations of corticosterone, CRP, and TNF-α were assayed using enzyme-linked immunosorbent assay kits (from USCN Life Science and Technology, Wuhan, China). The sensitivities (minimum detectable concentration) of the assays for corticosterone, CRP, and TNF-α were 0.4 ng/ml, 0.2 μg/ml, and 5.0 pg/ml, respectively.
Histology and immunohistochemistry
Cryosections of 5 μm in thickness were stained with 0.3% Oil Red O solution in 60% isopropanol for 10 min at room temperature to visualize lipid components. Paraffin sections were used for immunohistochemical analysis except for the anti-rabbit macrophage (RAM11) antibody, which was tested in cryosections. For paraffin sections, antigen retrieval was performed by heating the sample in citrate buffer (10 mM sodium citrate, pH 6.0) to boiling point. Samples were incubated with 3% H2O2 for 10 min to quench endogenous peroxidase activity and blocked with 5% bovine serum albumin. The following primary antibodies were used: RAM11 (1:200), anti-TNF-α (1:100), anti-human CRP (1:200), anti-human monocyte chemoattractant protein-1 (MCP-1, 1:200), anti-human macrophage migration inhibitory factor (MIF, 1:200), and anti-human intercellular adhesion molecule-1 (ICAM-1, 1:100). All of the antibodies were purchased from Abcam (Cambridge, UK) and Santa Cruz Biotechnology (Santa Cruz, California, USA). The cross-reactivity of these antibodies with rabbit antigens was tested in preliminary experiments and confirmed by negative control experiments in which saline was used instead of primary antibodies. The slides were visualized with 3,3′-diaminobenzidine (DAB) as chromogen.
Real-time polymerase chain reaction
To assess the effects of UCMS on vascular inflammation, we performed real-time polymerase chain reaction (PCR) analysis on expression levels of mRNAs for TNF-α, MCP-1, MIF, and CRP in the abdominal aorta. To evaluate effects on vascular endothelial function, we measured the mRNA expression levels of endothelial nitric oxide synthase (eNOS) and endothelin-1. Total RNA was extracted using Trizol reagent (Invitrogen, Carlsbad, CA, USA). The quality of samples was determined by agarose gel electrophoresis. Total RNA (1 μg) was reverse transcribed to single-strand cDNA using the TaqMan Gold real-time PCR kit and random hexamer in a total volume of 50 μl. The thermocycler settings were 25°C for 10 min, 48°C for 30 min, and 95°C for 5 min. Real-time PCR was performed in duplicates using 1 μl cDNA and SYBR Green Master Mix (SYBR Exscript, TaKaRa (Kyoto, Japan)) in a 25 μl reaction volume with an iQ5 thermocycler (Bio-Rad, Hercules, California, USA). The primer sequences used in this study are given in Supplementary . Glyceraldehyde-3-phosphate dehydrogenase was used as housekeeping gene.
Cell culture and treatment
Aortic smooth muscle cells were isolated from normal male C57/bl6 mice as described previously (Jiang et al. Citation2008). Briefly, the aorta was dissected into cold saline, cleaned of fat, and incubated in a digestion mixture containing collagenase I (1 mg/ml), elastase (0.5 mg/ml), and trypsin (1.25 mg/ml) in serum-free Dulbecco's modified Eagle's medium (DMEM; Invitrogen) at 37°C for 10 min. Then the adventitia was peeled off with forceps. The remaining tissue was transferred to a clean vial containing 1.5 ml of sterile digestion mixture and further incubated for 1 h at 37°C. The released smooth muscle cells were cultured in DMEM supplemented with 10% fetal bovine serum (FBS). Cells between passages 4 and 10 were used. The identity of the cells was determined by immunocytochemistry for smooth muscle α-actin. For treatment, cells of 90% confluence were washed with saline and incubated with DMEM in which the fetal calf serum was replaced with 20% or 40% serum collected from stressed (16-week group) or control rabbits for a varying time. In some experiments, the cells were pretreated with a neutralizing antibody against TNF-α (R&D systems, Minneapolis, Minnesota, USA), Catalog number: MAB2101), or inhibitors of p38 (SB202190) and c-Jun N-terminal kinase (JNK; SP600125, all purchased from Merck, Germany).
Western blot
Western blot analysis was performed as described previously (Jiang et al. Citation2008). Cells were lysed with cold lysis buffer containing 25 mM Tris (pH 7.5), 2 mM ethylenediaminetetraacetic acid (EDTA), 100 mM NaCl, 50 mM NaF, 1 mM Na3VO4, 1 mM dithiothreitol, 1% Triton-X 100, and the complete proteinase inhibitor cocktail (Roche), and the total protein was extracted by incubating the sample at 4oC for 1 h. The protein samples were separated by 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred onto a nitrocellulose membrane. The membrane was blocked with 5% nonfat milk powder in tris-buffered saline (pH 7.5) and hybridized overnight with anti-phospho-specific and total extracellular signal-regulated kinase (ERK), p38, and JNK antibodies (all from Cell Signaling Technology, Danvers, Massachusetts, USA). The antigen was detected with horseradish peroxidase-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories, (West Grove, Pennsylvania, USA) and visualized with an enhanced chemiluminescence (ECL)-plus kit (GE Healthcare, Piscataway, New Jersey, USA).
Statistical analysis
Data are presented as mean ± standard error of the mean (SEM). Mann–Whitney's U test was used to compare data from two groups: one-way analysis of variance (ANOVA) was used to analyze the effects of a single factor among multiple groups; two-way ANOVA was used to simultaneously analyze the effects of and interaction between two factors. SPSS 11.5 was used for statistical analysis. A P-value of < 0.05 was considered to be statistically significant.
Results
UCMS exposure resulted in depression-like symptoms in normal rabbits
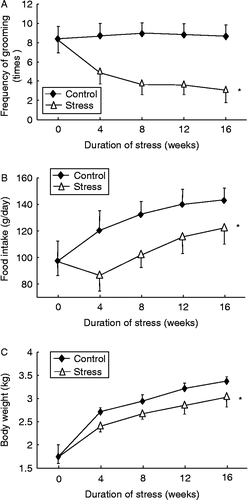
Throughout the 16-week period, the frequency of grooming in the splash test was not affected by time in the control group; in contrast, we found that stress exposure significantly reduced the frequency of grooming in a time-dependent manner (; two-way ANOVA, stress: F 257.7, df 1, 90, P < 0.01; time: F 12.7, df 4, 90, P < 0.01; interaction P < 0.01). All data presented hereafter are mean ± SEM. The discrete effects of time in each experimental group were also analyzed with one-way ANOVA. The average food intake and body weight increased time dependently in control rabbits, and both parameters were significantly decreased by stress exposure (; two-way ANOVA, stress: F 74.5, df 1, 90, P < 0.01; time: F 27.7, df 4, 90, P < 0.01; interaction P < 0.01; and , two-way ANOVA, stress: F 57.8, df 1, 90, P < 0.01; time: F 254.6, df 4, 90, P < 0.01; interaction P < 0.01).
Figure 1. Effects of chronic psychological stress on (A) frequency of grooming, (B) food intake, and (C) body weight gain in rabbits. Grooming frequency was measured by counting the number of attempts made to clean the fur by licking or scratching for 5 min after spraying a 10% sucrose solution on the dorsal coat. Data are mean ± SEM. *P < 0.01 vs. control group, two-way ANOVA (n = 9–10 per group/time point).
Chronic psychological stress disrupted the homeostasis of the hypothalamic–pituitary–adrenal axis
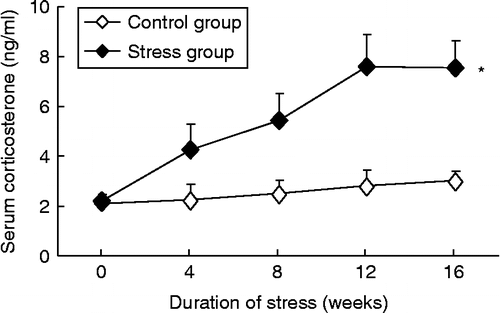
The serum corticosterone concentration did not significantly change with time in control rabbits. In response to chronic psychological stress, serum corticosterone increased in a time-dependent manner and reached the maximum at 12 weeks, which was maintained for 16 weeks (, two-way ANOVA, stress: F 311.6, df 1, 90, P < 0.01; time: F 49.4, df 4, 90, P < 0.01; interaction P < 0.01).
Figure 2. Effects of chronic psychological stress on serum corticosterone concentrations. Data are mean ± SEM. *P < 0.01 versus control group, two-way ANOVA (n = 9–10 per group/time point).
Chronic psychological stress disturbed systemic hemodynamics
There was a significant, time-dependent increase in SBP in control rabbits. UCMS exposure further enhanced the increase in SBP (; two-way ANOVA, stress: F 194.4, df 1, 90, P < 0.01; time: F 36.9, df 4, 90, P < 0.01; interaction P < 0.01). In contrast, the DBP and HR remained stable throughout the experiment in controls. UCMS significantly elevated DBP and HR as compared with untreated rabbits (; two-way ANOVA, DBP, stress: F 217.5, df 1, 90, P < 0.05; time: F 34.6, df 4, 90, P < 0.05; interaction P < 0.05; HR, stress: F 142.9, df 1, 90, P < 0.05; time: F 28.2, df 4, 90, P < 0.05; interaction P < 0.05).
Table II. Effects of psychological stress on systemic hemodynamic parameters.
Chronic psychological stress-induced vascular inflammation
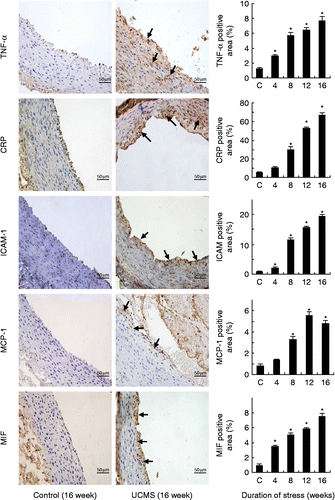
As shown in , psychological stress significantly increased the mRNA levels for TNF-α (two-way ANOVA, stress: F 1133.8, df 1, 90, P < 0.01; time: F 187.6, df 4, 90, P < 0.01; interaction P < 0.01), MCP-1 (two-way ANOVA, stress: F 1152.1, df 1, 90, P < 0.01; time: F 21.7, df 4, 90, P < 0.01; interaction P < 0.01), CRP (two-way ANOVA, stress: F 733.6, df 1, 90, P < 0.01; time: F 102.4, df 4, 90, P < 0.01; interaction P < 0.01), and MIF (two-way ANOVA, stress: F 587.9, df 1, 90, P < 0.01; time: F 63.6, df 4, 90, P < 0.01; interaction P < 0.01), and in a time-dependent manner, whereas time had no significant effects on these parameters in control rabbits. Consistent with the PCR results, we further demonstrated using immunohistochemistry analysis that the protein levels of TNF-α, CRP, MCP-1, and MIF were also increased in the stress group (; one-way ANOVA, TNF-α: F 52.5, df 4, 20, P < 0.05; CRP: F 154.8, df 4, 20, P < 0.05; MCP-1: F 65.9, df 4, 20, P < 0.05; MIF: F 52.5, df 4, 20, P < 0.05). Moreover, we found that the protein expression level of ICAM-1 was also elevated by stress exposure (; one-way ANOVA, F 265.7, df 4, 20, P < 0.05). We also demonstrated that eNOS mRNA expression in aorta was significantly lower in stressed rabbits as compared to controls (55.9 ± 7.2% and 32.1 ± 8.1% of control at 8 and 16 weeks, respectively, all P < 0.05, Mann–Whitney's U test, n = 9–10), whereas endothelin-1 mRNA expression was elevated after stress exposure (146.9 ± 6.7% and 174.1 ± 14.4% of control at 8 and 16 weeks, respectively, all P < 0.05, Mann–Whitney's U test, n = 9–10).
Figure 3. Expression levels of mRNAs for TNF-α, CRP, MCP-1, and macrophage MIF in the abdominal aorta in control and chronic variable stress groups measured by real-time PCR. Data are mean ± SEM. *P < 0.01 vs. control group, two-way ANOVA (n = 4–5 per group/time point).
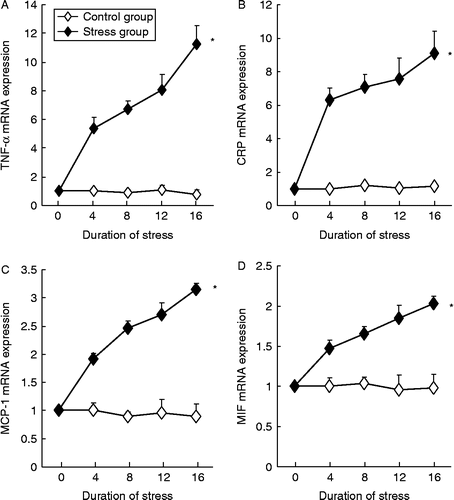
Figure 4. Chronic psychological stress increased the protein expression levels of TNF-α, CRP, ICAM-1, MCP-1, and macrophage MIF in the abdominal aorta. Antigens were detected with immunohistochemistry and visualized with DAB chromogen (brown color, arrows). Samples from the 4- and 12-week group showed a similar trend of upregulation of these inflammatory molecules (images not shown). Scale bar = 50 μm. The quantitative data for all treatment groups are shown as bar graphs on the right. Digital images were analyzed with Image-Pro Plus 6.0 software and results expressed as percentage of positively stained area. Data are mean ± SEM. *P < 0.05 vs. control (C, 16-week untreated animals), one-way ANOVA, n = 5 per group/time point.
Leukocyte infiltration and lipid accumulation in the vessel wall
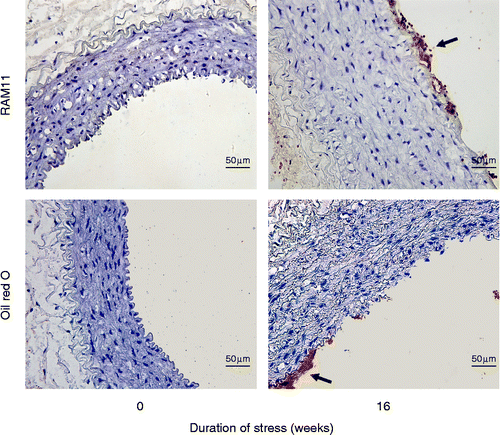
We observed that at 16 weeks, clusters of macrophages could be detected in the intima of aorta from stressed rabbits (), whereas no macrophage infiltration was detected in the media. No macrophage infiltration was observed in the aorta from control rabbits. Oil Red O staining in cryosections of the aorta, demonstrated signs of lipid accumulation in the subendothelial space only in stressed rabbits ().
Figure 5. Macrophage infiltration and lipid accumulation in the subendothelial space at 16-week poststress exposure. Macrophages were identified by the anti-rabbit macrophage (RAM1) antibody (arrow); lipids were detected with Oil Red O staining (arrow). The sections were counterstained with hematoxylin. Scale bar = 50 μm.
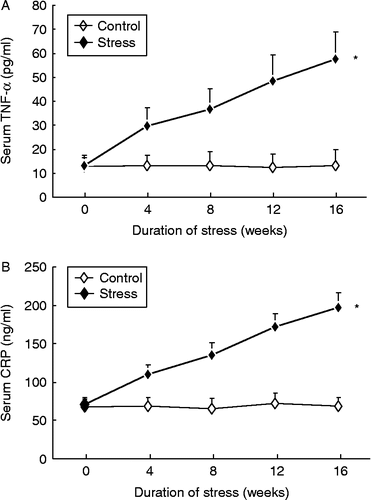
Effects of chronic psychological stress on lipid metabolism and systemic inflammatory markers
Stress had no statistically significant effects on serum concentrations of total cholesterol, triglyceride, high-density lipoprotein cholesterol, or low-density lipoprotein cholesterol (all P>0.05, two-way ANOVA; Supplementary Figure II). However, consistent with the increased inflammatory response in the aortic wall, we found that the serum concentration of TNF-α was significantly increased in the stress group in a time-dependent manner (; two-way ANOVA, stress: F 266.1, df 1, 90, P < 0.01; time: F 26.5, df 4, 90, P < 0.01; interaction P < 0.01). Moreover, the circulating concentration of CRP was also elevated by stress exposure (; two-way ANOVA, stress: F 611.7, df 1, 90, P < 0.01; time: F 68.1, df 4, 90, P < 0.01; interaction P < 0.01). Serum TNF-α or CRP concentrations did not change over time in control rabbits.
Figure 6. Effects of chronic psychological stress on serum TNF-α (A) and CRP (B) in control and stressed animals. Data are mean ± SEM. *P < 0.01 vs. control group, two-way ANOVA (n = 9–10 per group/time point).
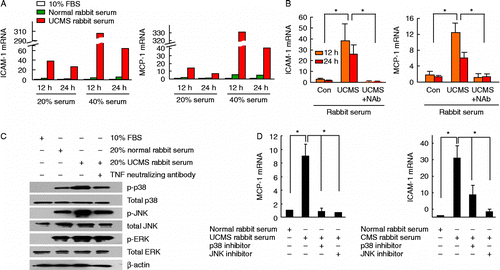
Roles of TNF-α and activation of p38 and JNK pathways in stress-induced vascular inflammation
Treatment of vascular smooth muscle cells in culture with 20% and 40% serum from stressed rabbits, as compared with those treated with serum from control rabbits, significantly increased the mRNA expression levels of ICAM-1 and MCP-1 in a dose-dependent manner (). Pretreatment with a neutralizing antibody against TNF-α blunted serum-induced upregulation of ICAM-1 and MCP-1 (; one-way ANOVA, ICAM-1 12 h: F 5.736, df 2, 6, P < 0.05; ICAM-1 24 h: F 9.563, df 2, 6, P < 0.05; MCP-1 12 h: F 15.51, df 2, 6, P < 0.01; MCP-1 24 h: F 7.456, df 2, 6, P < 0.05). Western blot analysis showed that stimulation with 20% rabbit serum from stressed rabbits increased phosphorylation of p38 and JNK, but not ERK, as compared with cells treated with serum from control rabbits ( and Supplementary Figure III). Phosphorylation of p38 and JNK was suppressed by the TNF-α neutralizing antibody (). Moreover, we demonstrated that treatment with specific p38 and JNK inhibitors SB202190 and SP600125 significantly inhibited MCP-1 and ICAM-1 expression stimulated by serum from stressed rabbits (; one-way ANOVA, MCP-1: F 21.50, df 3, 8, P < 0.01; ICAM-1: F 11.34, df 3, 8, P < 0.01).
Figure 7. Serum from rabbits exposed to UCMS-treated rabbits increased MCP-1 and ICAM-1 expression in cultured murine vascular smooth muscle cells via TNF-α and p38/JNK pathways. (A) Real-time PCR results showing that MCP-1 and ICAM-1 mRNAs were upregulated by incubation with serum from stressed rabbits in a dose-dependent manner, as compared with serum from untreated rabbits (averaged data from two experiments). FBS, fetal bovine serum. (B) Serum-induced upregulation of ICAM-1 and MCP-1 mRNAs was blunted by pretreatment with neutralizing antibody against TNF-α (NAb, 2 μg/ml; data expressed as fold of those from cells maintained in 10% FBS), n = 3. (C) Western blots showing that stimulation with 20% serum from stressed rabbits increased phosphorylation of p38 and JNK, but not ERK, as compared with cells treated with serum from control rabbits. Phosphorylation of p38 and JNK was suppressed by TNF-α neutralizing antibody (example from three independent experiments). (D) Treatment with specific p38 and JNK inhibitors SB202190 (1 μM) and SP600125 (1 μM) significantly inhibited MCP-1 and ICAM-1 mRNA expression stimulated by serum from stressed rabbits. Data are mean (A) or mean ± SEM (B, D). *P < 0.05, one-way ANOVA (n = 3).
Discussion
In this study, we tested whether chronic psychological stress exposure modulated vascular inflammation in the rabbit aorta. UCMS has been used to induce depression or depression-like symptoms in mice (Deussing Citation2006). To clarify whether this approach had similar effects in rabbits, we employed UCMS protocols as described previously in mice (Bernberg et al. Citation2009; d'Audiffret et al. Citation2010). There was a sustained increase in blood concentration of corticosterone during the stress treatment period, which validated our stress protocol. Then we demonstrated that UCMS also induced depression-like symptoms in rabbits, including reduced grooming activity during the splash test (psychomotor impairment), decreased food intake, and retardation of body weight gain, as reported in other animal models (Kumari et al. Citation2003; Herzog et al. Citation2009; d'Audiffret et al. Citation2010). These signs were consistently observed in all of the short-term (4-week) and long-term (16-week) treatment groups, indicating that the rabbits were not habituated to the stressors. Psychological stress significantly enhanced the levels of mRNAs for proinflammatory cytokines (MIF, TNF-α), chemokine (MCP-1), adhesion molecule (ICAM-1), and endothelium-derived vasoconstricting factor (endothelin-1) in the aortic wall. The increased protein expression of TNF-α, ICAM-1, and MCP-1 was not restricted to the intima and was also detected in the medial and adventitial layers (). These data are in line with those reported by Nation and colleagues (Citation2008) in Watanabe heritable hyperlipidemic rabbits, showing that unfavorable social environment-induced stress was associated with increased inflammation and vascular oxidative stress. Together, these observations indicate that chronic psychological stress may contribute to the development of a proinflammatory status in large arteries. However, our data are in contrast to those from a recent study in mice, which showed that UCMS had minor effects on the concentrations of circulating inflammatory markers (Isingrini et al. Citation2011). These authors, however, did not directly measure the expression levels of inflammatory molecules in the vascular tissue, leaving unclear the effects of psychological stress on localized vascular inflammation.
Blood vessels are in continuous contact with the blood. In this study, we found that the concentrations of TNF-α and CRP in blood were elevated by UCMS. This is in agreement with previous clinical studies showing that the presence of psychological stress is associated with higher concentrations of these inflammatory markers in the circulation (Miller et al. Citation2002; Empana et al. Citation2005; Janszky et al. Citation2005; Ladwig et al. Citation2005; Papageorgiou et al. Citation2006; Ranjit et al. Citation2007), which predict cardiovascular morbidity and mortality (Koenig et al. Citation2004). However, these factors are unlikely to be merely a systemic biomarker, but may be directly involved in modulating vascular inflammation. For example, recent studies have suggested that CRP may participate in the modulation of lipoprotein uptake, eNOS function, and nitric oxide (NO) generation, the activity of the proinflammatory transcription factor nuclear factor-κB, and the expression of adhesion molecules and angiotensin II receptors in vascular cells (Calabro et al. Citation2003; Szmitko et al. Citation2003; Inoue et al. Citation2005; Zwaka et al. Citation2001). Similarly, TNF-α is one of the most important proinflammatory mediators implicated in cardiovascular disease. In this study, we found that serum from stressed rabbits increased phosphorylation of p38 and JNK, and upregulated MCP-1 and ICAM-1 expression in cultured vascular smooth muscle cells. Moreover, these effects of serum were partially blocked by TNF-α neutralizing antibody and inhibitors of p38 and JNK. These results indicate that TNF-α and the activation of p38 and JNK pathways may have a pivotal role in UCMS-induced vascular inflammation.
Nonetheless, it is also noted that our study cannot rule out the involvement of other pathways in stress-induced vascular inflammation (Hansel et al. Citation2010). For example, chronic psychological stress induces hyperactivation of the sympathetic nervous system, leading to increased catecholamine release and stimulation of the renin-angiotensin system (Hansel et al. Citation2010; Groeschel and Braam Citation2011), which may stimulate vascular inflammation by modulating gene expression and cellular functions in both vascular and immune cells (Hansel et al. Citation2010; Groeschel and Braam Citation2011). In addition, psychological stress may blunt the function of the parasympathetic system, which has profound anti-inflammatory actions (Hansel et al. Citation2010). Moreover, chronic psychological stress may cause endothelial dysfunction (Ghiadoni et al. Citation2000), and this is consistent with our observation that expression of eNOS in the aorta was downregulated by stress. Therefore, more in vivo studies with different genetic or pharmacological interventions are warranted to further delineate the mechanisms of stress-induced vascular inflammation. Interestingly, we found that UCMS caused hypertension in rabbits. It is speculated that all of these stress-associated pathophysiological processes as mentioned above may contribute to the development of hypertension.
The hypothalamic–pituitary–adrenal axis responds to stress conditions by increasing the release of corticosteroids, which have general anti-inflammatory actions. We showed that UCMS treatment in rabbits resulted in sustained increase in the serum concentration of corticosterone. The elevation of corticosterone during stressor presentation evidences the induction of a systemic stress reaction, while the persistence of this increase indicates the absence of adaptation to the stressor. However, it is argued that chronic hyperactivity of the hypothalamic–pituitary–adrenal axis and prolonged exposure to high levels of glucocorticoids may have opposite effects on inflammation. For example, this may result in downregulation of the glucocorticoid receptors on leukocytes, thereby abolish the primary immunosuppressant effects of glucocorticoids, producing a proinflammatory condition (de Jonge et al. Citation2010).
To clarify whether the increased expression of inflammatory factors in the vessel wall was of functional importance, we performed an immunohistochemistry test using an anti-macrophage (RAM11) antibody in aortic sections. These experiments revealed that after 16 weeks of UCMS, there were infiltrating macrophages in the subendothelial space in the abdominal aorta, a pathological feature that is similar to early atherogenesis. Moreover, we performed Oil Red O staining in cryosections of the aorta, and demonstrated signs of lipid accumulation in the subendothelial space, presumably in infiltrating macrophages. However, gross examination of the aorta did not identify any typical atherosclerotic lesions. This may be because the rabbits were maintained on a standard chow without high-fat supplementation.
There is abundant experimental evidence showing that all of the proinflammatory factors mentioned above (i.e. MIF, TNF-α, MCP-1, and ICAM-1) may have important roles in the formation of atherosclerotic plaques. In the arterial wall, adhesion molecules and chemokines mediate subendothelial infiltration of leukocytes, the early pathological feature of atherosclerosis (Sheikine and Hansson Citation2004). Proinflammatory cytokines such as TNF-α and interferon released by activated vascular cells and infiltrating leukocytes may contribute to the perpetuation and intensification of local vascular inflammation, leading to plaque growth and destabilization (Hansson et al. Citation2006). Moreover, enhanced production of vasoconstrictive factors such as endothelin-1 may also promote the formation of atherosclerotic plaques (Little et al. Citation2008). Although we did not directly test the effects of psychological stress on plaque formation, in light of these lines of evidence, our results strongly support the notion that psychological stress may promote the development of atherosclerosis by inducing vascular inflammation.
In summary, this study demonstrated that rabbits exposed to UCMS showed a sustained inflammatory response in the aorta. This effect was associated with macrophage infiltration and lipid accumulation in the subendothelial space, indicating that psychological stress may contribute to atherogenesis by modulating vascular inflammation.
Supplementary Material
Download MS Word (72 KB)Acknowledgments
This study was partially supported by grants from National 973 Basic Research Program of China (No. 2010CB732605 and 2012CB518603), the National Natural Science Foundation of China (Nos 30801503, 30772810, 30873325, and 60971023), grants from Natural Science Foundation of Shandong Province (Nos Y2007C064 and 2009ZRB02129), and a grant from Independent Innovation Foundation of Shandong University (No. 2009TS128).
Declaration of interest : The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Abela GS, Picon PD, Friedl SE, Gebara OC, Miyamoto A, Federman M, Tofler GH, Muller JE. 1995. Triggering of plaque disruption and arterial thrombosis in an atherosclerotic rabbit model. Circulation. 91:776–784.
- Bernberg E, Andersson IJ, Tidstrand S, Johansson ME, Bergstrom G. 2009. Repeated exposure to stressors do not accelerate atherosclerosis in ApoE-/- mice. Atherosclerosis. 204:90–95.
- Black PH, Garbutt LD. 2002. Stress, inflammation and cardiovascular disease. J Psychosom Res. 52:1–23.
- Calabro P, Willerson JT, Yeh ET. 2003. Inflammatory cytokines stimulated C-reactive protein production by human coronary artery smooth muscle cells. Circulation. 108:1930–1932.
- Constantinides P, Booth J, Carlson G. 1960. Production of advanced cholesterol atherosclerosis in the rabbit. Arch Pathol. 70:712–724.
- Cullen P, Baetta R, Bellosta S, Bernini F, Chinetti G, Cignarella A, von Eckardstein A, Exley A, Goddard M, Hofker M, Hurt-Camejo E, Kanters E, Kovanen P, Lorkowski S, McPheat W, Pentikainen M, Rauterberg J, Ritchie A, Staels B, Weitkamp B, de Winther M. 2003. Rupture of the atherosclerotic plaque: Does a good animal model exist?. Arterioscler Thromb Vasc Biol. 23:535–542.
- Das S, O'Keefe JH. 2006. Behavioral cardiology: Recognizing and addressing the profound impact of psychosocial stress on cardiovascular health. Curr Atheroscler Rep. 8:111–118.
- d'Audiffret AC, Frisbee SJ, Stapleton PA, Goodwill AG, Isingrini E, Frisbee JC. 2010. Depressive behavior and vascular dysfunction: A link between clinical depression and vascular disease?. J Appl Physiol. 108:1041–1051.
- de Jonge P, Rosmalen JG, Kema IP, Doornbos B, van Melle JP, Pouwer F, Kupper N. 2010. Psychophysiological biomarkers explaining the association between depression and prognosis in coronary artery patients: A critical review of the literature. Neurosci Biobehav Rev. 35:84–90.
- Deussing JM. 2006. Animal models of depression. Drug Discov Today Dis Model. 3:375–383.
- Empana JP, Sykes DH, Luc G, Juhan-Vague I, Arveiler D, Ferrieres J, Amouyel P, Bingham A, Montaye M, Ruidavets JB, Haas B, Evans A, Jouven X, Ducimetiere P. 2005. Contributions of depressive mood and circulating inflammatory markers to coronary heart disease in healthy European men: The Prospective Epidemiological Study of Myocardial Infarction (PRIME). Circulation. 111:2299–2305.
- Ghiadoni L, Donald AE, Cropley M, Mullen MJ, Oakley G, Taylor M, O'Connor G, Betteridge J, Klein N, Steptoe A, Deanfield JE. 2000. Mental stress induces transient endothelial dysfunction in humans. Circulation. 102:2473–2478.
- Groeschel M, Braam B. 2011. Connecting chronic and recurrent stress to vascular dysfunction: No relaxed role for the renin-angiotensin system. Am J Physiol Renal Physiol. 300:F1–F10.
- Hansel A, Hong S, Camara RJ, von Kanel R. 2010. Inflammation as a psychophysiological biomarker in chronic psychosocial stress. Neurosci Biobehav Rev. 35:115–121.
- Hansson GK, Robertson AK, Soderberg-Naucler C. 2006. Inflammation and atherosclerosis. Annu Rev Pathol. 1:297–329.
- Herzog CJ, Czeh B, Corbach S, Wuttke W, Schulte-Herbruggen O, Hellweg R, Flugge G, Fuchs E. 2009. Chronic social instability stress in female rats: A potential animal model for female depression. Neuroscience. 159:982–992.
- Inoue T, Kato T, Uchida T, Sakuma M, Nakajima A, Shibazaki M, Imoto Y, Saito M, Hashimoto S, Hikichi Y, Node K. 2005. Local release of C-reactive protein from vulnerable plaque or coronary arterial wall injured by stenting. J Am Coll Cardiol. 46:239–245.
- Isingrini E, Belzung C, d'Audiffret A, Camus V. 2011. Early and late-onset effect of chronic stress on vascular function in mice: A possible model of the impact of depression on vascular disease in aging. Am J Geriatr Psychiatry. 19:335–346.
- Janszky I, Lekander M, Blom M, Georgiades A, Ahnve S. 2005. Self-rated health and vital exhaustion, but not depression, is related to inflammation in women with coronary heart disease. Brain Behav Immun. 19:555–563.
- Jiang F, Guo N, Dusting GJ. 2008. Modulation of nicotinamide adenine dinucleotide phosphate oxidase expression and function by 3′,4′-dihydroxyflavonol in phagocytic and vascular cells. J Pharmacol Exp Ther. 324:261–269.
- Kiank C, Holtfreter B, Starke A, Mundt A, Wilke C, Schutt C. 2006. Stress susceptibility predicts the severity of immune depression and the failure to combat bacterial infections in chronically stressed mice. Brain Behav Immun. 20:359–368.
- Koenig W, Lowel H, Baumert J, Meisinger C. 2004. C-reactive protein modulates risk prediction based on the Framingham score: Implications for future risk assessment: Results from a large cohort study in southern Germany. Circulation. 109:1349–1353.
- Kumari M, Grahame-Clarke C, Shanks N, Marmot M, Lightman S, Vallance P. 2003. Chronic stress accelerates atherosclerosis in the apolipoprotein E deficient mouse. Stress. 6:297–299.
- Ladwig KH, Marten-Mittag B, Lowel H, Doring A, Koenig W. 2005. C-reactive protein, depressed mood, and the prediction of coronary heart disease in initially healthy men: Results from the MONICA-KORA Augsburg Cohort Study 1984–1998. Eur Heart J. 26:2537–2542.
- Lichtman JH, Bigger JTJr, Blumenthal JA, Frasure-Smith N, Kaufmann PG, Lesperance F, Mark DB, Sheps DS, Taylor CB, Froelicher ES. 2008. Depression and coronary heart disease: Recommendations for screening, referral, and treatment: A science advisory from the American Heart Association Prevention Committee of the Council on Cardiovascular Nursing, Council on Clinical Cardiology, Council on Epidemiology and Prevention, and Interdisciplinary Council on Quality of Care and Outcomes Research: Endorsed by the American Psychiatric Association. Circulation. 118:1768–1775.
- Little PJ, Ivey ME, Osman N. 2008. Endothelin-1 actions on vascular smooth muscle cell functions as a target for the prevention of atherosclerosis. Curr Vasc Pharmacol. 6:195–203.
- Lombard JH. 2010. Depression, psychological stress, vascular dysfunction, and cardiovascular disease: Thinking outside the barrel. J Appl Physiol. 108:1025–1026.
- McCabe PM, Gonzales JA, Zaias J, Szeto A, Kumar M, Herron AJ, Schneiderman N. 2002. Social environment influences the progression of atherosclerosis in the watanabe heritable hyperlipidemic rabbit. Circulation. 105:354–359.
- Miller GE, Stetler CA, Carney RM, Freedland KE, Banks WA. 2002. Clinical depression and inflammatory risk markers for coronary heart disease. Am J Cardiol. 90:1279–1283.
- Nakamura M, Abe S, Tanaka M. 1998. Oral administration of NO synthase inhibitor failed to promote arteriosclerotic lesions in the aorta and the coronary arteries of rabbits fed cholesterol. Atherosclerosis. 141:53–60.
- Nation DA, Gonzales JA, Mendez AJ, Zaias J, Szeto A, Brooks LG, Paredes J, D'Angola A, Schneiderman N, McCabe PM. 2008. The effect of social environment on markers of vascular oxidative stress and inflammation in the Watanabe heritable hyperlipidemic rabbit. Psychosom Med. 70:269–275.
- Nerem RM, Levesque MJ, Cornhill JF. 1980. Social environment as a factor in diet-induced atherosclerosis. Science. 208:1475–1476.
- Neves VJ, Moura MJ, Tamascia ML, Ferreira R, Silva NS, Costa R, Montemor PL, Narvaes EA, Bernardes CF, Novaes PD, Marcondes FK. 2009. Proatherosclerotic effects of chronic stress in male rats: Altered phenylephrine sensitivity and nitric oxide synthase activity of aorta and circulating lipids. Stress. 12:320–327.
- Papageorgiou C, Panagiotakos DB, Pitsavos C, Tsetsekou E, Kontoangelos K, Stefanadis C, Soldatos C. 2006. Association between plasma inflammatory markers and irrational beliefs; the ATTICA epidemiological study. Prog Neuropsychopharmacol Biol Psychiatry. 30:1496–1503.
- Phinikaridou A, Hallock KJ, Qiao Y, Hamilton JA. 2009. A robust rabbit model of human atherosclerosis and atherothrombosis. J Lipid Res. 50:787–797.
- Ranjit N, Diez-Roux AV, Shea S, Cushman M, Seeman T, Jackson SA, Ni H. 2007. Psychosocial factors and inflammation in the multi-ethnic study of atherosclerosis. Arch Intern Med. 167:174–181.
- Sheikine Y, Hansson GK. 2004. Chemokines and atherosclerosis. Ann Med. 36:98–118.
- Shen BJ, Avivi YE, Todaro JF, Spiro A3rd, Laurenceau JP, Ward KD, Niaura R. 2008. Anxiety characteristics independently and prospectively predict myocardial infarction in men the unique contribution of anxiety among psychologic factors. J Am Coll Cardiol. 51:113–119.
- Sherwood A, Johnson K, Blumenthal JA, Hinderliter AL. 1999. Endothelial function and hemodynamic responses during mental stress. Psychosom Med. 61:365–370.
- Spieker LE, Hurlimann D, Ruschitzka F, Corti R, Enseleit F, Shaw S, Hayoz D, Deanfield JE, Luscher TF, Noll G. 2002. Mental stress induces prolonged endothelial dysfunction via endothelin-A receptors. Circulation. 105:2817–2820.
- Strawn WB, Bondjers G, Kaplan JR, Manuck SB, Schwenke DC, Hansson GK, Shively CA, Clarkson TB. 1991. Endothelial dysfunction in response to psychosocial stress in monkeys. Circ Res. 68:1270–1279.
- Strike PC, Steptoe A. 2004. Psychosocial factors in the development of coronary artery disease. Prog Cardiovasc Dis. 46:337–347.
- Szmitko PE, Wang CH, Weisel RD, de Almeida JR, Anderson TJ, Verma S. 2003. New markers of inflammation and endothelial cell activation: Part I. Circulation. 108:1917–1923.
- Williams JK, Kaplan JR, Manuck SB. 1993. Effects of psychosocial stress on endothelium-mediated dilation of atherosclerotic arteries in cynomolgus monkeys. J Clin Invest. 92:1819–1823.
- Willner P. 1997. Validity, reliability and utility of the chronic mild stress model of depression: A 10-year review and evaluation. Psychopharmacology (Berl). 134:319–329.
- Yalcin I, Aksu F, Belzung C. 2005. Effects of desipramine and tramadol in a chronic mild stress model in mice are altered by yohimbine but not by pindolol. Eur J Pharmacol. 514:165–174.
- Zwaka TP, Hombach V, Torzewski J. 2001. C-reactive protein-mediated low density lipoprotein uptake by macrophages: Implications for atherosclerosis. Circulation. 103:1194–1197.