Abstract
Extreme heat stress-induced gastrointestinal injury and dysfunction may occur during summer. We investigated possible mechanisms of heat stress-induced damage in the small intestine using male Sprague-Dawley rats subjected to 2 h of heat stress (40°C, 60% relative humidity) daily for 10 consecutive days. Rats were killed at specific times immediately following heat treatment to determine: morphological changes by optical and electron microscopy; intestinal permeability using fluorescein isothiocyanate–dextran; production of reactive oxygen species (ROS), malondialdehyde (MDA), and activities of superoxide-dismutase and glutathione-peroxidase by specific assays; phosphorylation of extracellular signal-regulated kinase 1/2 (ERK1/2), c-Jun N-terminal kinase (JNK), and p38 mitogen-activated protein kinase (MAPK) by immunocytochemistry and western-blot analysis. The rat intestinal epithelial cell line (IEC-6) and specific MAPK inhibitors were used for in vitro investigation of effects of activation of MAPKs by heat stress. Heat stress caused marked morphological damage to the small intestine and significantly increased intestinal permeability. Heat stress increased ROS and MDA production, and significantly reduced anti-oxidase activity. MAPK activity in small intestine was increased by heat stress. In vitro, heat stress caused damage and apoptosis in IEC-6 cells; inhibition of ERK1/2 activation (by U0126) exacerbated these effects, which were attenuated by inhibition of JNK (by SP600125) and p38 (by SB203580) activation. Hence, heat stress caused severe small intestine injury, increased oxidative stress, and activated MAPK signaling pathways. The in vitro studies indicated that ERK1/2 activation is anti-apoptotic, and JNK and p38 activation are pro-apoptotic in heat stressed intestinal epithelial cells.
Introduction
The intestinal epithelium is directly exposed to a wide variety of nutrients, as well as ingested microbes and exogenous pathogens. The critical role of the intestinal epithelium is to absorb water and nutrients, while simultaneously providing a protective barrier for the gastrointestinal (GI) tract, and acting as a sentinel for the mucosal immune system. External sources of stress, including radiation, lipopolysaccharides (LPS), various drugs, endotoxins, and heat stress are all reported to cause significant damage to the intestinal epithelium (Shen et al. Citation2009). Concerning heat stress, the injury sustained by the intestinal epithelium is the indirect result of elevated peripheral blood flow in order to dissipate internal body heat (Lambert et al. Citation2002). As a result, the GI tract experiences a degree of ischemia and subsequent injury, including increased permeability associated with unregulated diffusion of molecules into the blood circulation (Kregel et al. Citation1988; Wingo et al. Citation2010). Aside from heat stress, increased intestinal permeability can result from numerous pathophysiological conditions, including hemorrhage, diarrhea, and endotoxemia (Honzawa et al. Citation2011; Kim et al. Citation2011). As a protective adaptation, the intestinal epithelium has a rapid rate of cellular replacement, with damaged and differentiated epithelial cells shed into the lumen and continuously replaced by stem cells, maintaining physiological intestinal function (Booth and Potten Citation2000). Our previous investigations indicated that changes in extracellular signal-regulated kinase 1/2 (ERK1/2) activity, growth factor expression, epidermal growth factor receptor signaling, and miRNA profiles are responsible for regulating the changes in cellular proliferation, growth, differentiation, and migration of crypt stem cells in response to heat stress in small intestinal epithelial cells (Yu et al. Citation2010a, Citation2010b, Citation2011a).
Ischemia of the small intestine following hyperthermia has been found to promote formation of reactive oxygen species (ROS; Moriwaki et al. Citation1993). Elevated ROS levels negatively disrupt the balance between oxidation and antioxidant defense systems, increasing oxidative damage to intracellular proteins and DNA, and lipid peroxidation (LPO; Belozerskaia and Gessler Citation2007; Ahmad et al. Citation2010; Naziroglu et al. Citation2010), and inducing apoptosis of intestinal epithelial cells.
The mitogen-activated protein kinase (MAPK) family is a group of highly conserved proline-directed, serine/threonine kinases, involved in intracellular signaling in response to stressors including cytokines, irradiation, heat stress, ROS, ischemia, and osmotic stress (Porta et al. Citation2011). The family of MAPKs are considered to be stress-activated protein kinases, capable of regulating a large number of cellular processes, including cell survival, apoptosis, adaptation, differentiation, and proliferation in response to stress (Peter and Dhanasekaran Citation2003; Muthusamy and Piva Citation2010). The three most predominant members of the MAPK family include ERK, c-Jun N-terminal kinase (JNK), and p38 MAPK (Jorge et al. Citation2009).
Owing to the phenomenon of global warming, ambient temperatures are predicted to increase over the next decades, likely contributing to an increase in heat stress-induced GI injury and dysfunction during summer; however, the molecular mechanisms underlying the cellular changes remain undefined. This study sought to identify factors involved in the pathophysiology of heat stress-induced injury in the small intestine in vivo and in vitro. Previous studies have demonstrated oxidative stress and MAPK signaling pathways to be involved in stress-induced cellular and organ injury. It was, therefore, hypothesized that oxidative stress and MAPK signaling pathways are likely implicated in a model of heat stress-induced injury in the small intestine of rats.
Materials and methods
Animals
All protocols and procedures involving animals were approved by and conducted in accordance with the China Agricultural University Institutional Animal Care and Use Committee. Male Sprague-Dawley rats weighing 200 ± 20 g (mean ± SEM; obtained from Beijing Vital River Laboratory, Animal Technology Co., Beijing, China) were acclimatized to 25°C, 60% relative humidity (RH) and maintained under a 12-h light/dark (19:00–07:00 h) cycle. Food and water were provided ad libitum for 7 days. On the 8th day, rats were randomly assorted into control or heat-treatment groups, and housed in plastic cages (40 × 30 × 18 cm, six rats per cage) with a layer of soft woodchips. Feed (200 g) and water (400 mL) were provided daily. Other data from these rats subjected to the same treatment protocol described below have been published (Yu et al. Citation2011a).
Treatment protocol
Rats in the control group were housed under normal conditions (25°C, 60% RH). Rats in the heat treatment group were housed under control group conditions, with the addition of exposure to 40°C and 60% RH between 11:00 and 13:00 h daily for 10 consecutive days. On the 1st, 3rd, 6th, and 10th day, six rats from each group were killed by decapitation without anesthesia immediately following the 2 h heat exposure (Yu et al. Citation2011a).
Sampling
Rat rectal and body surface temperatures were recorded daily before and after heat exposure using a digital and infrared thermometer (Fluke 561, Evered, Washington, DC, USA; data in Yu et al. Citation2011a). On the 1st, 3rd, 6th, and 10th day, six rats from each group were killed by decapitation without anesthesia immediately after treatment. Following killing, trunk blood was collected and allowed to clot at room temperature; it was then centrifuged (3000g at 4°C) and the serum extracted and stored at − 80°C until required. The intestine was removed and immediately irrigated with physiological saline (0.9%) to remove all intestinal contents, before being sectioned into the duodenum (15 mm from the pylorus), the distal jejunum–ileum (half of the remaining small intestine up to the cecum), and the ileum (20 mm proximal to the cecum). Each intestinal section was divided into three pieces: one fixed for histological analysis; one for molecular analysis; and one stored at − 80°C. Total serum cortisol concentration was determined (data in Yu et al. Citation2011a).
Analysis of intestinal permeability
Rats were anesthetized using 2% isoflurane in oxygen delivered at 1000 mL/min to enable measurement of intestinal permeability on the 1st, 3rd, 6th, or 10th day. A midline laparotomy was performed in order to ligate a segment of distal small intestine using silk ties. Fluorescein isothiocyanate (FITC)-dextran (3.5 ml of 25 mg/ml; Sigma, St Louis, MO, USA) diluted in phosphate-buffered saline (PBS) was injected into the lumen of the isolated segment of intestine to determine permeability. The small bowel was then returned to the abdominal cavity and the abdominal wall closed. Rats were then returned to their specific treatment environment. On the 1st, 3rd, 6th, or 10th day, three rats from each group were killed immediately after treatment, as above, and a sample of systemic blood collected, centrifuged at 10,000g for 10 min and the serum obtained. The serum was analyzed for FITC-dextran using a fluorescence spectrophotometer (Synergy HT, Biotek, Winooski, Vermont, USA).
Histopathological examination
Samples of small intestine were excised, promptly rinsed with physiological saline and immediately fixed in 10% phosphate-buffered formalin. The formalin-fixed samples were embedded in paraffin and transversely sectioned (5 μm thick). After deparaffinization and dehydration, the sections of duodenum, jejunum, and ileum were stained with hematoxylin and eosin (Sigma). Microstructures of the small intestine were observed using a BH2 Olympus microscope (DP71, Olympus, Tokyo, Japan). Villus height and crypt depth of six well-oriented villi per rat were measured and recorded using an Olympus Image Analysis System (Olympus 6.0, Tokyo, Japan).
Scanning electron microscopy (SEM)
Rectangular sections of small intestine 1 mm thick and 3–8 mm2 were fixed overnight at 4°C with 2.5% glutaraldehyde in PBS and post-fixed with 1% osmium tetroxide. After dehydration in a graded series of ethanol solutions, samples were treated with isoamyl acetate overnight. Specimens were then dried in hexamethyl disilizane, sputter-coated with gold, and examined using a Hitachi S-3400N scanning electron microscope operated at 15 kV (S-3400N, Hitachi, Tokyo, Japan).
Transmission electron microscopy (TEM)
Sections (1 mm thick) of small intestine were fixed overnight at 4°C with 2.5% glutaraldehyde in PBS, gently rinsed in 0.1 M phosphate buffer (pH 7.2), post-fixed with 1% osmium tetroxide, and rinsed thoroughly in the same buffer. Subsequently, the tissue was dehydrated in a graded series of acetone, embedded in resin after tissue processing and polymerized at 60°C for 2 days. Ultra-thin sections (70 nm) were then made, stained with uranyl acetate and counterstained with lead citrate before examination under TEM (JEM-1230, JEOL, Tokyo, Japan) operating at 80 kV.
Immunocytochemistry
Intestinal sections to be subjected to immunohistochemical analysis were incubated for 15 min in 3% H2O2/methanol to quench endogenous peroxidase activity, and then rinsed for 20 min with PBS. Non-specific protein binding was attenuated by incubation of the tissue with 5% goat serum in PBS for 30 min (#KGSP04 histostain-plus kit, KeyGen Biotech, Nanjing, China). To localize phospho-ERK1/2, phospho-c-Jun, and phospho-p38 expression, antibodies (#4376, #9164 and #9215, CST, MA, USA) specific for each antigen were used at a dilution of 1:100. Antibodies were pre-diluted, stored in PBS containing bovine serum albumin and 0.05% sodium azide. Antibodies were applied directly to the sections and the slides incubated for 2 h at 37°C in a moist chamber. Sections were subsequently treated with a secondary antibody and immune complexes detected using Streptavidin peroxidase (#KGSP04 histostain-plus kit, KeyGen Biotech), each incubated for 30 min at room temperature. After washing three times in PBS, immunoreactivity was visualized using 0.1% 3,3-diaminobenzidine and 0.02% hydrogen peroxide for 5 min (DAB substrate kit, Vector Laboratories, Burlingame, CA, USA). After rinsing in distilled water, sections were counterstained with hematoxylin.
Quantification of ROS, LPO and anti-oxidative enzyme activity
The production of ROS in rat small intestine was determined by an OxiSelect™ in vitro ROS/reactive nitrogen species (RNS) assay kit (Green Fluorescence; Catalog #STA-347, Cell Biolabs, Inc., San Diego, CA, USA). Briefly, 50 mg of small intestinal tissue was homogenized in 1 ml of PBS on ice, centrifuged at 10,000g for 5 min, and the supernatant collected and stored at − 80°C until required. Fifty microliters of supernatant were added to each well of a 96-well plate suitable for fluorescence measurement, and combined with 50 μL of catalyst. The plate was incubated for 5 min at room temperature before 100 μL of dichlorofluorescein (DCFH) solution was added to each well, and incubated at room temperature for a further 30 min protected from light. Fluorescence was determined at 480 nm excitation and emission at 530 nm using a fluorescence spectrophotometer (Synergy HT, Biotek); the detection sensitivity limit of the kit was 10 pM for DCF and 40 nM for H2O2, respectively. Intracellular ROS in IEC-6 cells were determined with an OxiSelect™ ROS assay kit (Catalog # STA-342, Cell Biolabs, Inc.) according to the manufacturer's instructions. Briefly, IEC-6 cells were seeded in a black 96-well cell plate (Product #3603, Corning Inc., New York, USA), 100 μL of DCFH-DA/media solution was added to each well, and then incubated at 37°C for 30 min. The DCFH-DA loaded control cells were kept strictly at 37°C, while cells of the heat treatment group were exposed to 42°C for 3 h. After treatment, fluorescence of DCF-DA loaded control and heat treated IEC-6 cells was measured with an inverted fluorescence microscope (IX71/IX2, Olympus); the DCF detection sensitivity limit of the kit was 10 pM.
The concentration of malondialdehyde (MDA) and the activities of superoxide dismutase (SOD) and glutathione peroxidase (GSH-px) in tissue from the small intestine were determined using detection kits specific for each (#A003, #A001, and #A005-1 purchased from Nanjing Jiancheng Bioengineering Institute, Nanjing, China).
Western-blot analysis
Protein was extracted from rat small intestine and IEC-6 cells using a total protein extraction kit (#K3011010, Biochain, Hayward, CA, USA), and the concentration determined using a bicinchoninic acid protein assay (#23225, Pierce, Rockford, IL, USA). Thirty micrograms of total protein were loaded and run by electrophoresis for 120 min at 100 V, before being transferred onto nitrocellulose membranes (#88585, Pierce). Membranes were blocked overnight at 4°C in SuperBlock T20 (TBS) blocking buffer (#37536, Pierce). Phospho-ERK1/2, ERK1/2 (#4376, #4695, CST), phospho-c-Jun, c-Jun (#9164, #9165, CST), phospho-p38 MAPK, p38 MAPK (#9215, #9212, CST), and actin (#SC-1615, Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA) antibodies were added to the blocking buffer (diluted 1:1000) and incubated for 2 h under agitation. Blots were washed in PBS-T20 for 5 min with shaking. Goat anti-rabbit IgG-horseradish peroxidase (HRP; #SC-2004, Santa Cruz Biotechnology) and donkey anti-goat IgG-HRP (#SC-2033, Santa Cruz Biotechnology) secondary antibodies were applied to the membranes (diluted 1:5000) and incubated for 1 h under agitation. Blots were then washed in PBS-T20 for 5 min with shaking and visualized using a SuperSignal® West Pico Chemiluminescent substrate (#34080, Pierce) before exposure to X-ray film (Kodak, New York, USA).
In vitro studies
Cell culture
Rat IEC-6 cells (#CRL21592, purchased from Peking Union Medical College, Beijing, China) were cultured in Dulbecco's modified Eagle medium containing 5% (v/v) fetal bovine serum (HyClone, Logan, UT, USA), 2 mg/l insulin, 50 IU/ml penicillin and 50 μg/ml streptomycin and incubated at 37°C under 5% (v/v) CO2. The medium was replaced 24 h following initial cell plating.
Treatment, morphological examination, and apoptosis analysis
Control cells were kept strictly at 37°C under 5% CO2, while cells of the heat treatment group were exposed to 42°C for 3 h and 5% CO2. To inhibit specific intracellular agents, cells were pretreated with 20 μmol/ml U0126 (ERK1/2 inhibitor, #1144, Tocris Bioscience, Louis, MO, USA), 10 μmol/ml SP600125 (JNK inhibitor, #1496, Tocris Bioscience), and 20 μmol/ml SB203580 (p38 inhibitor, #1202, Tocris Bioscience) for 1 h before heat treatment, respectively. Changes in cell morphology following all treatments were observed using a phase-contrast inverted biological microscope (IX71/IX2, Olympus).
Cells (1.0 × 106) were collected from each treatment, using trypsin (without EDTA), centrifuged (1000g for 5 min), and washed with ice-cold PBS twice. The cells were then stained with fluorescein isothiocyanate (FITC)-labeled annexin V and propidium iodide (PI; Invitrogen, Carlsbad, CA, USA) and subjected to flow cytometry following the manufacturer's instructions (Beckman, Coulter, CA, USA).
Statistical analysis
Statistical comparisons of two groups were carried out by Student's t-test, or for more than two groups with analysis of variance (ANOVA) combined with the post-hoc Tukey test. Statistical analyses were carried out using the SPSS 12.0 software and graphs were created using Graphpad Prism version 5.0. All results are presented as the mean ± SEM, with a value of p < 0.05 considered statistically significant.
Results
Assessment of the rat heat stress model
The rat heat stress model was assessed in the same rats by increased serum corticosterone concentration and elevated body surface and rectal temperatures, as published (Yu et al. Citation2011a).
Histopathological examination and increased intestinal permeability following heat stress
Gross pathological changes in the small intestine were evident following heat treatment, including edema, hyperemia, and petechial hemorrhages in the mesenteries (). To further investigate the pathological changes within the small intestine caused by heat stress, sections of duodenum, jejunum, and ileum were stained with hematoxylin and eosin. Histopathological examination under light microscopy revealed significant structural alterations of the intestinal wall; specifically, bleeding in the intestinal villi, and desquamation at the tips of the intestinal villi and of the mucosal epithelial cells, exposing the lamina propria (). Villus height and crypt depth were significantly smaller in the heat stress group compared with control rats (p < 0.05, Student's t-test; n = 6 rats per treatment; six villi and crypts were measured per rat, and animal means calculated; Table S1). Heat-induced injury to the intestine was most severe on the 3rd day of treatment (). As a result of this finding, the small intestine tissue obtained from rats on the 3rd day of treatment was used for molecular analysis. Intestinal permeability was significantly increased after 2 h of heat treatment at all time points (1st, 3rd, 6th, and 10th day of treatment; F (7,16) = 17.9, p < 0.001; ).
Figure 1. Gross morphology of control and heat-treated rat small intestine on the 3rd day of heat treatment. (A) Edema, hyperemia, and petechial hemorrhages in the mesentery are clearly visible (arrow) in response to heat treatment (40°C for 2 h per day). (B) Photomicrographs of hematoxylin and eosin-stained sections of rat small intestine after 3 days of heat treatment. Upper panels are small intestine from control rats, lower panels are small intestine from heat-stressed rats. Panels (a) and (d) illustrate duodenum sections; (b) and (e) jejunum sections; (c) and (f) ileum sections. Abnormal microstructures are indicated by arrows with numbers. Severe damage to the intestinal villi by heat treatment is apparent, with hyperemia (indicated by 2, 3, 6, and 7) desquamation at the tips of the intestinal villi (1, 5, and 9), and exposure of the lamina propria (4 and 8). Scale bar represents 100 μm.
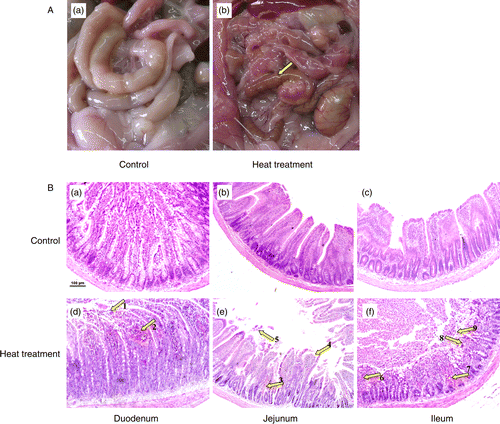
Figure 2. Intestinal permeability of control and heat-stressed rats was assessed using FITC-dextran in vivo on Days 1, 3, 6, and 10. Heat treatment comprised exposure to 40°C for 2 h, on 10 consecutive days. Data are mean ± SEM, n = 3 rats per group and day. *p < 0.05 compared to control, one-way ANOVA.
The effect of heat stress on epithelial morphology of the small intestine
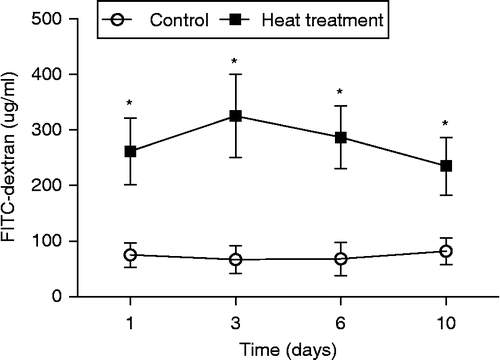
The mucosal surface of the small intestine (jejunum) after 3 days of heat treatment was visualized using a scanning electron microscope. As displayed in , the upper portions of the intestinal villi were lost following heat stress and, as a result, the residual epithelial cells ruptured and peeled off with the basement membrane. There was also evidence of epithelial cells becoming separated from the lamina propria, leaving a small gap. The reticular fibers were exposed after exuviation of intestinal epithelial cells, revealing the surface of the lamina propria in which erythrocytes flowed into the lumen.
Figure 3. SEM and TEM of the small intestine (jejunum) from rats subjected to control conditions or heat treatment (40°C for 2 h per day) for 3 days. Abnormal microstructures are indicated with arrows with numbers. (A) Mucosal surface of the small intestine was visualized using a scanning electron microscope (scale bar divisions represent 10 μm). Heat stress caused: loss of the upper portion of the intestinal villi (indicated by 2), some epithelial cells to separate from the lamina propria (1), the reticular fibers to become exposed after exuviation of intestinal epithelial cells (2), and erythrocytes were able to enter the lumen (3). (B) Ultrastructure of the jejunum examined using a transmission electron microscope. Changes in microvillus height (3, 7, and 8), mitochondria (6), endoplasmic reticulum and nuclei (1, 2, and 5) were apparent when compared to control sections. Vacuolization was also visible in the epithelium of heat-stressed rats (4), associated with a progressive loss of epithelial cells from the basement membrane. In (B), scale bars represent 20, 10, and 0.5 μm from left to right, respectively.
Morphological changes within the small intestine (jejunum) on the 3rd day of heat treatment were also examined by TEM. Examination of the ultrastructure revealed heat stress to have significantly reduced microvillus height of the intestinal epithelium when compared with control (1.5 ± 0.2 μm versus 3.1 ± 0.3 μm, p < 0.05, n = 6 villi in each rat, three different rats for each treatment, N = 18). Further examination revealed a greater number of swollen mitochondria with shortened internal cristae, as well as increased degranulation and vesicularization of the endoplasmic reticulum compared with non-stressed rats. Sections of intestine from heat-stressed rats at Day 3 also exhibited visible organelle debris within the lysosomes, and marked changes in the shape of the nucleus were also apparent. Furthermore, vacuolization was visible in the intestinal epithelium of heat-stressed rats, as well as a progressive loss of epithelial cells from the basement membrane ().
Ros/rns, Mda, And Antioxidant Activity
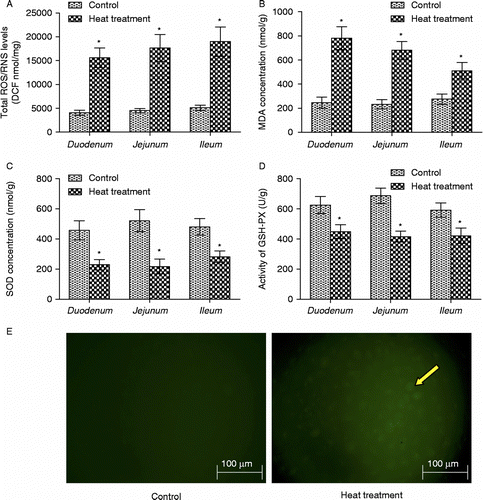
Heat stress is known to negatively alter the balance between the production of ROS, and the generation of antioxidants to counteract them. The production of ROS/RNS and concentration of MDA in the rat duodenum, jejunum, and ileum following 3 days of heat stress were significantly increased (p < 0.05, n = 6; ). Conversely, the activity of the antioxidant enzymes SOD and GSH-px were both significantly reduced in response to heat treatment (p < 0.05, n = 6; ). Intracellular ROS of intestinal epithelial cell (IEC-6) was also obviously increased after heat treatment at 42°C for 3 h ().
Figure 4. The oxidative state of rat small intestine following heat treatment was determined by measuring ROS and RNS, MDA, SOD, and GSH-px. The production of ROS/RNS (A) and the concentration of MDA (B) in rat small intestine (duodenum, jejunum, and ileum) were both significantly increased; activities of the antioxidant enzymes (C) SOD and (D) GSH-px were significantly reduced in response to heat treatment. Data are mean ± SEM, n = 6 per treatment. *p < 0.05 compared to control, Student's t-test. (E) Photomicrographs of intracellular ROS in rat IEC-6 cells. Increased green dichlorofluorescein (DCF, indicator of intracellular ROS production) fluorescence (right panel, arrow) indicates increased intracellular ROS production in IEC-6 cells after heat treatment at 42°C for 3 h. Scale bar represents 100 μm.
Heat stress stimulates ERK1/2, JNK, and p38 MAPK in the rat small intestine
The family of MAPKs is synonymous with regulation of cellular growth, survival, adaptation, apoptosis, and the response to stress in epithelial cells. Three days of heat stress was found to significantly increase the phosphorylation of ERK1/2, JNK, and p38 MAPK in the rat small intestine (p < 0.05, n = 6; ).
Figure 5. Immunohistochemical analysis of phospho-ERK1/2, phospho-JNK and phospho-p38 MAPK expression in the small intestine (jejunum) crypts of rats subjected to 3 days of heat treatment (40°C for 2 h per day) or control conditions. Phosphorylation of ERK1/2, JNK, and p38 MAPK were all increased (brown stain, arrows; sections counterstained with hematoxylin, light purple) in response to heat treatment above control levels. Scale bar represents 20 μm.
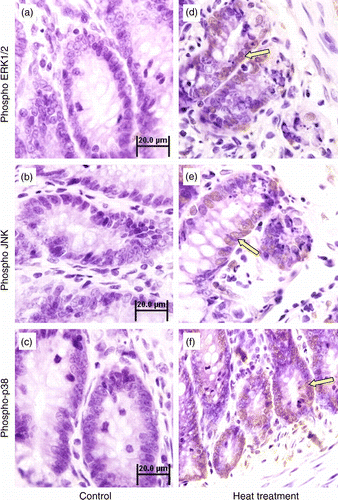
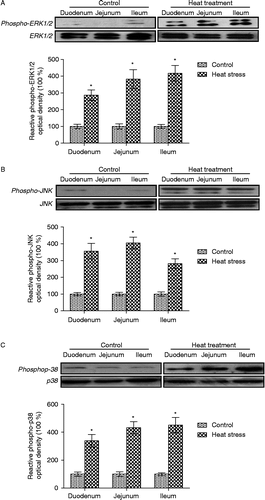
Figure 6. Quantification of western-blot determination of phospho-ERK1/2, phospho-JNK, and phospho-p38 MAPK in sections of small intestine collected from control and heat-treated rats on the 3rd day of treatment (40°C for 2 h per day). Phosphorylation of (A) ERK1/2, (B) JNK, and (C) p38 were all significantly elevated in heat-treated rats. Values are expressed as the percentage of control. Data are mean ± SEM, n = 6 per treatment. *p < 0.05 compared to control, Student's t-test.
The effects of ERK1/2, JNK, and p38 MAPK phosphorylation on heat stress-induced damage and apoptosis in intestinal epithelial cells (IEC-6)
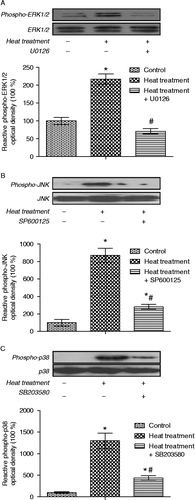
To investigate the effects of activation of MAPKs in intestinal epithelial cells in response to heat stress, specific inhibitors of MAPKs were employed in combination with heat treatment. Heat stress (42°C, 3 h) evidently increased damage and apoptosis, which were exacerbated when ERK1/2 was inhibited by pretreatment with U0126. In contrast, pretreating IEC-6 cells with the JNK or p38 MAPK inhibitors (SP600125 and SB203580, respectively) attenuated the heat stress-induced damage () and apoptosis, F (7,16) = 34.2, p < 0.001 (). Heat stress significantly increased the phosphorylation of ERK1/2, JNK, and p38 MAPK in IEC-6 cells, and this was blocked by specific inhibitors (p < 0.05, n = 6; ). These results indicate that ERK1/2 activation has an anti-apoptotic function, while both JNK and p38 MAPK are pro-apoptotic in the intestinal epithelium in response to heat stress.
Figure 7. Photomicrographs of rat IEC-6 cells demonstrating gross changes in cellular morphology in response to heat stress and in combination with specific MAPK inhibitors. Cellular morphology was markedly altered following heat treatment (42°C for 3 h), with changes in cell shape (arrows) and a greater number of dead cells in the supernatant. This effect was exacerbated by pretreating cells with U0126 (extracellular signal-regulated kinase, ERK1/2, inhibitor), but attenuated by pretreating cells with SP600125 (c-Jun N-terminal kinase, JNK, inhibitor) or SB203580 (p38 inhibitor). Scale bar represents 100 μm.

Figure 8. Apoptosis in rat IEC-6 cells. Apoptosis was determined by annexin V/PI staining and flow cytometry following heat stress and in combination with specific MAPK inhibitors. Cell apoptosis was strikingly increased after heat stress (42°C for 3 h). Heat stress induced-apoptosis was augmented by U0126 (extracellular signal-regulated kinase, ERK1/2, inhibitor) pretreatment, but attenuated by SP600125 (c-Jun N-terminal kinase, JNK, inhibitor) and SB203580 (p38 inhibitor) pretreatment. (A) Flow cytometry data per treatment. G1 (Annexin 744 V − /PI+) represents percentage of necrotic cells; G2 (Annexin V+/PI+) percentage of late apoptosis cells; G3 (Annexin V − /PI − ) percentage of live cells; G4 (Annexin V+/PI − ) percentage of early apoptotic cells. (B) Total apoptotic cells (percentage) = early apoptotic cells (G4) + late apoptosis cells (G2). Data are mean ± SEM, n = 3 per treatment. *p < 0.05 compared to control, # p < 0.05 compared to heat treatment, one-way ANOVA.
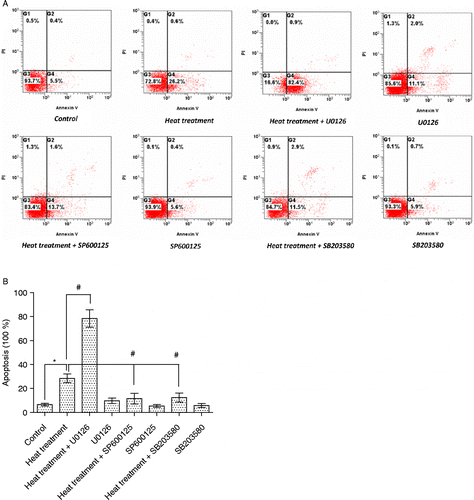
Figure 9. Quantification of immunoblot densities for phospho-ERK1/2, phospho-JNK and phospho-p38 MAPK in rat IEC-6 cells. Phosphorylation levels of ERK1/2, JNK, and p38 were significantly increased after heat treatment (42°C for 3 h); these increases were blocked by pretreatment with specific MAPK inhibitors (U0126, SP600125, and SB203580, respectively, inhibitors of ERK1/2, JNK, and p38). Values are percentage of control. Data are mean ± SEM, n = 6 per treatment. *p < 0.05 compared to control, # p < 0.05 compared to heat treatment, one-way ANOVA.
Discussion
This study investigated the effect of heat stress on the small intestine of rats in vivo and in vitro. Initial findings revealed heat stress to cause clear morphological damage associated with a significant increase in intestinal permeability (). Despite the treatment period extending for 10 days, heat stress-induced injury was found to be most severe on the 3rd day of treatment. Subsequently, this time point was used for all molecular and histological analyses. Microarray results in our recently published studies revealed that many genes associated with stress, hypoxia, oxidative stress, and MAPK signaling pathways were differentially expressed in rat small intestine in response to heat stress (Lu et al. Citation2011; Yu et al. Citation2011a). These findings are consistent with previous studies reporting oxidative stress and MAPK signaling to mediate the injury sustained after exposure to various environmental stressors (Kyriakis and Avruch Citation2001; Johnson and Lapadat Citation2002; Nabenishi et al. Citation2011). In this study, the potential regulatory actions of oxidative stress and activity of MAPKs in the pathophysiological response to heat stress were determined by measuring ROS production, LPO, and antioxidant enzyme activity; with MAPK expression determined by immunocytochemistry and western-blot analysis. Heat stress increased the production of ROS and LPO (MDA), and reduced antioxidase activity (SOD and GSH-px; ). Phosphorylation of ERK1/2, JNK, and p38 MAPK were all increased following heat stress (), supporting both our published microarray findings (Lu et al. Citation2011; Yu et al. Citation2011a), and previous reports indicating that heat stress-induced injury in the small intestine is associated with oxidative stress and activation of MAPK signaling pathways (Peter and Dhanasekaran Citation2003; Circu and Aw Citation2010; Muthusamy and Piva Citation2010). Furthermore, it is apparent that organelle damage in response to heat stress in this study is similar to that reported previously in response to oxidative stress (Kaneko et al. Citation2007; Koh et al. Citation2010). The rat intestinal epithelial cell line (IEC-6) is a homogenous population of epithelial-like cells commonly used as a model to elucidate the mechanism of intestinal epithelial cell survival, apoptosis, differentiation, growth, and wound healing. Thus, in this study, we used the rat IEC-6 and specific inhibitors of MAPKs for in vitro investigation of the effects of activation of MAPKs in response to heat stress. The results show that heat stress significantly induced damage and apoptosis in IEC-6 cells and that inhibition of ERK1/2 activation exacerbated the heat stress-induced damage and apoptosis; conversely, inhibition of JNK and p38 activation attenuated the heat stress-induced damage and apoptosis (). We, therefore, hypothesize that oxidation and the activation of JNK and p38 are likely to be one mechanism contributing to the cellular and morphological damage observed in response to heat stress in the small intestine, while the activation of ERK1/2 may be cytoprotective in heat stress.
The intestinal epithelium enables absorption of nutrients and water from the intestine, while simultaneously protecting the gut and circulation from ingested pathogens (Turner Citation2009). This protective barrier is referred to as the “GI barrier”. Dysfunction of the GI barrier results in increased intestinal permeability, which involves the passive, unregulated diffusion of both small and large molecules from the GI lumen into the blood. Increased intestinal permeability can result from numerous pathophysiological conditions, including hemorrhage and endotoxemia (Lambert et al. Citation2002; Generoso et al. Citation2010). Moreover, many environmental and biological stressors including radiation, LPS, various drugs, endotoxins, ROS and hyperthermia can also cause significant damage to the intestinal epithelium (Shen et al. Citation2009). Regarding hyperthermia (heat stress), significant injury to the intestine is sustained due to reduced blood flow to the gut (Lambert et al. Citation2002), resulting in greater dissipation of heat at the peripheral vasculature, and localized ischemia within the small intestine (Kregel et al. Citation1988; Hall et al. Citation2001; Deja et al. Citation2010). Ischemic injury is associated with ATP depletion, acidosis, and cellular dysfunction, causing necrosis and shedding of epithelial cells from the intestinal villi (Gisolfi Citation2000). This study found heat stress to cause severe damage in the small intestine, including shedding of epithelial cells at the tips of intestinal villi, resulting in exposure of the intestinal mucosa lamina propria (). Further examination of the ultrastructure revealed erosion and ulcerations on the mucosal surface, and multiple cellular organelles exhibited visible damage (). Furthermore, heat stress pathologically reduced microvillus height, caused mitochondrial swelling, degranulation and vesicularization of the endoplasmic reticulum, and increased the presence of lysosomes and vacuolization (). These findings are in agreement with previous studies (Liu et al. Citation2009; Yu et al. Citation2010a, Citation2010b).
Extreme heat stress can have a devastating effect on cellular metabolism, disrupting cellular homeostasis, and uncoupling major physiological processes (Zhang et al. Citation2003). A direct result of heat stress-induced cellular changes is disruption of an intricate balance between the formation and clearance of ROS (Nabenishi et al. Citation2011). Depending on their concentration, ROS can be beneficial or harmful to cells and tissues. At low physiological levels, ROS play a critical role in intracellular signaling and homeostasis, whereas an excess of ROS leads to oxidative damage of lipids, nucleic acids and proteins, abnormal aggregation of cytoskeletal proteins, and mitochondrial dysfunction, and promotes cell death (Circu and Aw Citation2010). Previous studies have demonstrated that excess ROS was a major contributor to LPS-induced vascular injury (Qian et al. Citation2009), ischemia–reperfusion-induced hepatocyte (CitationYu et al. Citation2011b) and heart injury (Luo et al. Citation2009; Di Lisa et al. Citation2011), lung injury (Wang et al. Citation2010; Tickner et al. Citation2011), and kidney injury (Ruggiero et al. Citation2011). In this study, the production of ROS and MDA was significantly increased, accompanied by decreased enzymatic activity of antioxidative SOD and GSH-px in rat small intestine after heat treatment (). Our recently published microarray results showed that many oxidative stress-related genes are differentially expressed in rat small intestine after heat treatment (Yu et al. Citation2011a). The above findings indicate that oxidative stress may be a major contributor to heat stress-induced injury in the small intestine; however, further study is needed to investigate whether inhibition of ROS production can protect intestinal epithelium from heat stress-induced injury.
MAPKs and their downstream targets include essential signaling pathways capable of translating environmental cues into a cellular response (Peter and Dhanasekaran Citation2003; Muthusamy and Piva Citation2010). MAPKs are activated by various stimuli, including cytokines, irradiation, heat, ROS, ischemic, and osmotic stress. Activated MAPKs can phosphorylate a diverse array of substrate proteins, enabling multiple signals to coordinate the most appropriate response (Kyriakis and Avruch Citation2001; Johnson and Lapadat Citation2002; Porta et al. Citation2011). Our recently published microarray results revealed heat stress to differentially regulate genes known to be associated with MAPK signaling pathways (Yu et al. Citation2011a). This finding was supported in this study at the post-translational level by both immunocytochemical and western-blot analysis, which revealed significantly increased phosphorylation of ERK1/2, JNK, and p38 MAPK following heat treatment (). Together, these findings strongly suggest MAPK signaling to play a critical role in the response to heat stress in the rat small intestine. ERK1/2 is described as a critical regulator of cellular differentiation, proliferation, and survival, with the activation of ERK1/2 reported to be a critical regulator of cellular growth in response to both physiological and pathological stimuli (Lu and Xu Citation2006). ERK1/2 has previously been shown to protect cells from a variety of cytotoxic insults, including oxidative stress, UV radiation and hypoxia (Kyriakis and Avruch Citation2001; Johnson and Lapadat Citation2002). In contrast, a growing number of studies indicate that under certain conditions, aberrant ERK1/2 activation is implicated in apoptotic cell death induced by glutamate, ROS, and RNS in several cell types (Johnson and Lapadat Citation2002; Zhang et al. Citation2004; Lu and Xu Citation2006). These disparate effects mediated by ERK1/2 are suggested to be dependent upon cell type, duration, and magnitude of the stimulus (Cagnol and Chambard Citation2010; Subramaniam and Unsicker Citation2010). To determine whether the heat stress-induced increase of ERK1/2 activation promoted cell survival or cell death in rat small intestine, we used the rat IEC-6 and a specific inhibitor of ERK1/2 (U0126) for in vitro investigation of the effects of ERK1/2 activation in response to heat stress. Our results show that inhibition of ERK1/2 activation (by U0126) exacerbated the heat stress-induced damage and apoptosis in intestinal epithelial cells, indicating that the activation of ERK1/2 may be cytoprotective in the small intestine in response to heat stress.
JNK and p38 MAPK are well established to be activated in response to a variety of environmental stressors or inflammatory stimuli, promoting apoptosis and inhibiting cell growth (Kyriakis and Avruch Citation2001; Johnson and Lapadat Citation2002). Indeed, oxidative stress, inflammation, hypoxia, irradiation, heat, and osmotic stress are all reported to induce apoptosis resulting in tissue injury (Jia et al. Citation2007; Choi et al. Citation2010; Muthusamy and Piva Citation2010; Chang et al. Citation2011). In this study, phosphorylation of JNK and p38 MAPK was significantly increased after heat treatment both in vivo (small intestine) and in vitro (IEC-6 cells); inhibition of JNK and p38 activation could attenuate the heat stress-induced damage and apoptosis. Based on these findings, we suggest that the elevations in JNK and p38 MAPK activity, observed in response to heat stress in this study, are key factors involved in promoting the heat-induced injury observed in the rat small intestine.
In summary, our model of extreme heat stress caused severe morphological damage to the rat small intestine and increased intestinal permeability. These effects were associated with an increase in oxidative stress and the activation of MAPK signaling pathways. Results from our in vitro studies indicated that the activation of JNK and p38 is likely to be one mechanism contributing to the cellular and morphological damage observed in response to heat stress in the small intestine, while the activation of ERK1/2 may be cytoprotective. Further investigation is required to test the specific relationships between oxidative stress and activation of MAPKs in heat stress-induced injury.
Supplementary Material
Download MS Word (34.5 KB)Acknowledgements
Our gratitude is extended to all members of the CAU-BUA TCVM teaching and research team. This work was supported by grants from National Twelve-Five Technological Supported Plan of China (No. 2011BAD34B01), PHR (IHLB), Research Fund for the Doctoral Program of Higher Education of China and Ministry of Agriculture, Public Service Sectors Agriculture Research Projects (No. 201003060-9/10).
Declaration of interest : The authors report no conflicts of interest. The authors alone are responsible for the content and production of this manuscript.
References
- Ahmad A, Rasheed N, Banu N, Palit G. 2010. Alterations in monoamine levels and oxidative systems in frontal cortex, striatum, and hippocampus of the rat brain during chronic unpredictable stress. Stress. 13:355–364.
- Belozerskaia TA, Gessler NN. 2007. Reactive oxygen species and the strategy of the antioxidant defense in fungi: A review. Prikl Biokhim Mikrobiol. 43:565–575.
- Booth C, Potten CS. 2000. Gut instincts: Thoughts on intestinal epithelial stem cells. J Clin Invest. 105:1493–1499.
- Cagnol S, Chambard JC. 2010. ERK and cell death: Mechanisms of ERK-induced cell death—apoptosis, autophagy and senescence. FEBS J. 277:2–21.
- Chang WT, Li J, Huang HH, Liu H, Han M, Ramachandran S, Li CQ, Sharp WW, Hamann KJ, Yuan CS, Hoek TL, Shao ZH. 2011. Baicalein protects against doxorubicin-induced cardiotoxicity by attenuation of mitochondrial oxidant injury and JNK activation. J Cell Biochem. 112:2873–2881.
- Choi WI, Syrkina O, Kwon KY, Quinn DA, Hales CA. 2010. JNK activation is responsible for mucus overproduction in smoke inhalation injury. Respir Res. 11:172.
- Circu ML, Aw TY. 2010. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic Biol Med. 48:749–762.
- Deja M, Ahlers O, Macguill M, Wust P, Hildebrandt B, Riess H, Kerner T. 2010. Changes in hepatic blood flow during whole body hyperthermia. Int J Hyperthermia. 26:95–100.
- Di Lisa F, Kaludercic N, Paolocci N. 2011. Beta-adrenoceptors, NADPH oxidase, ROS and p38 MAPK: Another ’radical’ road to heart failure?. Br J Pharmacol. 162:1009–1011.
- Generoso SV, Viana ML, Santos RG, Arantes RM, Martins FS, Nicoli JR, Machado JA, Correia MI, Cardoso VN. 2010. Protection against increased intestinal permeability and bacterial translocation induced by intestinal obstruction in mice treated with viable and heat-killed Saccharomyces boulardii. Eur J Nutr. 50:261–269.
- Gisolfi CV. 2000. Is the GI system built for exercise?. News Physiol Sci. 15:114–119.
- Hall DM, Buettner GR, Oberley LW, Xu L, Matthes RD, Gisolfi CV. 2001. Mechanisms of circulatory and intestinal barrier dysfunction during whole body hyperthermia. Am J Physiol Heart Circulat Physiol. 280:H509–H521.
- Honzawa Y, Nakase H, Matsuura M, Chiba T. 2011. Clinical significance of serum diamine oxidase activity in inflammatory bowel disease: Importance of evaluation of small intestinal permeability. Inflamm Bowel Dis. 17:E23–E25.
- Jia YT, Wei W, Ma B, Xu Y, Liu WJ, Wang Y, Lv KY, Tang HT, Wei D, Xia ZF. 2007. Activation of p38 MAPK by reactive oxygen species is essential in a rat model of stress-induced gastric mucosal injury. J Immunol. 179:7808–7819.
- Johnson GL, Lapadat R. 2002. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. 298:1911–1912.
- Jorge AA, Malaquias AC, Arnhold IJ, Mendonca BB. 2009. Noonan syndrome and related disorders: A review of clinical features and mutations in genes of the RAS/MAPK pathway. Horm Res. 71:185–193.
- Kaneko N, Sugioka T, Sakurai H. 2007. Aluminum compounds enhance lipid peroxidation in liposomes: Insight into cellular damage caused by oxidative stress. J Inorg Biochem. 101:967–975.
- Kim GS, Choi YK, Song SS, Kim WK, Han BH. 2005. MKP-1 contributes to oxidative stress-induced apoptosis via inactivation of ERK1/2 in SH-SY5Y cells. Biochem Biophys Res Commun. 338:1732–1738.
- Kim BI, Kim HJ, Park JH, Park DI, Cho YK, Sohn CI, Jeon WK, Kim HS, Kim DJ. 2011. Increased intestinal permeability as a predictor of bacterial infections in patients with decompensated liver cirrhosis and hemorrhage. J Gastroenterol Hepatol. 26:550–557.
- Koh G, Lee DH, Woo JT. 2010. 2-Deoxy-D-ribose induces cellular damage by increasing oxidative stress and protein glycation in a pancreatic beta-cell line. Metabolism. 59:325–332.
- Kregel KC, Wall PT, Gisolfi CV. 1988. Peripheral vascular responses to hyperthermia in the rat. J Appl Physiol. 64:2582–2588.
- Kyriakis JM, Avruch J. 2001. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev. 81:807–869.
- Lambert GP, Gisolfi CV, Berg DJ, Moseley PL, Oberley LW, Kregel KC. 2002. Selected contribution: Hyperthermia-induced intestinal permeability and the role of oxidative and nitrosative stress. J Appl Physiol. 92:1750–1761.
- Liu F, Yin J, Du M, Yan P, Xu J, Zhu X, Yu J. 2009. Heat-stress-induced damage to porcine small intestinal epithelium associated with downregulation of epithelial growth factor signaling. J Anim Sci. 87:1941–1949.
- Lu A, Wang H, Hou X, Li H, Cheng G, Wang N, Zhu X, Yu J, Luan W, Liu F, Xu J. 2011. Microarray analysis of gene expression profiles of rat small intestine in response to heat stress. J Biomol Screen. 16:655–667.
- Lu Z, Xu S. 2006. ERK1/2 MAP kinases in cell survival and apoptosis. Iubmb Life. 58:621–631.
- Luo X, Cai H, Ni J, Bhindi R, Lowe HC, Chesterman CN, Khachigian LM. 2009. c-JunDNAzymes inhibit myocardial inflammation, ROS generation, infarct size, and improve cardiac function after ischemia-reperfusion injury. Arterioscler Thromb Vasc Biol. 29:1836–1842.
- Moriwaki Y, Katamura H, Hashimoto K, Ichikawa Y, Sugiyama M. 1993. The worsening of tissue damage due to the transient release during the ischemia of rat small intestine—the relation with reactive oxygen species and lipid peroxidation. Nippon Geka Gakkai Zasshi. 94:1017–1021.
- Muthusamy V, Piva TJ. 2010. The UV response of the skin: A review of the MAPK, NFkappaB and TNFalpha signal transduction pathways. Arch Dermatol Res. 302:5–17.
- Nabenishi H, Ohta H, Nishimoto T, Morita T, Ashizawa K, Tsuzuki Y. 2011. The effects of cysteine addition during in vitro maturation on the developmental competence, ROS, GSH and apoptosis level of bovine oocytes exposed to heat stress. Zygote. 18:1–11 Published on-line: DOI: 10.1017/S0967199411000220.
- Naziroglu M, Akkus S, Soyupek F, Yalman K, Celik O, Eris S, Uslusoy GA. 2010. Vitamins C and E treatment combined with exercise modulates oxidative stress markers in blood of patients with fibromyalgia: A controlled clinical pilot study. Stress. 13:498–505.
- Peter AT, Dhanasekaran N. 2003. Apoptosis of granulosa cells: A review on the role of MAPK-signalling modules. Reprod Domest Anim. 38:209–213.
- Porta H, Cancino-Rodezno A, Soberon M, Bravo A. 2011. Role of MAPK p38 in the cellular responses to pore-forming toxins. Peptides. 32:601–606.
- Qian F, Deng J, Cheng N, Welch EJ, Zhang Y, Malik AB, Flavell RA, Dong C, Ye RD. 2009. A non-redundant role for MKP5 in limiting ROS production and preventing LPS-induced vascular injury. Embo J. 28:2896–2907.
- Ruggiero C, Ehrenshaft M, Cleland E, Stadler K. 2011. High-fat diet induces an initial adaptation of mitochondrial bioenergetics in the kidney despite evident oxidative stress and mitochondrial ROS production. Am J Physiol Endocrinol Metab. 300:E1047–E1058.
- Shen L, Su L, Turner JR. 2009. Mechanisms and functional implications of intestinal barrier defects. Dig Dis. 27:443–449.
- Subramaniam S, Unsicker K. 2010. ERK and cell death: ERK1/2 in neuronal death. FEBS J. 277:22–29.
- Tickner J, Fan LM, Du J, Meijles D, Li JM. 2011. Nox2-derived ROS in PPARgamma signaling and cell-cycle progression of lung alveolar epithelial cells. Free Radic Biol Med. 51:763–772.
- Turner JR. 2009. Intestinal mucosal barrier function in health and disease. Nat Rev Immunol. 9:799–809.
- Wang T, Chiang ET, Moreno-Vinasco L, Lang GD, Pendyala S, Samet JM, Geyh AS, Breysse PN, Chillrud SN, Natarajan V, Garcia JG. 2010. Particulate matter disrupts human lung endothelial barrier integrity via ROS- and p38 MAPK-dependent pathways. Am J Respir Cell Mol Biol. 42:442–449.
- Wingo JE, Low DA, Keller DM, Brothers RM, Shibasaki M, Crandall CG. 2010. Skin blood flow and local temperature independently modify sweat rate during passive heat stress in humans. J Appl Physiol. 109:1301–1306.
- Yu J, Liu F, Yin P, Zhu X, Cheng G, Wang N, Lu A, Luan W, Zhang N, Li J, Guo K, Yin Y, Wang H, Xu J. Integrating miRNA and mRNA expression profiles in response to heat stress-induced injury in rat small intestine. Funct Integr Genomics. 2011a; 11:203–213.
- Yu HC, Qin HY, He F, Wang L, Fu W, Liu D, Guo FC, Liang L, Dou KF, Han H. Canonical notch pathway protects hepatocytes from ischemia reperfusion injury by repressing ROS production through JAK2/STAT3 signaling. Hepatology. 2011b; 54:979–988.
- Yu J, Yin P, Liu F, Cheng G, Guo K, Lu A, Zhu X, Luan W, Xu J. Effect of heat stress on the porcine small intestine: A morphological and gene expression study. Comp Biochem Physiol A Mol Integr Physiol. 2010a; 156:119–128.
- Yu J, Yin P, Yin J, Liu F, Zhu X, Cheng G, Guo K, Yin Y, Xu J. Involvement of ERK1/2 signalling and growth-related molecules’ expression in response to heat stress-induced damage in rat jejunum and IEC-6 cells. Int J Hyperthermia. 2010b; 26:538–555.
- Zhang L, Pelech S, Uitto VJ. 2004. Bacterial GroEL-like heat shock protein 60 protects epithelial cells from stress-induced death through activation of ERK and inhibition of caspase 3. Exp Cell Res. 292:231–240.
- Zhang HJ, Xu L, Drake VJ, Xie L, Oberley LW, Kregel KC. 2003. Heat-induced liver injury in old rats is associated with exaggerated oxidative stress and altered transcription factor activation. FASEB J. 17:2293–2295.