Abstract
Epinephrine and norepinephrine are produced during psychological stress and can directly bind to cells to induce DNA damage. These effects may have more long-lasting consequences such as DNA mutations resulting in an increased potential for cellular transformation and/or tumor progression. This study examined the molecular effects of a chronic (24 h) in vitro exposure to these stress hormones on murine 3T3 cells. Long exposures (24 h) in dose–response experiments with norepinephrine or epinephrine induced significant increases in DNA damage in treated cells compared to that of untreated controls as measured by the alkaline comet assay. Pre-treatment with a blocking agent (the β-adrenergic receptor antagonist propranolol) eliminated this increase in damage. In addition, both norepinephrine and epinephrine increased cellular transformation, as assessed by growth in soft agar, and 3T3 cells pre-treated with either norepinephrine or epinephrine induced a more rapid onset of tumors and more aggressive tumor growth in nude mice. In summary, incubation of 3T3 cells with catecholamines results in long-term DNA damage as measured by increased transformed phenotypes and tumor progression, indicating that they are important mediators of stress effects on genomic instability and vulnerability to tumor formation.
Introduction
Considerable research has examined stress and its neuroendocrine sequelae as a factor in tumor progression and tumor initiation (Fox and Goldsmith Citation1976; Fox Citation1978, Citation1984; Ben-Eliyahu et al. Citation1999; Garssen and Goodkin Citation1999; Ben-Eliyahu Citation2003; Thaker et al. Citation2006). However, unique effects of stress hormones on modulation of cellular function related to increased cancer risk or cancer progression are not well understood. Production and release of epinephrine and norepinephrine are increased during psychological stress, and these hormones can bind directly to G protein-coupled adrenergic receptors on the surface of many cells (Perez-Sayans et al. Citation2010; Daly and McGrath Citation2011). The effects of these hormones are similar to those associated with neural activation during stress but may have more long-lasting consequences such as genomic instability leading to DNA damage, resulting in increased cell transformation and/or tumor formation and progression. Stress hormones may contribute to tumor progression and metastasis in essentially any tissue since almost all cell types express adrenergic receptors (Barnes Citation1995). Studies using cancer cell lines in mice have shown that stress can increase tumor development through β-adrenergic activation (Hasegawa and Saiki Citation2002). Norepinephrine can stimulate cell proliferation and promotes migration of breast (Slotkin et al. Citation2000) and colon cancer cell lines (Masur et al. Citation2001). Norepinephrine has also been shown to upregulate vascular endothelial growth factor in nasopharyngeal carcinoma tumor cells as well as in multiple myeloma-derived cell lines (Yang et al. Citation2006, Citation2008). β-Adrenergic stimulation has been reported to increase metastasis of prostate and breast cancer cell lines in xenograft models (Palm et al. Citation2006; Sloan et al. Citation2010). Administration of isoproterenol (a non-specific β-agonist) and terbutaline (a β2-agonist) has been shown to significantly increase mean tumor weights and number of tumor nodules in nude mice injected with HeyA8 ovarian cancer cells (Thaker et al. Citation2006). This suggests that β-adrenergic stimulation can increase the growth and metastasis of ovarian cancer. DNA damaging effects of epinephrine on human melanoma cells have also been reported (Hara et al. Citation2011).
We have shown previously that the stress hormones, norepinephrine and epinephrine, can induce significant DNA damage in a line of cultured murine fibroblasts (3T3 cells) following an acute (less than 30 min) exposure (Flint et al. Citation2007). We found that norepinephrine but not epinephrine interfered with the ability of the 3T3 cells to repair ultraviolet (UV) light-induced DNA damage. We also found that a 30-min exposure to norepinephrine can induce cellular transformation in vitro by the loss of contact inhibition and the ability to grow in soft agar in clonogenic assays. A recent study using a chronic stress murine model and murine cell lines has shown that β-adrenergic catecholamines act via both Gs-protein kinase A and β-arrestin-mediated signaling pathways to induce DNA damage and suppress p53 levels, respectively (Hara et al. Citation2011). These results suggest that stress hormones may have long-term effects on genomic stability. In this report, using NIH 3T3 cells, we test the hypothesis that stress, through the action of stress hormones, can result in permanent DNA damage, increasing the risk of cancer development and progression.
Materials and methods
Cell culture and hormone treatment
NIH 3T3 fibroblasts were obtained from ATCC and subcultured for less than 6 months prior to initiation of the described experiments. NIH 3T3 cells were grown in Dulbecco's Modified Eagle's Medium (DMEM) with 4 mM l-glutamine, 1.5 g/l sodium bicarbonate, 4.5 g/l glucose, and 10% fetal bovine serum (FBS). 3T3 cultures were synchronized in the G1 phase of the cell cycle by overnight serum starvation. 3T3 cells are known to possess surface β-adrenergic receptors but not α-adrenergic receptors (Sheppard Citation1977; Sheppard et al. Citation1977); therefore, we selected propranolol as a β-receptor antagonist.
Dose–response experiments
The cells were untreated or stimulated with either norepinephrine or epinephrine at concentrations of 10− 6, 10− 7, 10− 8, and 10− 9 M for 24 h. Following the 24-h exposure to these stress hormones, the cells were trypsinized and assayed for double-stranded DNA breaks by the comet assay. In certain experiments, the effects of norepinephrine or epinephrine were blocked by addition of propranolol (10− 6 M) 30 min before addition of stress hormones. Each experiment was performed in duplicate and replicated twice. All drugs were purchased from Sigma (St. Louis, MO, USA).
Overnight hormone treatment followed by 7-day rest period
The cells were untreated or stimulated with either norepinephrine or epinephrine at a concentration of 10− 7 M. Following the 24-h exposure period, the cells were washed and media replaced with DMEM containing 10% FBS. The cells were incubated at 37°C for 7 days. During that period, the cells were split as needed (at least once, but not more than twice). At the end of the 7-day rest period, the cells were trypsinized and analyzed by the comet assay, soft agar assay, and tumorigenicity studies.
Measurement of double-stranded DNA damage (comet assay)
To determine the effects of the stress hormones on double-stranded DNA damage, 3T3 cells were embedded in agarose in preparation for the comet assay, which is a single cell gel electrophoresis assay (Collins Citation2004). A commercial comet assay kit was used for these studies (Trevigen, Gaithersburg, MD), and the assay was performed according to the manufacturer's instructions. In brief, 2 × 104 cells were mixed in low-melting-point agarose in phosphate-buffered saline (PBS) and pipetted onto slides pre-coated with 1% agarose. The gels were allowed to set at 4°C in the dark for 30 min and then incubated for 1 h in lysing solution (containing 1% Triton X-100), followed by 1 h incubation in unwinding solution (1 mM EDTA, 300 mM NaOH, pH>13). The slides were then subjected to electrophoresis at 25 V (300 mÅ) for 30 min at 4°C. Following electrophoresis, the slides were dehydrated with ethanol and then stained with Sybr green (Trevigen) and analyzed using computerized image software (Comet IV, Perceptive Instruments, Suffolk, UK). Nucleoid DNA extends under electrophoresis to form “comet tails,” and the relative intensity of DNA in the tail reflects DNA break frequency. A total of 100 cells per slide were analyzed for a representative population of the cells. Comet tails were visualized using an epifluorescence microscope and the percentage tail intensity for each cell was computed using the Comet IV software. Each sample was run in duplicate and each experiment repeated once. All comet analyses were performed blinded by a single user.
Assessment of 3T3 transformation
3T3 cells were incubated with stress hormones at a concentration of 10− 7 M for 24 h and then evaluated for their capacity for anchorage-independent growth using a soft agar assay. In some experiments, the cells were allowed to rest for 7 days following the stress hormone treatment. Briefly, 1 × 105 3T3 cells were suspended in 4 ml of DMEM containing 10% FBS and 0.4% SeaKem GTG agarose (Lonza Rockland, Inc., ME, USA) and 1 ml plated in each of 4 wells of 12-well tissue culture dishes, pre-treated with 2 ml of 0.8% agarose in DMEM at 37°C. Cultures were fed with 0.1 ml of DMEM+10% FBS twice a week for 2–3 weeks, and colonies were stained with crystal violet and counted.
Animal tumor model
Six- to eight-week-old female BALB/c nude mice were housed in groups of four in standard cages. The animal room was maintained on a 12-h light/12-h dark diurnal cycle. Lights went on at 06:00 h and off at 18:00 h. All mice were given food and water ad libitum. All animal protocols were approved by the Institutional Animal Care and Use Community at the University of Pittsburgh and were carried out in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. The mice were housed in a noise-free environment 1 week prior to the experiment and were then handled once a day for 2 weeks to allow them to acclimatize to the environment and to the investigators. NIH 3T3 cells were treated with 10− 7 M of the stress hormones for 24 h, trypsinized, and 5 × 105 3T3 cells in 0.2 ml PBS were inoculated subcutaneously into the flanks of mice. All mice in a single cage received the same treatment. The mice were assessed between 09:00 and 12:00 h three times a week for development of tumors. Tumor sizes were recorded by measuring two perpendicular diameters by calipers. Tumor volume (v) was calculated as follows: v (cm3) = (width2 × length)/2. A tumor with a minimum size of 5 mm diameter was scored as positive. Any mouse with a tumor volume of 4 cm3 or greater was humanely euthanized.
Statistical analyses
For the comet analyses and soft agar assays, data are expressed as mean ± SD. For the in vitro studies, statistically significant differences between hormone-treated and control groups were determined by ANOVA, with Tukey's honestly significant difference as post-hoc test for multiple comparisons. For the tumor studies, results are presented as mean tumor volume ± SD. Significant differences were determined using a repeated measures ANOVA. A value of p < 0.05 was accepted as the level of statistical significance.
Results
Effects of stress hormones on double-stranded DNA damage
The effects of epinephrine or norepinephrine on double-stranded DNA damage were first evaluated immediately following a 24-h treatment. As shown in , there were significant dose-dependent increases in DNA damage in cells treated with either of the stress hormones [for epinephrine: F(8,891) = 38.44, p < 0.0001, ; for norepinephrine: F(8,891) = 147.8, p < 0.0001, . Previous studies investigating the role of β-adrenergic stimulation on different cell types have used concentrations of epinephrine or norepinephrine from 0.1 to 10 μM (10− 7 to 10− 5 M) (Palm et al. Citation2006; Thaker et al. Citation2006; Landen et al. Citation2007; Nilsson et al. Citation2007; Li et al. Citation2010; Hara et al. Citation2011), and DNA damage in our experiments was achieved using similar (0.1 μM) and lower (0.01 μM) concentrations to those reported in the literature studying β-adrenergic signaling (). The epinephrine- or norepinephrine-induced increases in DNA damage were eliminated by pre-treatment of the cells with 1 × 10− 6 M propranolol, a β-receptor antagonist (). ANOVA with Tukey's tests for multiple comparisons demonstrated significant differences between different dosages of both epinephrine and norepinephrine compared with the propranolol treatment alone, with the exception of the 10− 9 M treatment. shows photographs of 3T3 cells untreated or treated with propranolol, epinephrine, or norepinephrine. The comet tails caused by the movement of the damaged DNA away from the nucleus under electrophoresis can clearly be seen with epinephrine or norepinephrine treatment. These tails are absent in cells that were untreated or pre-treated with propranolol.
Figure 1. Effect of 24-h exposure to stress hormones on DNA damage. NIH 3T3 cells were incubated in the presence or absence of (A) epinephrine or (B) norepinephrine for 24 h, and DNA damage was measured by the comet assay. In some experiments, cells were pre-treated for 30 min with 10− 6 M propranolol prior to treatment with the stress hormones. Results are expressed as mean% tail intensity ± SD. Asterisk (*) indicates the significant increase compared to that of untreated control (p < 0.05) using a one-way ANOVA with Tukey's post-hoc analyses. Each experiment was performed twice, and 100 cells were analyzed in each comet assay. Prop, propranolol; Epi, epinephrine; NE, norepinephrine.
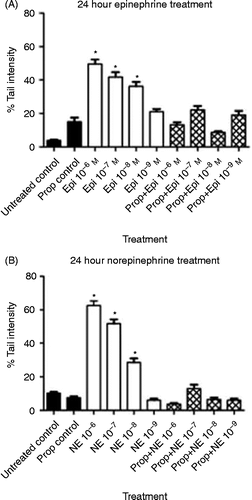
Figure 2. Photographs of NIH 3T3 cells following comet assay. NIH 3T3 cells were untreated or treated (24 h) with stress hormones (epinephrine or norepinephrine) at 10− 7 M in the presence or absence of 10− 6 M propranolol. Arrows point to examples of comet tails. All pictures were taken at 50 × . Bars equal 50 μm. Epi, epinephrine; Norepi, norepinephrine; Prop, propranolol.

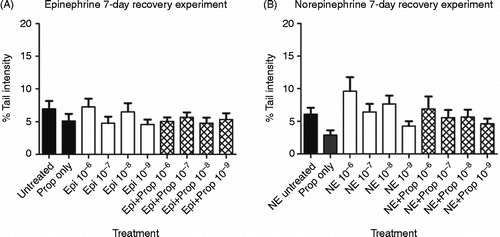
Next, we evaluated the amount of double-stranded DNA damage in 3T3 cells treated with epinephrine or norepinephrine for 24 h followed by a 7-day rest period. The purpose of these experiments was to determine whether residual DNA damage (double-stranded DNA breaks that are measured using the alkaline comet assay) was present after 7 days or whether the cells were able to repair the damage reported in . At the end of the 24-h hormone treatment, the cells were rinsed and media replaced with regular media (without stress hormones). During the 7-day rest period, the cells continued to grow and expand, resulting in the cells being trypsinized and split 1:2. At the end of the 7-day rest period, the cells were analyzed for double-stranded DNA damage using the alkaline comet assay. As seen in , there was no significant DNA damage remaining in cells treated with either epinephrine [F(8,891) = 1.037; p = 0.4063; or norepinephrine [F(8,916) = 1.448; p = 0.1725; compared with untreated cells.
Figure 3. Effect of 24-h exposure to stress hormones on DNA damage followed by a 7-day rest period. NIH 3T3 cells were incubated in the presence or absence of (A) epinephrine or (B) norepinephrine for 24 h, followed by a 7-day rest period, and DNA damage was measured by the comet assay. In some experiments, cells were pre-incubated for 30 min with 10− 6 M propranolol. Each experiment was performed twice, and 100 cells were analyzed in each comet assay. Results are expressed as mean% tail intensity ± SD. No significant differences were found with one-way ANOVA. Pro, propranolol; Epi, epinephrine; NE, norepinephrine.
Cellular transformation in 3T3 cells
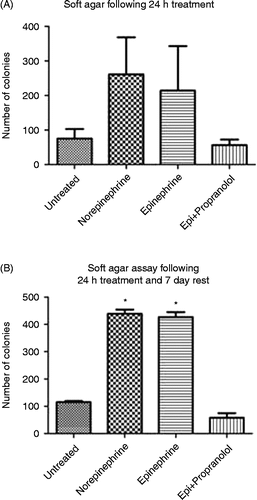
We determined whether the DNA damage caused by 24 h exposure of 3T3 cells to epinephrine or norepinephrine resulted in increased transformed phenotype. 3T3 cellular transformation was measured using the soft agar colony formation assay on cells treated for 24 h with both hormones (epinephrine or norepinephrine) as well as on cells that had been treated for 24 h and then allowed to rest for 7 days. As shown in , treatment of 3T3 cells for 24 h with epinephrine or norepinephrine did not result in a statistically significant increase in transformed phenotype (as measured by soft agar colony formation). Allowing the treated cells to rest for 7 days, however, resulted in a significant increase in transformed phenotype in cells treated with both hormones compared with untreated cells ; F(3,4) = 366.5; p < 0.0001]. This significant increase in cellular transformation was completely blocked by pre-treatment with propranolol.
Figure 4. Effect of 24-h exposure to stress hormones on cell transformation. NIH 3T3 cells were incubated in the presence or absence of epinephrine (Epi) or norepinephrine (10− 7 M) for (A) 24 h or (B) 24 h followed by a 7-day rest period, and the transformed phenotype was measured by a soft agar colony assay. In some experiments, cells were pre-incubated for 30 min with 10− 6 M propranolol. Each experiment was performed twice. Results are expressed as mean ± SD. Asterisk (*) indicates the significant increase compared with that of untreated control (p < 0.05) using a one-way ANOVA with Tukey's post-hoc analyses. Epi, epinephrine.
Hormone-induced tumor formation in mice
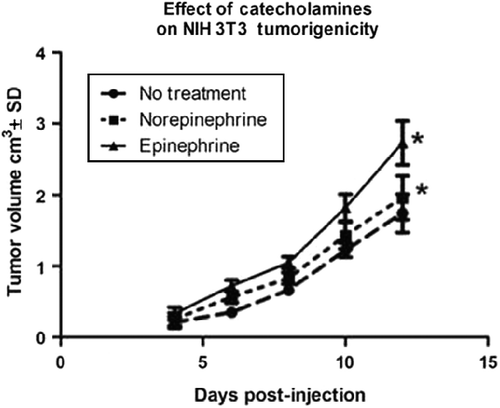
Tumor progression of 3T3 cells treated in vitro for 24 h with norepinephrine or epinephrine was evaluated by injection of the treated cells into the flanks of nude mice and measuring tumor development over time. summarizes the data for tumor growth (tumor volume/cm3). A repeated measures ANOVA indicated that pre-treatment of cells with the stress hormones, norepinephrine or epinephrine, was associated with more rapid and extensive tumor growth compared with 3T3 controls, F(2,7) = 6.84, p < 0.05. Treatment with epinephrine showed a slightly stronger but not statistically significant effect on tumor formation than treatment with norepinephrine. All groups showed strong significant effects of time, F(4,28) = 75.19, p < 0.001, but the tumor volume associated with hormone exposure was greater than controls after 4 days of growth.
Figure 5. Effect of stress hormones on tumor progression of NIH 3T3 cells. NIH 3T3 cells were incubated in the presence or absence of epinephrine or norepinephrine (10− 6 M) for 24 h, trypsinized, and 5 × 105 cells were injected subcutaneously into the flanks of Balb/c nude mice (four mice per group). Results are presented as mean tumor volume ± SD; mice were removed from study if tumor volume reached 4 cm3. Asterisk (*) indicates the significant increase compared with that of control (p = 0.01) using a repeated measures ANOVA.
Discussion
The development of cancer has been widely accepted as a multi-step process meaning that multiple changes to the DNA are required for advancement of the cell from normal to cancerous (Hanahan and Weinberg Citation2011). Damaged DNA is repaired by several mechanisms, depending on the type of damage. Thus mispaired DNA bases are repaired by a mismatch repair mechanism and DNA bases that are chemically altered are replaced by base excision repair (Lindahl and Barnes Citation2000; Jiricny Citation2006). Single-stranded breaks are repaired by single-stranded repair mechanism, and double-stranded breaks are repaired by non-homologous end-joining or homologous recombination (West Citation2003; Caldecott Citation2008). If any of these repair mechanisms fails to properly repair the DNA, a permanent mutation can result, the consequence of which is part of the multi-step process of carcinogenesis.
Environmental carcinogens such as tobacco smoke, pesticides, and UV radiation are known DNA mutagens increasing cancer risk. Psychological stress and the resulting hormones produced during stress (such as epinephrine and norepinephrine) have been reported in both epidemiological studies and through the use of animal models to increase cancer progression. This increased progression has been linked to immune suppression as well as increased angiogenesis (Ross Citation2008; Tilan and Kitlinska Citation2010; Lutgendorf et al. Citation2011). A recent study reported that administration of a β-blocker (propranolol) and a cyclo-oxygenase-2 inhibitor (etodolac) increases the rates of long-term survival following excision of a metastasizing syngeneic primary tumor in mice (Glasner et al. Citation2010). Less clear is the potential linkage between chronic stress and initiation or development of cancer. Epidemiological reports analyzing retrospective studies on the association of stressful life events and cancer development have yielded conflicting results. For example, Lillberg and coworkers (Citation2003), analyzing 10,808 women from the Finnish twin cohort, reported a role for stressful life events including divorce/separation, death of a husband, and death of a close relative or friend in an increased risk of breast cancer. In contrast, Michael et al. (Citation2009), analyzing 87,498 women enrolled in the Women's Health Initiative Study, reported no independent association between stressful life events and breast cancer risk. The reasons for these discrepancies are not clear but may include selection bias and interactions between stressful events and coping strategies or levels of social support. For these reasons, the use of animal and cell culture models is critical to provide a more defined environment to study the role of stress on tumor development. In vitro treatment of cell lines with stress hormones combined with animal models provides a method for investigating the direct effects of these hormones on cells leading to cellular transformation and tumor formation. The ability to treat the cells in vitro with the stress hormones permits the separation of stress hormone effects on individual cells from effects on immune responses.
The data presented in this report support our hypothesis that stress, through the action of stress hormones, can result in permanent DNA damage, increasing the risk of cancer development and progression. We have found that 3T3 cells treated with norepinephrine or epinephrine for 24 h have a significant increase in double-stranded DNA damage as measured by the alkaline comet assay. The ability to block these effects with propranolol demonstrates that the hormones are acting via the β-adrenergic receptor pathway. We have also demonstrated that treatment of 3T3 cells with these stress hormones increases both cellular transformation and tumor progression. Thus the DNA damage seen at 24 h with the comet assay results in permanent DNA damage. To our knowledge, this is the first demonstration that the stress hormones, epinephrine and norepinephrine, can contribute to development of tumorigenesis following the in vitro treatment. Studies using orthotopic mouse models have begun to show that chronic stress can promote tumor growth (Ben-Eliyahu Citation2003; Reiche et al. Citation2004; Thaker et al. Citation2006). Our data are consistent with studies using receptor-blocking technologies to show that β-adrenergic receptors play a role in tumor growth in a mouse model of ovarian carcinoma (Thaker et al. Citation2006). Our results also support recent translational studies demonstrating that β-adrenergic blockers can serve as therapeutic agents in treating cancer, particularly breast cancer (Powe and Entschladen Citation2011). In light of these findings, it is clear that further studies on the mechanism(s) driving the stress-induced DNA damage are needed.
Acknowledgment
This study is supported in part by a grant from the DOD (DAMD17-01-0373) and used the University of Pittsburgh Cancer Institute Animal Facility and was supported in part by award P30CA047904.
Declaration of interest : The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Barnes PJ. 1995. Beta-adrenergic receptors and their regulation. Am J Respir Crit Care Med. 152:838–860.
- Ben-Eliyahu S. 2003. The promotion of tumor metastasis by surgery and stress: Immunological basis and implications for psychoneuroimmunology. Brain Behav Immun. 17 Suppl. 1: S27–S36.
- Ben-Eliyahu S, Page GG, Yirmiya R, Shakhar G. 1999. Evidence that stress and surgical interventions promote tumor development by suppressing natural killer cell activity. Int J Cancer. 80:880–888.
- Caldecott KW. 2008. Single-strand break repair and genetic disease. Nat Rev Genet. 9:619–631.
- Collins AR. 2004. The comet assay for DNA damage and repair: Principles, applications, and limitations. Mol Biotechnol. 26:249–261.
- Daly CJ, McGrath JC. 2011. Previously unsuspected widespread cellular and tissue distribution of beta-adrenoceptors and its relevance to drug action. Trends Pharmacol Sci. 32:219–226.
- Flint MS, Baum A, Chambers WH, Jenkins FJ. 2007. Induction of DNA damage, alteration of DNA repair and transcriptional activation by stress hormones. Psychoneuroendocrinology. 32:470–479.
- Fox BH. 1978. Premorbid psychological factors as related to cancer incidence. J Behav Med. 1:45–133.
- Fox BH. 1984. Remote life-style causes of cancer. Cancer Detect Prev. 7:21–29.
- Fox BH, Goldsmith JR. 1976. Behavioral issues in prevention of cancer. Prev Med. 5:106–121.
- Garssen B, Goodkin K. 1999. On the role of immunological factors as mediators between psychosocial factors and cancer progression. Psychiatry Res. 85:51–61.
- Glasner A, Avraham R, Rosenne E, Benish M, Zmora O, Shemer S, Meiboom H, Ben-Eliyahu S. 2010. Improving survival rates in two models of spontaneous post-operative metastasis in mice by combined administration of a β-adrenergic antagonist and a cyclooxygenasae-2 inhibitor. J Immunol. 184:2449–2457.
- Hanahan D, Weinberg RA. 2011. Hallmarks of cancer: The next generation. Cell. 144:646–674.
- Hara MR, Kovacs JJ, Whalen EJ, Rajagopal S, Strachan RT, Grant W, Towers AJ, Williams B, Lam CM, Xiao K, Shenoy SK, Gregory SG, Ahn S, Duckett DR, Lefkowitz RJ. 2011. A stress response pathway regulates DNA damage through beta2-adrenoreceptors and beta-arrestin-1. Nature. 477:349–353.
- Hasegawa H, Saiki I. 2002. Psychosocial stress augments tumor development through beta-adrenergic activation in mice. Jpn J Cancer Res. 93:729–735.
- Jiricny J. 2006. The multifaceted mismatch-repair system. Nat Rev Mol Cell Biol. 7:335–346.
- Landen CNJr, Lin YG, Armaiz Pena GN, Das PD, Arevalo JM, Kamat AA, Han LY, Jennings NB, Spannuth WA, Thaker PH, Lutgendorf SK, Savary CA, Sanguino AM, Lopez-Berestein G, Cole SW, Sood AK. 2007. Neuroendocrine modulation of signal transducer and activator of transcription-3 in ovarian cancer. Cancer Res. 67:10389–10396.
- Li J, Yan B, Huo Z, Liu Y, Xu J, Sun Y, Liu Y, Liang D, Peng L, Zhang Y, Zhou ZN, Shi J, Cui J, Chen YH. 2010. Beta2- but not beta1-adrenoceptor activation modulates intracellular oxygen availability. J Physiol. 588:2987–2998.
- Lillberg K, Verkasalo PK, Kaprio J, Teppo L, Helenius H, Koskenvuo M. 2003. Stressful life events and risk of breast cancer in 10,808 women: A cohort study. Am J Epidemiol. 157:415–423.
- Lindahl T, Barnes DE. 2000. Repair of endogenous DNA damage. Cold Spring Harbor Symp Quant Biol. 65:127–133.
- Lutgendorf SK, DeGeest K, Dahmoush L, Farley D, Penedo F, Bender D, Goodheart M, Buekers TE, Mendez L, Krueger G, Clevenger L, Lubaroff DM, Sood AK, Cole SW. 2011. Social isolation is associated with elevated tumor norepinephrine in ovarian carcinoma patients. Brain Behav Immun. 25:250–255.
- Masur K, Niggemann B, Zanker KS, Entschladen F. 2001. Norepinephrine-induced migration of SW 480 colon carcinoma cells is inhibited by beta-blockers. Cancer Res. 61:2866–2869.
- Michael YL, Carlson NE, Chlebowski RT, Aickin M, Weihs KL, Ockene JK, Bowen DJ, Ritenbaugh C. 2009. Influence of stressors on breast cancer incidence in the Women's Health Initiative. Health Psychol. 28:137–146.
- Nilsson MB, Armaiz-Pena G, Takahashi R, Lin YG, Trevino J, Li Y, Jennings N, Arevalo J, Lutgendorf SK, Gallick GE, Sanguino AM, Lopez-Berestein G, Cole SW, Sood AK. 2007. Stress hormones regulate interleukin-6 expression by human ovarian carcinoma cells through a Src-dependent mechanism. J Biol Chem. 282:29919–29926.
- Palm D, Lang K, Niggemann B, Drell TLIV, Masur K, Zaenker KS, Entschladen F. 2006. The norepinephrine-driven metastasis development of PC-3 human prostate cancer cells in BALB/c nude mice is inhibited by beta-blockers. Int J Cancer. 118:2744–2749.
- Perez-Sayans M, Somoza-Martin JM, Barros-Angueira F, Diz PG, Gandara Rey JM, Garcia-Garcia A. 2010. Beta-adrenergic receptors in cancer: Therapeutic implications. Oncol Res. 19:45–54.
- Powe DG, Entschladen F. 2011. Targeted therapies: Using beta-blockers to inhibit breast cancer progression. Nat Rev Clin Oncol. 8:511–512.
- Reiche EM, Nunes SO, Morimoto HK. 2004. Stress, depression, the immune system, and cancer. Lancet Oncol. 5:617–625.
- Ross K. 2008. Mapping pathways from stress to cancer progression. J Natl Cancer Inst. 100:914–915 917.
- Sheppard JR. 1977. Catecholamine hormone receptor differences identified on 3T3 and simian virus-transformed 3T3 cells. Proc Natl Acad Sci USA. 74:1091–1094.
- Sheppard JR, Gormus R, Moldow CF. 1977. Catecholamine hormone receptors are reduced on chronic lymphocytic leukaemic lymphocytes. Nature. 269:693–695.
- Sloan EK, Priceman SJ, Cox BF, Yu S, Pimentel MA, Tangkanangnukul V, Arevalo JMG, Morizono K, Karanikolas BDW, Wu L, Sood AK, Cole SW. 2010. The sympathetic nervous system induces a metastatic switch in primary breast cancer. Cancer Res. 70:7042–4052.
- Slotkin TA, Zhang J, Dancel R, Garcia SJ, Willis C, Seidler FJ. 2000. Beta-adrenoceptor signaling and its control of cell replication in MDA-MB-231 human breast cancer cells. Breast Cancer Res Treat. 60:153–166.
- Thaker PH, Han LY, Kamat AA, Arevalo JM, Takahashi R, Lu C, Jennings NB, Armaiz-Pena G, Bankson JA, Ravoori M, Merritt WM, Lin YG, Mangala LS, Kim TJ, Coleman RL, Landen CN, Li Y, Felix E, Sanguino AM, Newman RA, Lloyd M, Gershenson DM, Kundra V, Lopez-Berestein G, Lutgendorf SK, Cole SW, Sood AK. 2006. Chronic stress promotes tumor growth and angiogenesis in a mouse model of ovarian carcinoma. Nat Med. 12:939–944.
- Tilan J, Kitlinska J. 2010. Sympathetic neurotransmitters and tumor angiogenesis – link between stress and cancer progression. J Oncol. 2010:539706.
- West SC. 2003. Molecular views of recombination proteins and their control. Nat Rev Mol Cell Biol. 4:435–445.
- Yang EV, Sood AK, Chen M, Li Y, Eubank TD, Marsh CB, Jewell S, Flavahan NA, Morrison C, Yeh PE, Lemeshow S, Glaser R. 2006. Norepinephrine up-regulates the expression of vascular endothelial growth factor, matrix metalloproteinase (MMP)-2, and MMP-9 in nasopharyngeal carcinoma tumor cells. Cancer Res. 66:10357–10364.
- Yang EV, Donovan EL, Benson DM, Glaser R. 2008. VEGF is differentially regulated in multiple myeloma-derived cell lines by norepinephrine. Brain Behav Immun. 22:318–323.