Abstract
Prenatal maternal stress (PMS) programs dysregulation of the hypothalamus–pituitary–adrenal axis (HPAA) in postnatal life, though time periods vulnerable to PMS, are still unclear. We evaluated in pregnant sheep the effect of PMS during early gestation [30–100 days of gestation (dGA); term is 150 dGA] or late gestation (100–120 dGA) on development of fetal HPAA function. We compared the effects of endogenous cortisol with synthetic glucocorticoid (GC) exposure, as used clinically to enhance fetal lung maturation. Pregnant sheep were exposed to repeated isolation stress twice per week for 3 h in a separate box with no visual, tactile, or auditory contact with their flock-mates either during early (n = 7) or late (n = 7) gestation. Additional groups received two courses of betamethasone (BM; n = 7; 2 × 110 μg kg− 1 body weight, 24 h apart) during late gestation (106/107 and 112/113 dGA, n = 7) or acted as controls (n = 7). Fetal cortisol responses to hypotensive challenge, a physiological fetal stressor, were measured at 112 and 129 dGA, i.e. before and during maturation of the HPAA. Hypotension was induced by fetal infusion of sodium nitroprusside, a potent vasodilator. At 112 dGA, neither PMS nor BM altered fetal cortisol responses. PMS, during early or late gestation, and BM treatment increased fetal cortisol responses at 129 dGA with the greatest increase achieved in stressed early pregnant sheep. Thus, development of the HPAA is vulnerable to inappropriate levels of GCs during long periods of fetal life, whereas early gestation is most vulnerable to PMS.
Introduction
Exposure to inappropriate levels of glucocorticoids (GCs) at critical stages of fetal development seems to have a lifelong impact on the function of hypothalamus–pituitary–adrenal axis (HPAA) which is regarded as one of the major mechanisms of fetal programming of health and disease in later life (Cottrell and Seckl Citation2009). In particular, the programming effects of prenatal exposure to maternal stress and excess synthetic GCs on HPAA function are regarded as significant contributors to the etiology of neuropsychiatric and cardiovascular diseases (Van den Bergh et al. Citation2005; Alexander Citation2006; Beydoun and Saftlas Citation2008; Nuyt Citation2008; Glover et al. Citation2010). Prenatal stress as well as prenatal administration of synthetic GCs program hyperactivity of the HPAA has been shown in human and animal studies (Sloboda et al. Citation2002; Coe et al. Citation2003; Glover et al. Citation2005; Van den Bergh et al. Citation2008; Tegethoff et al. Citation2009). Notably, synthetic GCs are prescribed in late gestation in about 10% of pregnancies to enhance fetal lung maturation in women threatened with premature labor (Polyakov et al. Citation2007).
Programming effects of prenatal GCs appear to vary with respect to time of exposure, although knowledge concerning exact periods of vulnerability is limited. Despite the finding that the HPAA matures at the end of gestation (Challis et al. Citation2001) and development of glucocorticoid receptors (GRs) begins in mid-gestation (Matthews et al. Citation1995), Van den Bergh et al. (Citation2008) have shown that in humans, maternal anxiety during early pregnancy is more strongly associated with increased HPAA activity in the 15-year-old offspring than is stress during mid- and late gestation. However, validation in an experimental model is essential as effects of maternal stress during pregnancy are confounded by maternal care after birth. As in prenatal stress, programming effects of synthetic GCs depend on the time of administration during gestation as well as dose. Experimental studies in fetal sheep, the model in which prenatal GC therapy was developed (Liggins and Howie Citation1972), have shown that the fetal HPAA is vulnerable to dexamethasone in early pregnancy leading to increased basal cortisol concentrations near term. Sloboda et al. (Citation2002) reported that a single injection of betamethasone (BM) to pregnant sheep at mid-gestation induced elevated basal values and stimulated cortisol concentrations even at 1 year of age. However, three additional BM injections, each 1 week apart, did not have the same effect on basal cortisol concentrations.
Determining the developmental window for greatest stress sensitivity is essential to provide maximum preventive care and targeted therapy in terms of functional programming of the HPAA. Yet, the effects of maternal stress on the fetus with respect to the time of stress exposure are largely unknown. We hypothesized that (i) maternal stress during early pregnancy (i.e. during the first and second-trimester of gestation) and late pregnancy (i.e. during the last trimester) has different programming effects on fetal HPAA function in sheep. Furthermore, we hypothesized that (ii) administration of synthetic GCs in late pregnancy, when administration is clinically relevant, may have similar effects on fetal HPAA function as maternal stress during late pregnancy. Hence, pregnant sheep, a major animal model for fetal physiology, were repeatedly isolated from the flock during early or late gestation. Isolation, constituting a typical major ovine stressor, was used because it is known to increase maternal cortisol concentrations reliably in sheep (Roussel et al. Citation2005). Another group of sheep were exposed to synthetic GCs at the pregnancy stage and dose used clinically to enhance fetal lung maturation. The effects of maternal stress or GC treatment on fetal HPAA function were tested by a hypotensive challenge as a common fetal stressor (Low Citation2004), which causes quantifiable and reproducible cortisol responses (Wood Citation1986). Hypotensive challenges were performed before and during maturation of the fetal HPAA in late gestation to examine whether maternal stress affects fetal HPAA development or rather resets HPAA function.
Materials and methods
All procedures were approved by the Animal Welfare Commission for animal research of the Free State of Thuringia and are in accordance with the European Communities Council Directives. For the purpose of this study, 39 Long-Wool Merino × Blackheaded Mutton were cross-bred on a single occasion. Following breeding, the 39 pregnant ewes were divided randomly into control, early and late stress, and a BM group. The four groups were held in different paddocks at the same facility. Ewes were fed 100% of the nutritional requirement throughout pregnancy, and water was provided ad libitum. During pregnancy environmental temperature was natural as outside ambient temperature. Studies were done during springtime.
Maternal stress protocol
Eighteen pregnant sheep followed a repeated maternal stress protocol between 30 and 100 ± 2 (mean ± SEM) days of gestation (dGA; early stress; term = 150 dGA) and seven pregnant sheep between 100 and 120 ± 2 dGA (late stress). Maternal stress was induced by isolating pregnant ewes in a well-lit box (3.0 m × 3.0 m × 1.4 m, w × l × h) with no visual, tactile, or auditory contact with flock-mates. During isolation, ewes had no access to food or water supply.Ewes were isolated for 2 days per week (Monday to Friday) with at least 2 days recovery time between the particular isolation bouts. Duration of isolation was 3 h. Isolation was performed either between 07:00 and 10:00 h, 11:00 and 14:00 h, or 15:00 and 18:00 h. Day of the week and time of isolation as well as the isolation box were changed randomly within the mentioned parameters to reduce habituation.
Maternal blood samples were collected by puncturing the jugular vein at 30, 44, 59, 72, and 89 dGA (early stress), and 110 dGA (late stress) before and during isolation to estimate maternal cortisol concentrations and the degree of habituation. Pregnant control and BM-treated ewes, were held under the same conditions as stressed ewes but did not undergo isolation procedures and blood sampling.
Surgical instrumentation
Following the repeated stress procedure, seven of the early stressed ewes and all seven of the late stress ewes were transported to the surgery facilities at least 3 days before undergoing surgery for chronic fetal instrumentation at 104 ± 1 dGA (controls, early stress, and BM) or 124 ± 1 dGA (late stress). Ewes were kept in rooms with controlled light/dark cycles (14 h light and 10 h dark with lights on from 07:00 to 21:00 h) and air conditioning (18°C and 50% relative air humidity). Hay and water were provided ad libitum. Before the surgical procedure, which is described in detail elsewhere (Schwab et al. Citation2001), ewes received no food for a period of 24 h. Thereafter, the ewes were sedated with 1 g ketamine intramuscularly (Ketamin 10, Atarost, Twistringen, Germany), and general anesthesia was induced by inhalation of 4% isoflurane (Isoflurane®, AstraZeneca, Wedel, Germany) via a face mask. Ewes were intubated, and anesthesia was maintained with 1.5% isoflurane in 100% oxygen. Ewes were instrumented with catheters (Rüschelit, Rüsch, Kernen, Germany) inserted into the carotid artery and the jugular vein to draw blood samples and for postoperative administration of antibiotics, respectively. Laparotomy and hysterotomy were performed for chronic fetal instrumentation. To provide the same intrauterine conditions for all fetuses during instrumentation, we removed the smaller fetus in twin pregnancies. The smaller fetus was identified by comparing the vertex-to-nose length through the unopened uterus. Directly after removal, the smaller fetus was euthanized using 360 mg of intracardiac sodium pentobarbital (Narcoren, Merial, Hallbergmoos, Germany) while still under general anesthesia with isoflurane. Fetuses were instrumented with catheters inserted into the left common carotid artery for fetal arterial blood pressure (FBP) recording and blood sampling and into the left external jugular vein for sodium nitroprusside (SNP) injection. To correct FBP for hydrostatic pressure, a third catheter was placed in the amniotic cavity to record the amniotic pressure. For electrocardiogram recording, stainless steel wire electrodes (LIFYY, Metrofunk Kabel-Union, Berlin, Germany) were implanted in the left suprascapular muscles, right shoulder muscles, and in the sternum cartilage. All catheters and electrode wires were exteriorized from the uterus and the abdomen through the maternal flank, and both uterus and abdomen were then closed with sutures. Catheters were maintained patent using a continuous infusion of heparinized saline (12.5 UI/ml; 0.5 ml/h). After surgery, ewes were returned to their home cage and received intravenous 0.5 g ampicillin (Ampicillin-ratiopharm®, Ratiopharm, Ulm, Germany) twice a day for the next 3 days. Fetuses received the same dose of ampicillin via the amniotic sac. Analgesia was ensured by i.v. administration of metamizol (Arthripur®, Atarost, Twistringen, Germany) twice a day (30–50 mg kg− 1) for as long as necessary.
BM treatment
Seven pregnant sheep that had not been stressed received two courses of BM at 106 and 107 dGA and at 112 and 113 dGA. Each course of BM consisted of two doses of 110 μg kg− 1 BM phosphate administered 24 h apart (Celestan, Essex, Munich, Germany). This dose is equivalent to the dose used clinically to enhance fetal lung maturation (8 mg BM weight adjusted for a 70 kg woman).
Fetal stress response (hypotensive challenge)
Fetal cortisol was examined in response to a hypotensive challenge at 112 ± 1 dGA (controls, early stress, and BM group before the second course of BM treatment) and at 129 ± 2 dGA (all groups).The hypotensive challenge was induced in the morning hours by infusion of SNP (Sigma-Aldrich, Hamburg, Deisenhofen, Germany; SNP acts as a donor of nitric oxide which is a potent vasodilator); 10 μg ml− 1 SNP was infused into the fetal jugular vein after filling the dead space of the catheter. SNP infusion rate started at 0.1 ml min− 1 and was increased stepwise for every 2 min. After 12 min, i.e. at a rate of 3.2 ml min− 1, SNP infusion was stopped. Fetal and maternal arterial blood samples were taken prior to the stress responses to analyze blood gases and oxygen saturation using a blood gas analyzer (ABL600, Radiometer, Copenhagen, Denmark; measurements corrected to 39°C). Plasma cortisol concentrations were determined 30 min before, at the end (0 min), 15, 45, and 75 min following the completion of SNP infusion; 1 ml of arterial blood samples was collected in chilled ethylenediaminetetraacetic acid tubes, and the plasma was separated by centrifugation at 4°C for 10 min with 3000g, flash frozen, and stored at − 80°C.
Hormone analyses
Fetal plasma cortisol was measured using a commercially available radioimmunoassay (RIA; DPC Coat-A-Count Radioimmunoassay, Diagnostic Products, Los Angeles, CA, USA) according to the manufacturer's protocol. RIA sensitivity was 5.4 nmol l− 1. For plasma pools measuring 27.6 and 138 nmol l− 1, intraassay coefficients of variation were 6.91% and 6.94%, respectively.
Statistical analysis
Normal distribution of all data was tested by the Shapiro–Wilk test. Following this, within-group analysis was done by Friedman's ANOVA and/or Wilcoxon paired test applied to maternal cortisol responses, maternal area under the curve (AUC) values as well as cortisol concentrations during the hypotensive challenge. Differences in hormone values between the treatment and control groups within the respective hypotensive challenges and differences between fetal AUC values were tested for significance by Kruskal–Wallis ANOVA followed by post-hoc analysis with Mann–Whitney U-test. P-values were adjusted using the Bonferroni–Shaffner method. Significance was assumed at a P value < 0.05. All values are given as mean ± SEM. The integrated cortisol response was estimated using the AUC in accordance with the formula:
Results
Maternal stress response to isolation
Maternal cortisol concentrations differed at baseline (Friedman's ANOVA: χ2 = 19.87, df = 4, P < 0.001, ). Maternal cortisol concentrations were elevated prior to the first bout of isolation at 30 dGA compared to the following isolation bouts (Wilcoxon: all P < 0.001, ). Maternal baseline cortisol concentrations did not differ between 44 and 110 dGA (). Repeated isolation resulted for each isolation bout in an immediate (Wilcoxon: all P < 0.001, − 30 vs. +15 min, ) and sustained (Wilcoxon: all P < 0.001, isolation vs. baseline, ) maternal cortisol response. For stress in early gestation, the first isolation bout was associated with the greatest maternal cortisol increase (Wilcoxon: all P < 0.001, AUC of first vs. following isolation bouts, ) which, however, decreased over time (Wilcoxon: all P < 0.001, 30 dGA vs. 88 dGA, ). In the late stress group, maternal cortisol concentrations at 110 dGA were similar to concentrations at 72 and 88 dGA in the early stress group ().
Figure 1. Maternal cortisol response profiles in venous blood samples from pregnant sheep exposed to repeated isolation stress for 3 h twice a week in early gestation [30–100 days gestation (dGA), n = 18]. Data are mean ± SEM; *P < 0.001, 30 dGA vs. 88 dGA; # P < 0.001, 30 dGA vs. the following dGA; $ represents all groups, P < 0.001, − 30 min vs. 15 min; Friedman's ANOVA and/or Wilcoxon paired test.
![Figure 1. Maternal cortisol response profiles in venous blood samples from pregnant sheep exposed to repeated isolation stress for 3 h twice a week in early gestation [30–100 days gestation (dGA), n = 18]. Data are mean ± SEM; *P < 0.001, 30 dGA vs. 88 dGA; # P < 0.001, 30 dGA vs. the following dGA; $ represents all groups, P < 0.001, − 30 min vs. 15 min; Friedman's ANOVA and/or Wilcoxon paired test.](/cms/asset/16a19b5f-30cd-4daf-aa1d-1dc1a7ff5a77/ists_a_686541_f0001_b.gif)
Figure 2. Integrated maternal cortisol response in pregnant sheep exposed to repeated isolation stress for 3 h twice a week at selected isolation bouts in early [30–100 days gestation (dGA), n = 18] and late (100–120 dGA, n = 7) gestation. Data are mean ± SEM; *P < 0.001 compared to baseline; # P < 0.001 compared to 30 dGA; § P < 0.001 compared to later gestational ages; Wilcoxon paired test.
![Figure 2. Integrated maternal cortisol response in pregnant sheep exposed to repeated isolation stress for 3 h twice a week at selected isolation bouts in early [30–100 days gestation (dGA), n = 18] and late (100–120 dGA, n = 7) gestation. Data are mean ± SEM; *P < 0.001 compared to baseline; # P < 0.001 compared to 30 dGA; § P < 0.001 compared to later gestational ages; Wilcoxon paired test.](/cms/asset/b1eb4d43-2b42-4f0f-a098-d85f9176aefa/ists_a_686541_f0002_b.gif)
Fetal stress response to hypotensive challenge
There were no differences in levels of arterial blood gases and pH values between the groups at 112 and 129 dGA. Moreover, both parameters were within the physiological range (). FBP increased and fetal heart rate (FHR) decreased in the control, early stress, and BM groups with regard to the development of the cardiovascular system between 112 and 129 dGA (Wilcoxon: all P < 0.05, ). However, basal FHR and basal FBP did not differ among the groups ().
Table I. Fetal physiological parameters before hypotensive challenge.
Fetal basal cortisol concentrations were similar between 112 and 129 dGA in the control, early stress, and BM groups and did not differ among the four groups at 112 and 129 dGA (). Exposure to SNP produced an immediate decrease in FBP compared to baseline in all groups at 112 and 129 dGA, which was associated with an immediate increase in FHR (Wilcoxon: all P < 0.05, ). Minimum FBP was slightly lower in early stressed fetuses at 112 dGA and in early and late stressed fetuses at 129 dGA (Mann–Whitney: all P < 0.05, ), but not in BM-treated fetuses compared to controls. FHR increase after SNP exposure was greater in early stressed fetuses at 112 dGA and in early and late stressed fetuses at 129 dGA compared to controls (Mann–Whitney: all P < 0.05, ).
Figure 3. Fetal carotid arterial blood cortisol responses to hypotensive challenge induced by fetal jugular vein infusion of SNP at 112 and 129 days gestation (dGA) in controls (n = 7), early (30–100 dGA, n = 7), and late (100–120 dGA, n = 7) isolation stressed ewes, and following maternal BM administration at 106/107 and 112/113 dGA (n = 7). Data are mean ± SEM; *P < 0.05 compared to controls; § P < 0.05 compared to baseline; # P < 0.05 compared to control, late stress, or BM groups, 15 min after the end of SNP infusion; Kruskal–Wallis ANOVA, Mann–Whitney U-test, and/or Wilcoxon paired test.
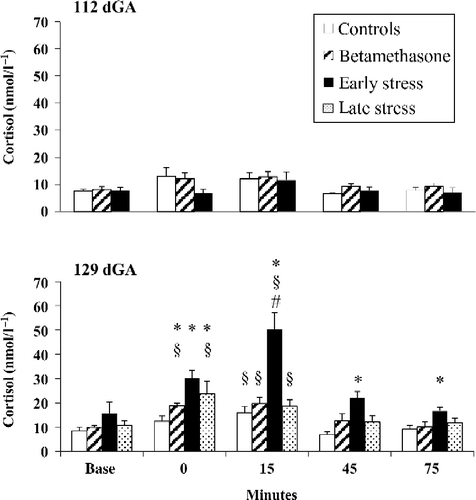
Table II. Fetal physiological parameters during hypotensive challenge.
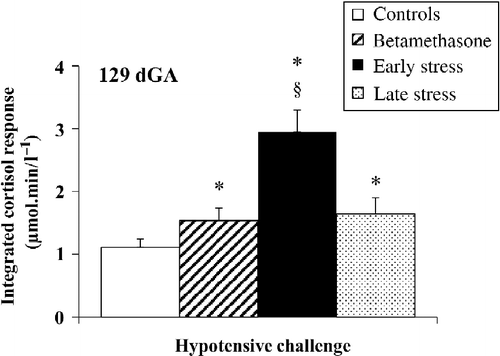
At 112 dGA, administration of SNP did not lead to increased cortisol concentrations (). In contrast to the values at 112 dGA, SNP led to an increase of plasma cortisol concentrations in all groups at 129 dGA compared to baseline (Wilcoxon: all P < 0.05, baseline vs. 15 min, ). Moreover, at the end of SNP administration, the cortisol increase was greater in the early and late stress, and BM groups compared to that of controls (Mann–Whitney: all P < 0.05, ). Indeed, 15 min after the SNP infusion, fetal cortisol concentrations differed among the four groups [Kruskal–Wallis: H(3) = 15.91, P = 0.012], and the early stress group showed the greatest cortisol response (Mann–Whitney: all P < 0.05, ). This exaggerated cortisol response in the early stressed group was prolonged for up to 75 min after the end of the infusion procedure (Mann–Whitney: P < 0.05, early stress vs. controls, ). In agreement with this, the integrated fetal cortisol response differed among the four groups [Kruskal–Wallis: H(3) = 16.04, P = 0.001, and was greater in the early and late stress, and BM groups than in controls with the highest integrated cortisol response found in the early stress group (Mann–Whitney: all P < 0.05, ).
Figure 4. Integrated fetal carotid arterial blood cortisol responses to hypotensive challenge at 129 days gestation (dGA) induced by fetal jugular vein infusion of SNP in controls (n = 7), early (30–100 dGA, n = 7), and late (100–120 dGA, n = 7) isolation stressed ewes, and following BM administration at 106/107 and 112/113 dGA (n = 7). Data are mean ± SEM; *P < 0.05 compared to controls; § P < 0.05 compared to late stress or BM; Kruskal–Wallis ANOVA and Mann–Whitney U-test.
Discussion
The aim of this study was to examine the development of fetal HPAA function after the exposure of sheep to repeated isolation stress during early and late pregnancy. We have shown that repeated stress during pregnancy leads to an increased fetal cortisol response at the end of gestation, which is dependent on the timing of the stress experience. Early repeated stress during the first and second trimester (30–100 dGA) had a more pronounced effect on the increase in fetal cortisol concentrations than late stress or BM administration during the third trimester (100–120 dGA). Our data showed that exposure to maternal stress and synthetic GCs alters the set point of HPAA function and increases HPAA activity at the end of gestation in an analogous manner.
We used isolation as a typical stress paradigm for inducing early and late stress in pregnant ewes. The significant and manifest increase in maternal circulating cortisol concentrations during each stress time period was at least sevenfold compared to non-stressed ewes. However, with advancing pregnancy, we found a slight decrease in maternal cortisol responses. This decreased response is in accordance with other studies (Coppinger et al. Citation1991; Roussel et al. Citation2004) and signifies either habituation to isolation stress or a pregnancy-related decrease of sensitivity to stress. A comparison between the cortisol responses of the late stress group at 110 dGA (10 days after the start of stress) and of the early stress group at 88 dGA, the last time point of cortisol assessment (58 days after the start of stress), showed essentially no difference. This indicates that the reduced maternal cortisol response is more likely to be pregnancy-related rather than an effect of habituation. Indeed, pregnant ewes show a decrease in acute anxiety over time (Vierin and Bouissou Citation2001). Similarly, the maternal response of the HPA axis to stressful challenges also decreases with advancing pregnancy in humans (de Weerth and Buitelaar Citation2005).
The baseline cortisol measurement of the first isolation bout was elevated compared to the following baselines, reflecting the novelty stress of blood sampling. The rapid adaptation to the blood sampling procedure compared to the low adaptation to the isolation stress is also reflected in the finding that the third blood sampling during the first isolation bout did not provide additional stress for the ewes (). In the light of the total amount of isolation stress perceived, the contribution of stress induced by blood sampling was likely to be negligible.
We applied SNP to induce a hypotensive challenge in order to measure HPAA responses in fetuses. Fetal exposure to hypotension is a clinically relevant challenge to human fetuses that can occur during birth, through intrauterine infection, and in high-risk pregnancies (Low Citation2004). Induction of hypotension in fetal sheep using SNP to test HPAA activity has also been used successfully in other studies (Wood Citation1986; Frasch et al. Citation2007).
FBP increased and FHR decreased slightly over the experimental period, indicating physiological maturation of the cardiovascular system, and this is in line with patterns of change reported by others (Unno et al. Citation1999). SNP led to a significant FBP decrease in all four study groups, which was slightly more pronounced in early and late stressed fetuses than in controls and BM-treated fetuses. Consecutively, early and late stressed fetuses showed a slightly larger reflex increase in FHR. The somewhat more pronounced decrease of FBP in response to SNP in the stressed ewes could have led to a stronger HPAA response (Wood Citation1986), with a consecutively greater cortisol increase. But, as early and late stressed fetuses did not show a similar increase in cortisol concentrations, the slightly more pronounced FBP decrease did not substantially contribute to the higher cortisol increase.
Our data are in agreement with clinical studies showing that development of HPAA function is vulnerable to maternal stress during fetal life, and where early stress seems to generate more pronounced effects than late stress (Van den Bergh et al. Citation2008). This is of particular interest, as fetal HPAA function develops during late gestation (Challis et al. Citation2001), and GRs including binding sites and GR mRNA are not detectable before 70 dGA (Rose et al. Citation1985; Yang et al. Citation1990; Matthews et al. Citation1995). The early stress period in our study lay between early and mid-gestation (30–100 dGA). Hence, either the effects of prenatal stress on the development of the HPAA are mediated via unknown mechanisms or only the late phase of our early stress period sufficiently affected HPAA development. The former assumption is supported by the study of Braun et al. (Citation2009) who showed that dexamethasone treatment at 40 dGA leads to increased HPAA activity at the end of gestation. In that study, the effects of dexamethasone treatment within 36 h contradict the notion that HPAA hyperactivation is a consequence of the prolonged impact of stress (70 days in early vs. 20 days in late stressed fetuses). This view is supported by the finding that 3 weeks of repeated isolation stress during late gestation causes similar effects as two doses of BM twice, each 1 week apart.
The effects of early stress on the development of the HPAA were not detectable at 112 dGA but at 129 dGA, which is probably due to the commencement of physiological maturation of the HPAA occurring after 112 dGA (Challis et al. Citation2001). Early stress did not lead to an earlier beginning of HPAA maturation indicating that early stress alters the set point of the HPAA rather than affecting its maturation. In a previous study, we have shown that resetting the set point occurs at the central level of the HPAA (CitationSchwab et al., in press). Mechanisms responsible for this resetting are thought to be associated with changes in the expression of GR, involving negative feedback regulation of the HPAA at the level of the limbic system, hypothalamus, or pituitary (Matthews Citation2002), which likely result from epigenetic changes in gene methylation (Glover et al. Citation2010). However, the exact mechanism as to how GCs contribute to GR gene methylation is still unclear.
Our data show that prenatal stress and BM treatment during late pregnancy have similar effects on HPAA activity. This is remarkable as about 90% of maternal cortisol, but not BM, is inactivated to cortisone by placental 11β-hydroxysteroid dehydrogenase (11β-HSD2). Thus, wide fluctuations in maternal cortisol concentrations are translated into minor fluctuations in fetal cortisol load (Benediktsson et al. Citation1997). As the fetal adrenals are inactive until maturation of the HPAA at the end of gestation (Challis et al. Citation2001), the remaining 10% of maternal cortisol crossing the placenta results in sufficient fetal cortisol concentrations to instigate HPAA hyperactivation at the end of gestation. Admittedly, a recent study showed that repeated stress during pregnancy reduces placental 11β-HSD2 in rats (Mairesse et al. Citation2007). Yet, the differing pharmacologic and metabolic characteristics of BM and cortisol make it difficult to directly compare their effects on the HPAA. In contrast to cortisol, BM crosses the placenta without appreciable inactivation (Blanford and Murphy Citation1977; Kajantie et al. Citation2004); its potency is 30 times higher than cortisol (Axelrod Citation1976); and it binds exclusively to GR, whereas cortisol binds to GR and mineralocorticoid receptors (Schwab et al. Citation2000).
Maternal stress effects on the fetus may not only be transferred by GCs that cross the placenta (Van den Bergh et al. Citation2005), but also by an impaired uterine artery blood flow, possibly by catecholamine-mediated vasoconstriction (Teixeira et al. Citation1999). This potentially reduces fetal oxygen supply and may affect indirectly HPAA development. In summary, the current study bridges research relating to the impact of prenatal maternal stress (PMS) and synthetic GC treatment on the development of the HPAA, in that it combines both a stressor and exogenous GC exposure in one experimental setting. In line with other clinical studies, our data show that development of HPAA function, which matures during late gestation, is sensitive to inappropriate levels of GCs during preceding periods of fetal life. Earlier periods of gestation are particularly vulnerable. Exposure to endogenous or synthetic GCs during late gestation has similar effects, although transfer of endogenous GCs through the placental barrier is extremely limited. Finally, GCs reset the set point of the HPAA rather than having an effect on its maturation.
Acknowledgment
This work was supported by the Max Kade Foundation and HL068649.
Declaration of interest : The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Alexander BT. 2006. Fetal programming of hypertension. Am J Physiol Regul Integr Comp Physiol. 290:R1–R10.
- Axelrod L. 1976. Glucocorticoid therapy. Medicine (Baltimore). 55:39–65.
- Benediktsson R, Calder AA, Edwards CR, Seckl JR. 1997. Placental 11 beta-hydroxysteroid dehydrogenase: A key regulator of fetal glucocorticoid exposure. Clin Endocrinol (Oxf). 46:161–166.
- Beydoun H, Saftlas AF. 2008. Physical and mental health outcomes of prenatal maternal stress in human and animal studies: A review of recent evidence. Paediatr Perinat Epidemiol. 22:438–466.
- Blanford AT, Murphy BE. 1977. In vitro metabolism of prednisolone, dexamethasone, betamethasone, and cortisol by the human placenta. Am J Obstet Gynecol. 127:264–267.
- Braun T, Li S, Sloboda DM, Li W, Audette MC, Moss TJ, Matthews SG, Polglase G, Nitsos I, Newnham JP, Challis JR. 2009. Effects of maternal dexamethasone treatment in early pregnancy on pituitary–adrenal axis in fetal sheep. Endocrinology. 150:5466–5477.
- Challis JR, Sloboda D, Matthews SG, Holloway A, Alfaidy N, Patel FA, Whittle W, Fraser M, Moss TJ, Newnham J. 2001. The fetal placental hypothalamic–pituitary–adrenal (HPA) axis, parturition and post natal health. Mol Cell Endocrinol. 185:135–144.
- Coe CL, Kramer M, Czeh B, Gould E, Reeves AJ, Kirschbaum C, Fuchs E. 2003. Prenatal stress diminishes neurogenesis in the dentate gyrus of juvenile rhesus monkeys. Biol Psychol. 54:1025–1034.
- Coppinger TR, Minton JE, Reddy PG, Blecha F. 1991. Repeated restraint and isolation stress in lambs increases pituitary–adrenal secretions and reduces cell-mediated-immunity. J Anim Sci. 69:2808–2814.
- Cottrell EC, Seckl JR. 2009. Prenatal stress, glucocorticoids and the programming of adult disease. Front Behav Neurosci. 3:19.
- de Weerth C, Buitelaar JK. 2005. Physiological stress reactivity in human pregnancy – a review. Neurosci Biobehav Rev. 29:295–312.
- Frasch MG, Muller T, Wicher C, Weiss C, Lohle M, Schwab K, Schubert H, Nathanielsz PW, Witte OW, Schwab M. 2007. Fetal body weight and the development of the control of the cardiovascular system in fetal sheep. J Physiol. 579:893–907.
- Glover V, Miles R, Matta S, Modi N, Stevenson J. 2005. Glucocorticoid exposure in preterm babies predicts saliva cortisol response to immunization at 4 months. Pediatr Res. 58:1233–1237.
- Glover V, O'Connor TG, O'Donnell K. 2010. Prenatal stress and the programming of the HPA axis. Neurosci Biobehav Rev. 35:17–22.
- Kajantie E, Raivio T, Janne OA, Hovi P, Dunkel L, Andersson S. 2004. Circulating glucocorticoid bioactivity in the preterm newborn after antenatal betamethasone treatment. J Clin Endocrinol Metab. 89:3999–4003.
- Liggins GC, Howie RN. 1972. A controlled trial of antepartum glucocorticoid treatment for prevention of the respiratory distress syndrome in premature infants. Pediatrics. 50:515–525.
- Low JA. 2004. Determining the contribution of asphyxia to brain damage in the neonate. J Obstet Gynaecol Res. 30:276–286.
- Mairesse J, Lesage J, Breton C, Breant B, Hahn T, Darnaudery M, Dickson SL, Seckl J, Blondeau B, Vieau D, Maccari S, Viltart O. 2007. Maternal stress alters endocrine function of the feto-placental unit in rats. Am J Physiol Endocrinol Metab. 292:E1526–E1533.
- Matthews SG. 2002. Early programming of the hypothalamo–pituitary–adrenal axis. Trends Endocrinol Metab. 13:373–380.
- Matthews SG, Yang K, Challis JR. 1995. Changes in glucocorticoid receptor mRNA in the developing ovine pituitary and the effects of exogenous cortisol. J Endocrinol. 144:483–490.
- Nuyt AM. 2008. Mechanisms underlying developmental programming of elevated blood pressure and vascular dysfunction: Evidence from human studies and experimental animal models. Clin Sci (Lond). 114:1–17.
- Polyakov A, Cohen S, Baum M, Trickey D, Jolley D, Wallace EM. 2007. Patterns of antenatal corticosteroid prescribing 1998–2004. Aust N Z J Obstet Gynaecol. 47:42–45.
- Rose JC, Kute TE, Winkler L. 1985. Glucocorticoid receptors in sheep brain tissues during development. Am J Physiol. 249:E345–E349.
- Roussel S, Hemsworth PH, Boissy A, Duvaux-Ponter C. 2004. Effects of repeated stress during pregnancy in ewes on the behavioural and physiological responses to stressful events and birth weight of their offspring. Appl Anim Behav Sci. 85:259–276.
- Roussel S, Boissy A, Montigny D, Hemsworth PH, Duvaux-Ponter C. 2005. Gender-specific effects of prenatal stress on emotional reactivity and stress physiology of goat kids. Horm Behav. 47:256–266.
- Schwab M, Roedel M, Anwar MA, Muller T, Schubert H, Buchwalder LF, Walter B, Nathalielsz W. 2000. Effects of betamethasone administration to the fetal sheep in late gestation on fetal cerebral blood flow. J Physiol. 528:619–632.
- Schwab M, Schmidt K, Roedel M, Mueller T, Schubert H, Anwar MA, Nathaniels PW. 2001. Non-linear changes of electrocortical activity after antenatal betamethasone treatment in fetal sheep. J Physiol. 531:535–543.
- Schwab M, Coksaygan T, Rakers F, Nathaniels PW. Glucocorticoid exposure of sheep at 0.7 to 0.75 gestation augments late gestation fetal stress responses. Am J Obstet Gynecol. in press doi:10.1016/j.ajog.2011.11.006.
- Sloboda DM, Moss TJ, Gurrin LC, Newnham JP, Challis JR. 2002. The effect of prenatal betamethasone administration on postnatal ovine hypothalamic–pituitary–adrenal function. J Endocrinol. 172:71–81.
- Tegethoff M, Pryce C, Meinlschmidt G. 2009. Effects of intrauterine exposure to synthetic glucocorticoids on fetal, newborn, and infant hypothalamic–pituitary–adrenal axis function in humans: A systematic review. Endocr Rev. 30:753–789.
- Teixeira JM, Fisk NM, Glover V. 1999. Association between maternal anxiety in pregnancy and increased uterine artery resistance index: Cohort based study. BMJ. 318:153–157.
- Unno N, Wong CH, Jenkins SL, Wentworth RA, Ding XY, Li C, Robertson SS, Smotherman WP, Nathanielsz PW. 1999. Blood pressure and heart rate in the ovine fetus: Ontogenic changes and effects of fetal adrenalectomy. Am J Physiol. 276:H248–H256.
- Van den Bergh BR, Mulder EJ, Mennes M, Glover V. 2005. Antenatal maternal anxiety and stress and the neurobehavioural development of the fetus and child: Links and possible mechanisms. A review. Neurosci Biobehav Rev. 29:237–258.
- Van den Bergh BR, Van Calster B, Smits T, Van Huffel S, Lagae L. 2008. Antenatal maternal anxiety is related to HPA-axis dysregulation and self-reported depressive symptoms in adolescence: A prospective study on the fetal origins of depressed mood. Neuropsychopharmacol. 33:536–545.
- Vierin M, Bouissou MF. 2001. Pregnancy is associated with low fear reactions in ewes. Physiol Behav. 72:579–587.
- Wood CE. 1986. ACTH, cortisol, and renin responses to arterial hypotension in sheep. Am J Physiol. 251:R18–R22.
- Yang K, Jones SA, Challis JR. 1990. Changes in glucocorticoid receptor number in the hypothalamus and pituitary of the sheep fetus with gestational age and after adrenocorticotropin treatment. Endocrinology. 126:11–17.