Abstract
Although stress-induced hyperlipidemia and increased oxidative stress have been reported and implicated in etiology of atherosclerosis, experimental evidence for stress-induced atherosclerotic development concomitant with these alterations is lacking. In this study, exposure of adult male albino Wistar rats (Rattus norvegicus) to restraint for 1 h and after a gap of 4 h to forced swimming for 15 min every day for 2, 4, or 24 weeks resulted in a duration of exposure-dependent hyperlipidemia as shown by significant increases in concentrations of blood cholesterol, low-density lipoproteins, and triglycerides and decrease in high-density lipoprotein concomitant with increased oxidative stress as indicated by decrease in hepatic antioxidant enzyme activities and increase in lipid peroxidation in the liver, kidney, and heart. These alterations were accompanied by development of fibrous layer, formation of foam cells, reduction in elastic fibers, and accumulation of Oil-Red-O-positive lipid droplets in the intima of thoracic aorta following 24 weeks of stress exposure, but not after 4 weeks. The study demonstrates for the first time that (i) chronic stress-induced hyperlipidemia and oxidative damage are coupled with atherosclerotic development in rats fed with normal diet and (ii) chronic stress effects prevail even after the cessation of stress exposure as indicated by high concentration of blood cholesterol and reduced hepatic superoxide dismutase activity 20 weeks after 2 or 4 weeks of stress. This study exemplifies long-term allostatic regulation leading to a pathological state, with long-term hyperlipidemia and oxidative damage from chronic stress resulting in atherosclerosis.
Introduction
Stress is a state of altered homeostasis, and the ability to cope with stress is a crucial determinant of health and disease. Chronic stress is known to alter the blood lipid profile. A significant elevation in circulating concentrations of cholesterol, low-density lipoproteins (LDLs), very-low-density lipoproteins (VLDL), and triglycerides (TGs) following immobilization in rabbits (Lata et al. Citation2004) and apoE− / − mice (Gu et al. Citation2009) has been reported. Decreased high-density lipoprotein (HDL) concentration following exposure to immobilization (Ruiz de Gordoa et al. Citation1994), chronic unpredictable stressors (Nayanatara et al. Citation2009) and restraint (Akhtar et al. Citation2008) in rats has been found. Similarly, emotional stressors such as workload problems, depression, anger, and social isolation cause alterations in the blood lipid profile in humans (Richards et al. Citation2000). In addition, preferential expression of genes related to lipid metabolism following immobilization in rats has also been reported (Sato et al. Citation2006). Furthermore, stress also causes leakage of cytoplasmic enzymes, glutamate pyruvate transaminase (GPT), and glutamate oxaloacetate transaminase (GOT) into the blood in humans (Kayashima et al. Citation1995) and laboratory animals (Sen et al. Citation1992; D'Souza et al. Citation2004), indicating tissue damage. Significant increase in the weight of the heart and adrenal glands, heart rate, and activities of serum glutamate pyruvate transaminase (SGPT) and serum glutamate oxaloacetate transaminase (SGOT) was observed following restraint (Sen et al. Citation1992) and isolation or immobilization (D'Souza et al. Citation2004) in rats.
Moreover, stress also causes an increase in the production of free oxygen radicals (Lakshmi et al. Citation2009). An imbalance between oxidants and antioxidants has been implicated in various disorders including neurodegenerative diseases, gastric ulcerogenesis, and impaired cognitive function (Nadeem et al. Citation2006). Several studies have shown altered antioxidant status due to stress. For instance, restraint, forced swimming, exposure to flashing light, isolation (Manoli et al. Citation2000), and repeated restraint (Nayanatara et al. Citation2005; Bhat et al. Citation2007; Zafir and Banu Citation2009) increase lipid peroxidation and decrease activities of antioxidant enzymes in rat brain. However, Bhattacharya et al. (Citation2001) reported increased superoxide dismutase (SOD) activity though there was a decrease in the activities of catalase and glutathione peroxidase (GPx) following foot shock in the brain of rats. In addition, an increase in the concentration of thiobarbituric acid reactive substances in the cerebral cortex (Fontella et al. Citation2005) and levels of plasma peroxides (Nadeem et al. Citation2006) in rats has been observed following restraint and immobilization.
Adaptive responses of the body to stressors have been termed “allostasis” (McEwen Citation2002), which involves compensatory alterations in physiological variables, overriding local feedback (Sterling Citation2004). Allostatic regulation is through mediators produced by the hypothalamic–pitutary–adrenal (HPA) axis, autonomic nervous system, and immune system. With prolonged stress, the mediators of allostasis are considered to produce wear and tear of the body, or “allostatic load” (McEwen Citation2002). This includes stress-induced alterations in blood lipid profile and oxidative damage. However, whether long-term alterations in lipid profile concomitant with altered antioxidant status lead to severe consequence such as atherosclerosis is not clear, although hyperlipidemia (Rainey et al. Citation1992) and oxidative stress (Lakshmi et al. Citation2009) induced by conditions other than stress can cause atherosclerosis. In earlier studies, stressors such as isolation in cynomolgus monkeys (Macaca fascicularis; Shively et al. Citation1989) and social deprivation in male Apo− / − mice for 20 weeks (Bernberg et al. Citation2008) resulted in atherosclerotic development. However, stress alone was not implicated in atherosclerotic development in these studies because the monkeys were fed an atherogenic diet coupled with isolation, and the mice were of a mutant strain. Kaplan et al. (Citation1983) reported development of coronary arterial atherosclerosis, without alterations in serum lipids following social stress in monkeys and suggested that psychosocial factors may influence atherogenesis in the absence of hyperlipidemia. However, whether oxidative stress was also evident in chronically stressed monkeys was not investigated (Kaplan et al. Citation1983). Furthermore, rats subjected to chronic unpredictable stress for 15 days showed hypertrophy of the intima and tunica media of thoracic aorta, increasing serum lipid concentration and atherogenic index without changes in HDL concentration (Neves et al. Citation2009), although there was no evidence for atherosclerotic plaques and oxidative damage was not investigated.
This study investigated the hypothesis that chronic intermittent stress can induce alterations in blood lipid profile and antioxidant status that are dependent on duration of stressor exposure and accompanied by atherosclerotic development in the thoracic aorta. The study tested whether stress-induced changes in lipid profile and antioxidant status are reversible.
Materials and methods
Animals
Adult male Wistar rats weighing 180–200 g obtained from the central animal facility, University of Mysore, were housed two or three rats per cage in a 12:12-h light–dark cycle (lights on 07:00–19:00 h), temperature 27 ± 2°C. The rats had free access to standard rat chow and water ad libitum. All experiments were conducted in accordance with standard ethical guidelines and were approved by the institutional animal ethics committee.
Stressors
Two kinds of stressors were used (Grissom et al. Citation2008): (i) restraint in a Plexiglas cylindrical restrainer of 22.3 cm long, 6.7 cm diameter, closed with a perforated lid, for 1 h per episode and (ii) forced swimming for 15 min per episode in a glass jar of 45.7 cm high, 22.2 cm outer diameter, water depth of 30 cm, and temperature of 27 ± 2°C.
Experimental plan and procedures
At the commencement of the experiment, 37 adult male rats were randomly chosen from the colony and weighed (). Five randomly chosen rats were killed by exposing to ether (anesthetic ether, I.P. Hydroquinone 0.002% w/v) to obtain baseline data (initial controls). The thorax and abdomen were opened, a blood sample was collected with a syringe from the vena cava, and the liver, kidney, heart, and adrenal glands were removed. Blood was transferred to an eppendorf tube containing 0.1% ethylenediaminetetraacetic acid, and plasma was separated by centrifugation. Biochemical analyses were carried out to obtain baseline data. Remaining rats were randomly divided into two groups, the no stress group (controls; n = 05) and the stress group (n = 25). Rats in the stress group were restrained for 1 h, and 4 h later they were subjected to forced swimming for 15 min at random times between 07:00 and 19:00 h, every day for 24 weeks. To minimize habituation, the sequence of stressors and timing of stress exposure were randomly changed every day. Five rats after 2 weeks exposure, and five rats after 4 weeks exposure in the stress group were killed, as above. The remaining rats were killed after 24 weeks exposure to the stressors, and the no stress controls were killed at the same time. Five rats exposed to the stressors for 2 weeks and five rats exposed for 4 weeks were then maintained without exposure to stressors (recovery groups) until the 24th week when they were killed. Blood samples and tissues were collected as above. Postmortem body weight and adrenal gland weight were recorded. Rats were killed 16 h after the last exposure to a stressor.
Figure 1. Experimental groups showing period of stress exposure or recovery. Blood and tissue samples were collected at autopsy.
Biochemical analyses
Blood samples were centrifuged at 604 g for 15 min, and plasma was separated and stored at − 20°C. Plasma concentrations of cholesterol (assay sensitivity 0.14 μmol/l), HDL (sensitivity 2.5 mg/dl) and TGs (sensitivity 0.4 mmol/l) and activities of plasma GOT (sensitivity 350 U/l) and lactic dehydrogenase (LDH; sensitivity 2400 U/l) were determined using kits (Agappe diagnostics, Kerala, India). Activity of plasma glutamate pyruvate transaminase was determined following the method of Segal et al. (Citation1962; sensitivity 0.30 U/l). Plasma concentrations of LDL and VLDL were determined by the method of Friedewald et al. (Citation1972). Adrenal glands were homogenized to estimate the activity of 3β-hydroxysteroid dehydrogenase (3β-HSDH) by measuring the rate of conversion of pregnenolone into progesterone (Shivanandappa and Venkatesh Citation1997; sensitivity 0.04 nmol/mg of protein). Liver homogenate (10%) was used to determine the activities of γ-glutamyl transferase (GGT; Rosalki and Tarlow Citation1974; sensitivity 9 U/l), SOD (Marklund and Marklund Citation1974; sensitivity 16 unit/mg protein), glutathione-S-transferase (GST; Habig et al. Citation1974; sensitivity 11 U/mg of protein), glutathione reductase (GR; Carlberg and Mannervik Citation1985; sensitivity 250 U/mg of protein), catalase (Aebi Citation1984; sensitivity 0.25–4 nmol/min/ml), and GPx (Tappel Citation1978; sensitivity 0.5 mU/ml). The concentration of malondialdehyde in the liver, heart, and kidney was determined (Ohkawa et al. Citation1979; sensitivity 4 nmol/mg protein). Intra- and interassay variability tests were conducted for all the assays, and variability was in the range of 3–5%.
Histology of aorta
A portion of the thoracic aorta (aortic root) was removed at autopsy and fixed in 10% buffered formalin. The fixed aorta was stained with Sudan IV to reveal sudanophilic plaques (Holman et al. Citation1958). After Sudan IV staining the aorta was embedded in paraffin wax, 5-μm-thick transverse sections were cut and stained with hematoxylin–eosin. Lipid deposition and elastic fibers and collagen in the blood vessel were detected using Oil Red O (Preece Citation1972) and van Gieson staining (Culling Citation1963), respectively.
The measurements of intima thickness (IT), media thickness (MT), total intima–media thickness (IMT), and wall thickness were made in hematoxylin–eosin stained sections of thoracic aorta under a microscope (100 × magnification; Olympus BX41, Tokyo, Japan) using ProgRes® CapturePro 2.7 software. The intima limit was the zone between the luminal surface and the internal elastic lamina, and a lesion comprised the entire intima including lipid cores and fibrotic components. The distance between the lumen–intima interface and the media–adventitia interface was the IMT. The total thickness of the aortic wall was the distance between the endothelium lining and the adventitia. Thickness of the different layers was measured in five randomly chosen areas per section of the blood vessel (i.e. five readings/section/layer) and from 10 randomly selected sections per rat.
Statistical analysis
Each parameter was expressed as the group mean ± SEM. One-way analysis of variance (ANOVA) followed by Duncan's multiple test was used to detect significant differences among groups; level for significance was set at p < 0.05.
Results
Body and adrenal weights
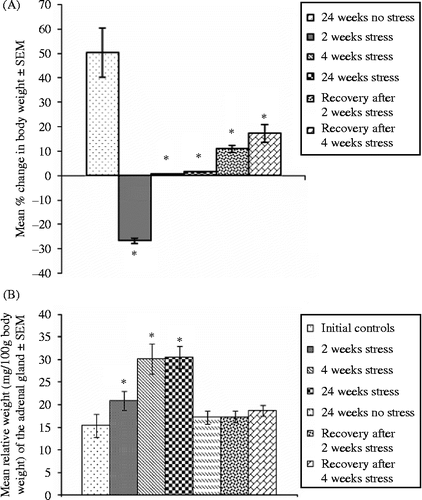
Gain in the body weight of rats exposed to stress for 4 or 24 weeks was significantly lower than controls, and there was a loss in body weight after 2 weeks stress (F = 11.12, df = 6, 34, p < 0.05, ). Rats in the recovery group gained body weight, but significantly less than controls (). There was a significant increase in relative adrenal gland weight (weight/100 g body weight) (F = 17.75, df = 6, 34, p < 0.05, ) at 2, 4, and 24 weeks of stress exposure compared to unstressed controls. The actual weight of the adrenal gland showed a significant increase in 4 and 24 weeks stressed rats, but not in rats stressed for 2 weeks or in the recovery groups, compared with controls (adrenal weight, mean ± SEM, mg, were initial controls, 25.6 ± 1.2, 2 weeks stress, 36.2 ± 1.3, 4 weeks stress, 52.4 ± 3.6, 24 weeks stress, 57.2 ± 6.3, recovery after 2 weeks stress, 32.3 ± 1.4, recovery after 4 weeks stress, 36.6 ± 2.5, no stress, 33.0 ± 4.7, F value = 10.65, df = 6, 34, p < 0.05).
Figure 2. Vertical bars are mean ± SEM. Percentage change in body weight compared to initial body weight (A) and relative adrenal weight (B) in control, stressed, and recovery groups, n = 5 rats per group. *p < 0.05 versus controls, ANOVA, and Duncan's test.
Adrenal 3β-HSDH activity
Compared to controls (basal or no stress for 24 weeks), there was a significant (ANOVA, p < 0.05; ) increase in adrenal 3β-HSDH activity in rats exposed to stress for 2 weeks, and further significant increases after 4 or 24 weeks of stressor exposure; in the recovery groups (no stress after 2 or 4 weeks exposure) adrenal 3β-HSDH activity at 24 weeks did not differ from that in the unstressed controls at 24 weeks.
Table I. Stress and adrenal 3β-HSDH activity and plasma lipid profile.
Blood lipid profile
HDL concentration was significantly decreased after stress exposure for 4 or 24 weeks compared to the 24 weeks no stress or recovery groups (ANOVA, Duncan's, p < 0.05) (). There was a significant increase in total cholesterol and LDL () concentrations after 2 weeks stressor exposure, with further significant increases after 4 or 24 weeks exposure (ANOVA, Duncan's, p < 0.05). In the recovery groups, total cholesterol and LDL concentrations were significantly less than in the group stressed for 24 weeks but were greater than in the unstressed controls (ANOVA, Duncan's, p < 0.05). TGs and VLDL concentrations were significantly increased after stress exposure for 2, 4, and 24 weeks compared to the initial control, no stress, and recovery groups, with no significant differences among groups stressed for 2, 4, or 24 weeks (ANOVA, Duncan's, p < 0.05).
Activities of hepatic antioxidant enzymes and GGT
Hepatic CAT activity was significantly decreased after stress exposure for 2 weeks and was further reduced after 4 or 24 weeks treatment compared to initial controls or the group unstressed for 24 weeks (); in the recovery groups, CAT activity was not different from the group not exposed to stress (ANOVA, Duncan's, p < 0.05). Hepatic GPx activity was significantly reduced after 24 weeks of stress exposure compared to initial controls or the group not exposed to stress for 24 weeks (ANOVA, Duncan's, p < 0.05), but was not changed after stress for 2 or 4 weeks, and was similar to controls in the recovery groups. GST activity decreased progressively with duration of stress exposure (ANOVA, Duncan's, p < 0.05) and returned to normal in the recovery groups. GR activity was significantly and similarly reduced in the groups stressed for 2, 4, or 24 weeks, as compared with initial controls and the group not exposed to stress for 24 weeks (ANOVA, Duncan's, p < 0.05); GR activity in the groups recovering from stress increased to or above the control levels. SOD activity decreased progressively with the duration of stress exposure and did not return to normal in the recovery groups (ANOVA, Duncan's, p < 0.05). GGT activity was significantly increased after stress exposure for 2 or 4 weeks compared to controls and was further increased after 24 weeks (ANOVA, Duncan's, p < 0.05). GGT activity was normalized at 24 weeks after cessation of exposure at 4 weeks, but not at 2 weeks.
Table II. Stress and activities of hepatic antioxidant enzymes and GGT.
Plasma aminotransferase and LDH activities
Plasma LDH and plasma GOT activities were significantly increased after exposure to stress for 4 weeks or 24 weeks compared to initial controls and the group unstressed for 24 weeks (ANOVA, Duncan's, p < 0.05) (); activities were normalized at 24 weeks after cessation of exposure at 2 or 4 weeks. Plasma GPT activity was significantly increased after stress exposure for 2 or 4 weeks, and further increased after 24 weeks, compared to the controls (ANOVA, Duncan's, p < 0.05). In the recovery groups, plasma GPT activity was significantly reduced compared with the stress groups but remained greater than in the controls (ANOVA, Duncan's, p < 0.05).
Table III. Stress and aminotransferases and LDH activities.
Malondialdehyde concentrations
Malondialdehyde (MDA) concentrations in the liver, heart, and kidneys progressively increased in stressed rats compared to the control groups (ANOVA, Duncan's, p < 0.05) (). In the recovery groups, MDA concentrations were decreased compared with the stress groups (ANOVA, Duncan's, p < 0.05) and normalized in liver, but in heart and kidney remained greater than in the controls.
Table IV. Stress and lipid peroxidation.
Histology of aorta
There were no marked histological changes in the thoracic aorta of rats exposed to stress for 4 weeks, but at 24 weeks histopathological changes were evident (). The aorta of controls (24 weeks not exposed to stress) did not show prominent lipid deposition following Sudan IV staining, and the sections revealed endothelial lining and intima with little collagen (). The stressed rats (24 weeks) showed dark red spots in the aortic vessel following Sudan IV staining, and the sections revealed thickening of the vessel wall and the appearance of fibrous layer in the intima (). Strikingly, foam cells were evident in the intima of the aorta of stressed rats (). The foam cells were large and irregular in shape with eccentric nuclei containing prominent nucleoli and contained minute pink granules in the cytoplasm (). Oil Red O staining showed a higher lipid content in the stressed rats compared to the control group (). van Gieson staining showed higher levels of elastin in the aorta of the control group than that of the stressed rats, as revealed by the intense blackish-brown colored staining () than in the aorta of stressed rats (). In addition, the elastic structure was disrupted and pinkish-stained collagen fibers were present in the aorta of the stressed rats (). These changes lead to the formation of atherosclerotic plaques, which consisted of a fibrous layer () beneath the endothelium () followed by a fatty streak containing foam cells () and extracellular lipids. There was a significant increase in mean thickness of the intima, media, total intima–media, and total aortic wall in rats stressed for 24 weeks, but not 4 weeks, compared to the control groups (). Of seven rats exposed to stressors for 24 weeks, two died in the 22nd week, four rats showed formation of atherosclerotic plaques (thickening of intima–media and foam cells), and the remaining rat showed slight thickening of intima without foam cells.
Figure 3. Transverse sections of similar areas of thoracic aorta of a control rat (24 weeks no stress; A and B), and a rat stressed for 24 weeks (C and D) and a part of the aorta of a stressed rat under high magnification to show a portion of an atherosclerotic plaque (E) and foam cells (arrow). Note the thickening of wall of the aorta and a fibrous layer in the vessel of the stressed rat. B and D are higher magnification images of A and C, respectively. Hematoxylin–eosin stain; AD, adventitia; EN, endothelium; FB, fibrous layer; FC, foam cells; ME, media.
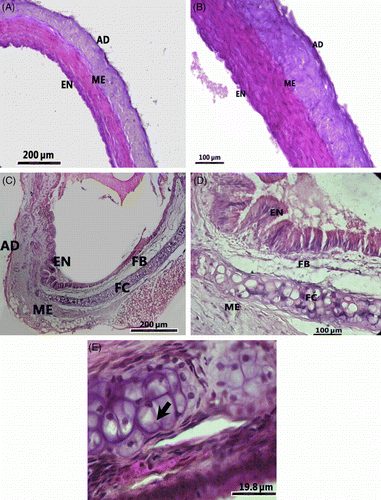
Figure 4. Transverse sections of the thoracic aorta of a control rat (24 weeks no stress; A), and a rat stressed for 24 weeks (B) stained with Oil Red O for localization of lipids. Note the Oil-Red-O-positive lipid deposition (arrow heads) in the section from a stressed rat (B). Transverse sections of the thoracic aorta of a control rat (24 weeks no stress; C), and a rat stressed for 24 weeks (D) stained with van Gieson technique to demonstrate elastic fibers (arrow head) and collagen (solid arrow). Note the presence of abundant elastic fibers in the control rat aorta (C) and depletion of elastic fibers and abundant collagen deposition in the stressed rat aorta (D).
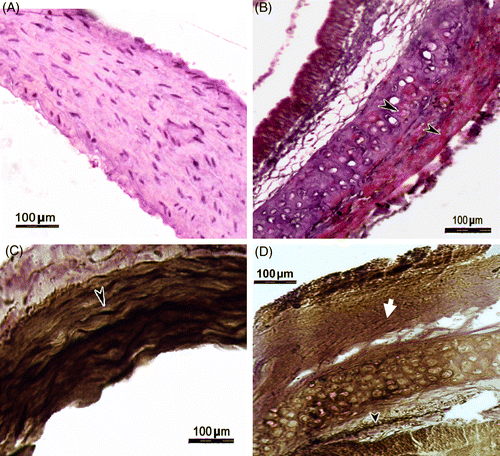
Table V. Stress and thickness of layers of thoracic aorta wall.
Discussion
In this study, exposing rats to restraint followed by forced swimming every day for 2, 4, or 24 weeks increased adrenal 3β-HSDH activity, a key enzyme in adrenal steroidogenesis, and the weight of the adrenal gland indicating stress-induced activation, with expected increased corticosterone secretion, driven by the hypothalamus and adrenocorticotrophic hormone (ACTH) (Tsigos and Chrousos Citation2002). Hence, the alterations in the blood lipid profile and antioxidant enzyme activities and lipid peroxidation that were found were associated with adrenocortical stress responses. In contrast to earlier studies, in which alterations in lipid profile (Ruiz de Gordoa et al. Citation1994; Richards et al. Citation2000; Lata et al. Citation2004; Akhtar et al. Citation2008; Gu et al. Citation2009; Nayanatara et al. Citation2009) and antioxidant status (Manoli et al. Citation2000; Fontella et al. Citation2005; Nayanatara et al. Citation2005; Nadeem et al. Citation2006; Bhat et al. Citation2007; Zafir and Banu Citation2009) were studied after a relatively shorter duration of stress exposure (maximum 30 days), this study clearly indicates that the HPA axis as well as other physiological mechanisms continued to respond to the stressors for a long period despite repeated daily exposure. As prolonged stress induces decreased food intake and loss in body weight (Zardooz et al. Citation2006), the reduced gain in body weight in rats after 4 or 24 weeks of stress exposure in this study further supports a long-term stress response.
This study clearly demonstrates that stress due to repeated restraint and forced swimming increased blood concentrations of cholesterol, TG, LDL, and VLDL and decreased HDL. Decreased activities of hepatic antioxidant enzymes coupled with increased MDA concentrations in heart, liver, and kidney at all the stress durations studied indicate hyperlipidemia and compromised antioxidant status. While forced swimming can be considered an exercise model, it is considered as a stressor because of emotional factors associated with forced exercise (Nagaraj and Jeganathan Citation1999). Hence, the metabolic alterations might be a combined effect of physical exercise and stress.
Exposure of laboratory animals to a variety of stressors (Lata et al. Citation2004; Sato et al. Citation2006; Nayanatara et al. Citation2009) and emotional stress in humans (Richards et al. Citation2000) cause an increase in serum cholesterol, TG, LDL, VLDL, and decrease in HDL concentrations. Though results of this study are similar to earlier reports, they clearly demonstrate that stress-induced increases in blood cholesterol and LDL and decrease in HDL are duration dependent, hence the alterations became more severe with increase in exposure period, which was not reported in earlier studies. It was also observed that all the components of the lipid profile did not show the same response to stress. Although TG and VLDL concentrations showed significant increases with repeated stress, the levels did not vary according to the duration of exposure. The findings revealed that the changes in TG and VLDL induced by stress are evident within a short duration of exposure to stress for 2 weeks and remain at the same level throughout the exposure period of 24 weeks. An increase in the blood concentration of cholesterol with stress might reflect release of cholesterol from adipose tissue or increased synthesis in the liver or decreased excretion; similarly, increased circulating TG levels with chronic stress might reflect lipolysis in adipose tissue, with the released fatty acids serving as substrate for TG synthesis (Luoma Citation2010).
There was a reduction in the antioxidant status with the chronic stress paradigm used here, as indicated by reduced activities of hepatic primary (CAT, SOD, GPx) and secondary (GR, GST, G6PDH) antioxidant enzymes, coupled with increased lipid peroxidation in the liver, heart, and kidney, in accordance with the earlier studies (Manoli et al. Citation2000; Fontella et al. Citation2005; Nayanatara et al. Citation2005; Nadeem et al. Citation2006; Zafir and Banu Citation2009). However, the present data indicate that antioxidant potential remains compromised even after a long exposure period of 24 weeks, which was not evident in earlier studies. Reduction in activities of primary antioxidants might be due to cross-linking or exhaustion of enzymes (Salo et al. Citation1990). In addition, increase in hepatic GGT activity following chronic stress in this experiment also indicates increased oxidative stress as GGT is involved in superoxide anion generation (Pompella et al. Citation2004), and high-GGT activity is found in human populations with increased risk for cardiovascular diseases (Emdin et al. Citation2005). Tissue injuries are expected due to high level of oxidants under stressful conditions. Indeed, in this study there was an increase in the activity of plasma GOT, plasma GPT, and LDH which leak into blood following tissue injury (Arakawa et al. Citation1997) and thereby indicate organ damage, including heart.
The unwanted effect of increased secretion of glucocorticoids (GCs) due to stress includes overproduction of reactive oxygen species (Bjelakovic et al. Citation2007), dysregulation of physiological processes, and increased lipolysis (Brindley Citation1995). GCs decrease concentration of low density lipoprotein cholesterol (LDL-C) receptors on hepatocytes (Rainey et al. Citation1992), leading to higher LDL-C concentration and increased production and secretion of VLDL in the liver (Brindley Citation1995). As effects of GCs on lipid metabolism persist for a long period (Eisenstein Citation1973), it is possible that alterations persist even after the challenge is past, resulting in chronic hyperlipidemia. In addition, GCs increase reactive oxygen radical generation in cells (McIntosh and Sapolsky Citation1996; Bjelakovic et al. Citation2007), reduce expression of primary antioxidant, SOD, on chronic exposure (McIntosh et al. Citation1998), and deplete glutathione (GSH) (Slater et al. Citation1995). Here, adrenal enlargement and increased activity of a GC synthesizing enzyme (3β-HSDH) provide evidence for increased GC production with the present chronic stress model. Such increased GC production likely provides a common pathway in this model that alters both lipid and antioxidant status, as hyperlipidemia and reduced antioxidant potential were observed in chronically stressed rats in this study, in contrast to earlier studies which either focused on lipid profile changes (Ruiz de Gordoa et al. Citation1994; Richards et al. Citation2000; Lata et al. Citation2004; Akhtar et al. Citation2008; Gu et al. Citation2009; Nayanatara et al. Citation2009) or oxidative damage (Manoli et al. Citation2000; Fontella et al. Citation2005; Nayanatara et al. Citation2005; Nadeem et al. Citation2006; Zafir and Banu Citation2009) following chronic stress exposure.
Several risk factors are implicated in the development of atherosclerosis including increased oxidized LDL-C and endothelial dysfunction induced by reactive oxygen species (Ross Citation1999) and induced by endothelial dysfunction reactive oxygen species (Ross Citation1999). HDL is a protective factor for atherosclerotic development as it reduces oxidation of LDL and helps in reverse transport of cholesterol. Hence, any factor that simultaneously causes oxidative stress and hyperlipidemia might induce atherosclerosis. Stress is most likely a factor as it results in high levels of GCs which cause dyslipidemia as well as oxidative damage as discussed above. However, though stress is implicated in pathogenesis of atherosclerosis, thus far there is no experimental evidence to link stress alone to the development of atheroma because earlier studies have used other factors along with stress to induce atherosclerosis. For instance, though monkeys showed atherosclerosis following stress due to isolation, they were fed an atherogenic diet during experimentation (Shively et al. Citation1989). Likewise, mice that developed atherosclerosis due to isolation were deficient in apolipoprotein (Bernberg et al. Citation2008). In contrast, in this study chronic exposure to stressors for different durations resulted in a significant elevation in circulating cholesterol, LDL, and TG and decrease in HDL, concomitant with increased oxidative stress (lipid peroxidation). Hence, the stress protocol used in this study was effective in simultaneously inducing both risk factors, i.e. lipid profile and oxidative damage for the development of atherosclerosis. Indeed, there was an atherosclerotic development in the aorta after 24 weeks stress exposure. A typical atherosclerotic plaque consists of a fibrous cap, accumulation of lipid droplets, a large number of foam cells in the intima and depletion of smooth muscle (Libby Citation2006). The macrophages that enter the intima region during pathogenesis of atherosclerosis accumulate oxidized LDL to become foam cells and are the hallmark of atherosclerotic lesions (Witztum and Steinberg Citation1991), whereas the normal blood vessel wall does not contain these cells. In rats stressed for 24 weeks, the aorta showed the development of a fibrous layer beneath the endothelium and a fatty streak, consisting of lipid droplets and foam cells, coupled with reduction in elastic fibers and smooth muscle and appearance of collagen fibers. Hence, our observations correspond with the description of a typical atherosclerotic plaque. Significant increases in IT, MT, and IMT and thickness of the aortic wall in rats exposed to stress for 24 weeks in this study further support atherosclerotic development as IMT is also a characteristic of atherosclerosis (Bots et al. Citation2007). Thus, this study is the first to provide experimental evidence, of a link between stress, compromised antioxidant status, and hyperlipidemia in the rat for the development of atherosclerosis. In addition, the stress protocol developed provides an animal model for studying pathogenesis and prevention of atherosclerosis. Although rats are generally considered an atherosclerosis-resistant species due to their hypo-responsiveness to dietary cholesterol, combination of a cholesterol-rich diet with other factors can induce atherosclerosis (Moghadasian Citation2002). For instance, hyperlipidemia and atherogenesis are induced by high-cholesterol/high-fat diet containing cholic acid and thiouracil (Joris et al. Citation1983; Moghadasian Citation2002; Jawien et al. Citation2004). Hence, the combined effect of prolonged hyperlipidemia due to long-term stress exposure and oxidative damage might have contributed to atherosclerotic development in rats in this study.
Normalization of stress-induced changes especially dyslipidemia, reduced antioxidant status after the cessation of the exposure, and the duration required for normalization are the important factors to be considered because of their involvement in atherosclerosis. In this study, in the recovery group rats studied 20–22 weeks after the cessation of 4 or 2 weeks of stress exposure, respectively, the blood lipid levels, i.e. HDL, TG, and VLDL, did not differ from controls, whereas cholesterol and LDL concentrations remained higher than in controls. Likewise, activities of hepatic catalase, GPx, GR, GST, and GGT and hepatic MDA concentration in the recovery groups did not differ from controls. However, lipid peroxidation (MDA concentration) in heart and kidney and hepatic SOD and plasma GOT activities remained increased compared with controls in the recovery groups. Thus, the present experimental results importantly reveal that though the majority of the parameters of lipid metabolism and the anti-oxidant system are restored to normalcy after cessation of stress exposure, some (cholesterol, LDL, lipid peroxidation) remain elevated even after a long withdrawal period (20 weeks). This observation gains importance because of pathological consequences of high concentrations of LDL-C and oxidative stress.
This study provides an example of long-term allostatic regulation leading to a pathological state. Lack of adaptation to repeated stress is a feature of allostasis (McEwen Citation2002), and many diseases are related to long-term chronic stress responses, when allostasis shifts from a healthy condition to a pathological state (Goldstein and McEwen Citation2002; McEwen Citation2002). The stress paradigm used here evidently did not result in habituation, as the stressed rats continued to show elevated adrenocortical activity, hyperlipidemia, and compromised antioxidant status, even after 24 weeks, and these changes were accompanied by atherosclerotic development (allostatic load) in thoracic aorta. These alterations may be mediated by GCs as they act as allostatic mediators (McEwen Citation2002). Another feature of allostasis is the inability to shut off allostatic responses after the challenge (stressor) is past (McEwen Citation2002). The finding here that the blood concentrations of LDL and cholesterol and hepatic MDA level remained significantly greater than in controls, even 20 weeks after cessation of exposure to the stressors, not only confirms a model of allostasis but also provides new evidence for the persistence of stress effects for a long period. With a similar regime of stress exposure in our earlier study (Nirupama et al. Citation2012), hyperglycemia remained for several weeks in rats after the cessation of stress exposure. Together, these findings support the validity of our animal model for studying allostasis and allostatic load.
Declaration of interest: The work was supported by a grant from Ministry of Human Resource Development, Government of India, through the University Grants Commission to University of Mysore, under the Institution of Excellence scheme. The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Aebi H. 1984. Catalase in vitro. Methods Enzymol. 105:121–126.
- Akhtar J, Begum S, Islam N, Begum N, Ara S. 2008. Study on restrain stress induced changes in serum lipid profile and haemoconcentration and effect of sympathetic blocker on it. Mymensingh Med J. 17 2 suppl.: S16–S21.
- Arakawa H, Kodama H, Matsuoka N, Yamaguchi I. 1997. Stress increases plasma enzyme activity in rats: Differential effects of adrenergic and cholinergic blockades. J Pharmacol Exp Ther. 280 3: 1296–1303.
- Bernberg E, Andersson IJ, Gan LM, Naylor AS, Johansson ME, Bergstrom G. 2008. Effects of social isolation and environmental enrichment on atherosclerosis in ApoE-/- mice. Stress. 11 5: 381–389.
- Bhat MS, Rao G, Murthy KD, Bhat PG. 2007. Housing in pyramid counteracts neuroendocrine and oxidative stress caused by chronic restraint in rats. Evid Based Complement Alternat Med. 4 1: 35–42.
- Bhattacharya A, Ghosal S, Bhattacharya SK. 2001. Anti-oxidant effect of Withania somnifera glycowithanolides in chronic footshock stress-induced perturbations of oxidative free radical scavenging enzymes and lipid peroxidation in rat frontal cortex and striatum. J Ethnopharmacol. 74 1: 1–6.
- Bjelakovic G, Beninati S, Pavlovic D, Kocic G, Jevtovic T, Kamenov B, Saranac LJ, Bjelakovic B, Stojanovic I, Basic J. 2007. Glucocorticoids and oxidative stress. J Basic Clin Physiol Pharmacol. 18 2: 115–127.
- Bots ML, Baldassarre D, Simon A, de Groot E, O'Leary DH, Riley W, Kastelein JJ, Grobbee DE. 2007. Carotid intima-media thickness and coronary atherosclerosis: Weak or strong relations?. Eur Heart J. 28 4: 398–406.
- Brindley DN. 1995. Role of glucocorticoids and fatty acids in the impairment of lipid metabolism observed in the metabolic syndrome. Int J Obes Relat Metab Disord. 19 Suppl. 1: S69–S75.
- Carlberg I, Mannervik B. 1985. Glutathione reductase. Methods Enzmol. 113:484–490.
- Culling CFA. 1963. Handbook of histopathological techniques. London: Butterworths207.
- D'Souza UJA, Nagaraja HS, D'Souza VM, D'Souza A, Jeganathan PS. 2004. Effect of ethanol on stress induced cardiovascular alterations in rats. The Thai J Pharm Sci. 17:80–85.
- Eisenstein A. 1973. Effects of adrenal cortical hormones on carbohydrate, protein and fat metabolism. Am J Clin Nutr. 26 1: 113–120.
- Emdin M, Pompella A, Paolicchi A. 2005. Gamma-glutamyltransferase, atherosclerosis, and cardiovascular disease: Triggering oxidative stress within the plaque. Circulation. 112 14: 2078–2080.
- Fontella FU, Siqueira IR, Vasconcellos AP, Tabajara AS, Netto CD, Dalmaz C. 2005. Repeated restraint stress induces oxidative damage in rat hippocampus. Neurochem Res. 30 1: 105–111.
- Friedewald WT, Levy RI, Fredrickson DS. 1972. Estimation of the concentration of low-density lipoprotein cholesterol in plasma, without use of the preparative ultracentrifuge. Clin Chem. 18 6: 499–502.
- Goldstein DS, McEwen B. 2002. Allostasis, haemostats and the nature of stress. Stress. 5 1: 55–58.
- Grissom N, Kerr W, Bhatnagar S. 2008. Struggling behavior during restraint is regulated by stress experience. Behav Brain Res. 191 2: 219–226.
- Gu H, Tang C, Peng K, Sun H, Yang Y. Effects of chronic mild stress on the development of atherosclerosis and expression of toll-like receptor 4 signaling pathway in adolescent apolipoprotein E knockout mice. J Biomed Biotechnol. 20091–13.
- Habig WH, Pabst MJ, Jakoby WB. 1974. Glutathione s-transferase. The first enzymatic step in mercapturic acid formation. J Biol Chem. 249 22: 7130–7139.
- Holman RL, McGill HC, Strong JP, Geer JC. 1958. Technics for studying atherosclerotic lesions. Lab Invest. 7 1: 42–47.
- Jawien J, Nastalek P, Korbut R. 2004. Mouse models of experimental atherosclerosis. J Physiol Pharmacol. 55 3: 503–517.
- Joris I, Zand T, Nunnari JJ, Kralikowks FJ, Majno G. 1983. Studies on the pathogenesis of atherosclerosis I Adhesion and emigration of mononuclear cells in the aorta of hypercholesterolemic rats. Am J Pathol. 133 3: 341–358.
- Kaplan JR, Manuck SB, Clarkson TB, Lusso FM, Taub DM, Miller EW. 1983. Social stress and atherosclerosis in normocholesterolemic monkeys. Science. 220 4598: 733–735.
- Kayashima S, Ohno H, Fujioka T, Taniguchi N, Nagata N. 1995. Leucocytosis as a marker of organ damage induced by chronic strenuous physical exercise. Eur J Appl Physiol Occup Physiol. 70 5: 413–420.
- Lakshmi SV, Padmaja G, Kuppusamy P, Kutala VK. 2009. Oxidative stress in cardiovascular disease. Ind J Biochem Biophys. 46 6: 421–440.
- Lata H, Ahuja GK, Narang APS, Walia L. 2004. Effect of immobilization stress on lipid peroxidation and lipid profile in rabbits. Ind J Clin Biochem. 19 2: 1–4.
- Libby P. 2006. Atherosclerosis: disease biology affecting the coronary vasculature. Am J Cardiol. 98 12A: 3Q–9Q.
- Luoma PV. 2010. Gene activation regresses atherosclerosis, promotes health, and enhances longevity. Lipids Health Dis. 9:67.
- Manoli LP, Gamaro GD, Silveira PP, Dalmaz C. 2000. Effect of chronic variate stress on thiobarbituric—acid reactive species and on total radical trapping potential in distinct region of rat brain. Neurochem Res. 25 7: 915–921.
- Marklund S, Marklund G. 1974. Involvement of the superoxide anion radical in the autoxidation of pyrogallol and a convenient assay for superoxide dismutase. Eur J Biochem. 47 3: 469–474.
- McEwen BS. 2002. Sex, stress and the hippocampus: Allostasis, allostatic load and the aging process. Neurobiol Aging. 23 5: 921–939.
- McIntosh LJ, Sapolsky RM. 1996. Glucocorticoids increase the accumulation of reactive oxygen species and enhance adriamycin-induced toxicity in neuronal culture. Exp Neurol. 141 2: 201–206.
- McIntosh LJ, Hong KE, Sapolsky RM. 1998. Glucocorticoids may alter antioxidant enzyme capacity in the brain: Baseline studies. Brain Res. 791 1-2: 209–214.
- Moghadasian MH. 2002. Experimental atherosclerosis: A historical overview. Life Sci. 70 8: 855–865.
- Nadeem A, Masood A, Masood N, Gilani RA, Shah AZ. 2006. Immobilization stress causes extra-cellular oxidant-antioxidant imbalance in rats: Restoration by L-NAME and vitamin E. Eur Neuropsychopharmacol. 16 4: 260–267.
- Nagaraj HS, Jeganathan PS. 1999. Forced swimming stress induced changes in the physiological and biochemical parameters in albino rats. Indian J Physiol Pharmacol. 45 1: 53–59.
- Nayanatara AK, Nagaraja HS, Anupama BK. 2005. The effect of repeated swimming stress on organ weights and lipid peroxidation in rats. Thai J Pharm Sci. 18 1: 3–9.
- Nayanatara AK, Nagaraja HS, Ramaswamy C, Bhagyalakshmi K, Bhat RM, Gowda DKM, Mantur SV. 2009. Effect of chronic unpredictable stressors on some selected lipid parameters and biochemical parameters in Wistar rats. J Chinese Clin Med. 4 2: 92–97.
- Neves VJ, Moura MJ, Tamascia ML, Ferreira R, Silva NS, Costa R, Montemor PL, Narvaes EA, Bernardes CF, Novaes PD, Marcondes FK. 2009. Proatherosclerotic effects of chronic stress in male rats: Altered phenylephrine sensitivity and nitric oxide synthase activity of aorta and circulating lipids. Stress. 12 4: 320–327.
- Nirupama R, Devaki M, Yajurvedi HN. 2012. Chronic stress and carbohydrate metabolism: Persistent changes and slow return to normalacy in male albino rats. Stress. 15 3: 262–271.
- Ohkawa H, Ohishi N, Yagi K. 1979. Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal Biochem. 95 2: 351–358.
- Preece A. 1972. A manual for histologic technicians. Boston, MA: Little, Brown and Company259–260.
- Pompella A, Emdin M, Passino C, Paolicchi A. 2004. The significance of serum gamma-glutamyltransferase in cardiovascular diseases. Clin Chem Lab Med. 42 10: 1085–1091.
- Rainey WE, Rodgers RJ, Mason JI. 1992. The role of bovine lipoproteins in the regulation of steroidogenesis and HMG-CoA reductase in bovine adrenocortical cells. Steroids. 57 4: 167–173.
- Richards JC, Hof A, Alvarenga M. 2000. Serum lipids and their relationships with hostility and angry affect and behaviors in men. Health Psychol. 19 4: 393–398.
- Rosalki SB, Tarlow D. 1974. Optimized determination of gamma-glutamyltransferase by reaction-rate analysis. Clin Chem. 20 9: 1121–1124.
- Ross R. 1999. Atherosclerosis—an inflammatory disease. N Engl J Med. 340 2: 115–126.
- Ruiz de Gordoa JC, Santafé J, Segarra Domenech J, Santisteban A. 1994. Modification of rat plasma lipoproteins induced by acute immobilization stress. Psychosom Med. 56 6: 486–492.
- Salo DC, Pacifici RE, Lin SW, Giulivi C, Davies KJ. 1990. Superoxide dismutase undergoes proteolysis and fragmentation following oxidative modification and inactivation. J Biol Chem. 265 20: 11919–11927.
- Sato T, Yamamoto H, Sawada N, Nashiki K, Tsuji M, Muto K, Kume H, Sasaki H, Arai H, Nikawa T, Taketani Y, Takeda E. 2006. Restraint stress alters the duodenal expression of genes important for lipid metabolism in rat. Toxicology. 227 3: 248–261.
- Segal HL, Beattie DS, Hopper S. 1962. Purification and properties of liver glutamic-alanine transaminase from normal and corticoid-treated rats. J Biol Chem. 237 6: 1914–1920.
- Sen P, Maiti PC, Puri S, Ray A, Audulov NA, Valdman AV. 1992. Mechanisms of anti-stress activity of Ocimum sanctum Linn, eugenol and Tinospora malabarica in experimental animals. Ind J Exp Biol. 30 7: 592–596.
- Shivanandappa T, Venkatesh S. 1997. A colorimetric assay method for 3beta-hydroxy delta5-steroid dehydrogenase. Anal Biochem. 254 1: 57–61.
- Shively CA, Clarkson TB, Kaplan JR. 1989. Social deprivation and coronary artery atherosclerosis in female cynomolgus monkey. Atherosclerosis. 77 1: 69–76.
- Slater AF, Nobel CS, Maellaro E, Bustamante J, Kimland M, Orrenius S. 1995. Nitrone spin traps and a nitroxide antioxidant inhibit a common pathway of thymocyte apoptosis. Biochem J. 306 Pt 3: 771–778.
- Sterling P. 2004. Principles of allostasis: Optimal design, predictive, regulation, pathophysiology and rational therapeutics. Cambridge: Cambridge University press1–36.
- Tappel AL. 1978. Glutathione peroxidase and hydroperoxidase. Methods Enzymol. 52:506–513.
- Tsigos C, Chrousos GP. 2002. Hypothalamic–pituitary–adrenal axis, neuroendrocrine factors and stress. J Psychosom Res. 53 4: 865–971.
- Witztum JL, Steinberg D. 1991. Role of oxidized low density lipoprotein in atherogenesis. J Clin Invest. 88 6: 1785–1792.
- Zafir A, Banu N. 2009. Induction of oxidative stress by restraint stress and corticosterone treatments in rats. Ind J Biochem Biophys. 46 1: 53–58.
- Zardooz H, Asl SZ, Naseri MKG, Hedayati M. 2006. Effect of chronic restraint stress on carbohydrate metabolism in rat. Physiol Behav. 89 3: 373–378.