
Abstract
The cortisol awakening response (CAR) is the most prominent, dynamic and variable part of the circadian pattern of cortisol secretion. Despite this, its precise purpose is unknown. Aberrant patterns of the CAR are associated with impaired physical and mental health and reduced cognitive function, suggesting that it may have a pervasive role or roles. It has been suggested that the CAR primes the brain for the expected demands of the day but the mechanisms underlying this process are unknown. We examined temporal covariation of the CAR and rapid transcranial magnetic stimulation (rTMS)-induced long term depression (LTD)-like responses in the motor cortex. Plasticity was evaluated across 180 measures from five time points on four sessions across nine healthy researcher participants, mean age 25 ± 2.5 years. Plasticity estimates were obtained in the afternoon after measurement of the CAR on 4 days, at least 3 days apart. As both CAR magnitude and rTMS-induced responses are variable across days, we hypothesized that days with larger than individual average CARs would be associated with a greater than individual average plasticity response. This was confirmed by mixed regression modelling where variation in the CAR predicted variation in rTMS-induced responses (df: 1, 148.24; F: 10.41; p = 0.002). As the magnitude of the CAR is regulated by the “master” circadian CLOCK, and synaptic plasticity is known to be modulated by peripheral “slave” CLOCK genes, we suggest that the CAR may be a mediator between the master and peripheral circadian systems to entrain daily levels of synaptic plasticity.
Introduction
There is good evidence that sustained exposure to high levels of glucocorticoids evokes neuronal cell damage and impairs synaptic plasticity (Joels, Citation2008; Sapolsky et al., Citation1990; Suri & Vaidya, Citation2013). However, it has recently become evident that the circadian rhythm of glucocorticoid secretion may promote internal homeostasis and optimal brain function (Nader et al., Citation2010). For example, animal studies indicate that healthy circadian glucocorticoid oscillations boost learning-dependent synaptic formation and maintenance (Liston et al., Citation2013). It is clear that disrupted circadian patterns (not just sustained high levels) of glucocorticoid secretion are associated with cognitive deficits (Cho et al., Citation2000; Evans et al., Citation2011; Gibson et al., Citation2010) as well as a wide range of neuropsychiatric diseases (Jagannath et al., Citation2013; Menet & Rosbash, Citation2011; Wulff et al., Citation2010).
In healthy animals, including humans, glucocorticoid hormones have a marked underlying circadian pattern with characteristically low levels during sleep, peak levels soon after awakening, followed by a gradual decline (Edwards et al., Citation2001). This circadian pattern is regulated by the central master CLOCK: the hypothalamic suprachiasmatic nucleus (SCN) (Perreau-Lenz et al., Citation2003). Furthermore, it has increasingly become recognized that glucocorticoids adjust the circadian rhythm and function of the ubiquitous peripheral CLOCKs. Dysfunction or dysregulation in either circadian system alters internal homeostasis and causes pathologic changes virtually in all tissues, including the brain (Nader et al., Citation2010).
In healthy humans, the initial burst of cortisol secretion that occurs on awakening (cortisol awakening response: CAR) is a distinct aspect of the circadian pattern of cortisol secretion (Clow et al., Citation2010; Edwards et al., Citation2001; Fries et al., Citation2009; Pruessner et al., Citation1997) and exhibits notable day-to-day variability (Hellhammer et al., Citation2007; Law et al., Citation2013). A study in one healthy young male revealed that daily CAR magnitude varied tenfold and was associated with anticipated obligations in the coming day (Stalder et al., Citation2009). Although this was consistent with the idea that the CAR may prime the brain for the expected demands of the day ahead (Fries et al., Citation2009), the mechanisms underlying this process have not been elucidated.
We set out to examine the CAR in relation to levels of synaptic plasticity measured in motor cortex some 6–7 h after awakening. We chose to use a model of synaptic plasticity previously used to explore associations with levels of cortisol in healthy intact participants: rapid transcranial magnetic stimulation (rTMS)-induced long term depression (LTD)-like responses in the motor cortex (Pitcher et al., Citation2012; Sale et al., Citation2008). We used this measure as a representative indicator of effects caused by any factor that affects the whole brain. We hypothesized that one function of the CAR is to regulate the sensitivity of synaptic plasticity, known to be modulated by peripheral CLOCK genes (Wang et al., Citation2009) during the coming day and that this could be one mechanism whereby the CAR could influence a wide range of behaviours. We predicted that day-to-day variation in the CAR would correlate with day-to-day variation in rTMS-induced synaptic plasticity of the motor cortex.
Materials and methods
Design
In this study, we sought to relate temporal variation within a large sample (N = 180) of plasticity estimates to temporal variation of the magnitude of the CAR. The design was entirely within-participant with estimates collected at five fixed time points repeated over four sessions and replicated over nine healthy, normal BMI researcher participants from the Universities of Adelaide and South Australia (78% female; mean age 25 ± 2.5 years), chosen to ensure rigorous adherence to the demanding protocol.
Procedure
The protocol was in accordance with the Declaration of Helsinki and was approved by the University of Adelaide Human Ethics Committee. Participants gave written informed consent prior to testing and were screened for any conditions that would contraindicate TMS (Rossi et al., Citation2009). Testing sessions were never <3 days apart to minimize carry over effects from the rTMS protocols (Goldsworthy et al., Citation2012; Hamada et al., Citation2013). On each study day, a CAR was determined upon awakening and plasticity estimates were assessed on the same afternoon.
Estimation of the cortisol awakening response
Saliva samples were collected using Salivettes (Sarstedt Ltd., Leicester, UK), immediately on awakening and at 15-, 30- and 45-min post-awakening (samples 1–4, respectively) on each study day. Sampling accuracy was recorded by electronic monitoring. Awakening times and sleep efficiency were determined using wrist-worn Actiwatches (Actiwatch-Score, Cambridge, UK). These are piezoelectric motion sensors that distinguish sleep and awakening periods by reduced and increased activity, respectively. Saliva sampling times were verified using Medical Event Monitoring (MEMS) (The Aardex Group, Sion, Switzerland) caps as described by Smyth et al. (Citation2013). During the saliva collection period, protocol instructions were to take nil by mouth other than water, and to refrain from brushing teeth to avoid abrasion and micro-vascular leakage. Cortisol analyses were carried out using a standard enzyme-linked immunosorbent assay protocol (Salimetrics, State College, PA). The limit of detection of the assay was 0.33 nmol/L.
Adherence to the saliva sampling protocol was excellent. No sampling time deviated >5 min from the requested saliva collection times relative to verified awakening (Smyth et al., Citation2013). About 4 of the 144 saliva samples were below the limit of detection of the assay: three awakening samples, and one 30-min sample. Undetectable samples were treated as missing data, and all other cortisol measures were included in the final analysis. CAR magnitude was calculated as the mean cortisol increase (MnInc) from 0 to 45 min: sample 2 + sample 3 + sample 4)/3 − sample 1.
Transcranial magnetic stimulation
All TMS sessions were completed in the afternoon (at 2 or 3 pm) in order to minimize time of day influences (Sale et al., Citation2007). Muscle contractions in the hand in response to TMS stimulation of the motor cortex were recorded as electromyographic (EMG) activity in the right first dorsal interosseous (FDI) using surface electrodes placed in a belly-tendon configuration. The EMG signal was amplified (×1000; CED 1902 amplifier, CED, UK), band pass filtered (20–1000 Hz) and digitized at a sampling rate of 2 kHz (CED 1401 interface, CED, UK). A Magstim-200 stimulator (Magstim Co., Whitland, UK) generated single-pulse stimuli, delivered through a figure-of-eight coil (90 mm diameter) placed tangentially to the scalp with the handle pointing backward at a 45° angle away from the midline. Suprathreshold pulses were delivered over the left M1 at numerous sites in order to identify the optimal site for consistently evoking motor evoked potential (MEPs) in the relaxed right FDI and this site was marked on the scalp. The TMS intensity that elicited MEPs of ∼1 mV (SI1 mV) in the relaxed FDI was determined (for each testing session) at baseline and was used to examine changes in MEP amplitude after each protocol. Although we did not use individual neuronavigation to place the coil, it is likely that there was little change in its position from day to day since the intensity employed for evoking baseline test MEPs, and the amplitude of these (1 mV) test MEPs was not significantly different between testing days. Any minor change in position or angle would have been random and could not contribute to the effects we observed. Two blocks of 15 single-pulse TMS trials, with an inter-trial interval of 7 s (±10%), were delivered at baseline and one block of 15 single-pulse TMS trials was then delivered 0, 5, 10, 20 and 30 min after rTMS. Individual MEP data trials were excluded if EMG activity was present in the 100 ms immediately prior to TMS. The peak-to-peak MEP amplitude (in mV) was measured for each trial to give an index of the size of the muscle twitch and the mean amplitude at each post-rTMS time point was expressed as a ratio of the mean of the two baseline samples. This provided an index of the change in the size of the muscle response to the same brain stimulus after rTMS relative to baseline prior to rTMS: the MEP ratio or neuroplasticity index.
The rTMS protocol adopted was continuous theta burst stimulation (cTBS) which was delivered using a figure of eight-shaped Double-Cooled-Coil-System coil (70 mm, Magstim, Whitland, UK). Bursts of three pulses were delivered at 50 Hz every 200 ms continuously for 40 s (Huang et al., Citation2005). TBS intensity was set to 80% of active motor threshold (AMT); AMT was defined as the minimum intensity required to elicit a MEP in FDI of at least 200 µV in at least 5 out of 10 consecutive trials when performing a low-level voluntary contraction of FDI (10% of maximal voluntary contraction) and was determined for each testing session. This paradigm is known to induce long term depression (LTD)-like effects resulting in a smaller muscle response (hence a reduced MEP ratio) post-rTMS.
Data analysis
Data were analyzed using mixed regression modelling (Blackwell et al., Citation2006) of variation in the 180 MEP-ratio estimates. CAR magnitude was included as the principal covariate, and was participant-centred since absolute differences in participants’ average CAR magnitude was not the focus of this study. Participant centring expresses exclusively within-participant variation (i.e. participants’ deviations from their own study means). We modelled time-point within session (at 0, 5, 10, 20 and 30 min) and session number (1–4) as fixed factors. Intercept effects were modelled as both fixed and random effects. Finally, further modelling was undertaken to check that any findings from initial modelling were not confounded by associations with awakening time and level of cortisol.
Results
presents descriptive data for the study. As expected, following rTMS, the average MEP ratio was <1(0.9 ± 0.29). In other words, as expected, the peak-to-peak MEP amplitude (in mV) post-rTMS was less than at base, indicating the induction of LTD-like synaptic plasticity. The average CAR across days showed a mean increase of 2.72 nmol/l cortisol in the 45 min after waking.
Table 1. Descriptives of MEP-ratios (mean of 0, 5, 10, 20 and 30 min after rTMS relative to base) cortisol concentrations (nmols/l) from 0- to 45-min post-awakening, CAR mean increase (MnInc) and awakening time.
shows F-ratios and significances for the parameters in the modelled data. There was no significant difference between the MEP ratios measured at the five post-rTMS time points (sample numbers 1–5), allowing us summarize the effect of rTMS as a single overall mean ratio (as shown in ). Similarly, there were no differences in the mean MEP ratios on each of the 4 days (session numbers 1–4), suggesting that there was no adaptation to the procedure over the period of testing.
Table 2. F-ratios, df and significances associated with parameters in mixed regression modelling of MEP ratio data.
The magnitude of the CAR was significantly associated with their mean MEP ratios collected on the same day. Calculation of the estimate coefficient (−0.013) suggests that for each single nmol/l above their own average CAR on any testing day, the predicted MEP ratio would be 0.013 points (∼1.4%) lower than average. Since lower MEP ratios reflect a larger response to the rTMS protocol, the finding suggests that larger than average CARs in the morning predict greater neuroplasticity measured later in the afternoon.
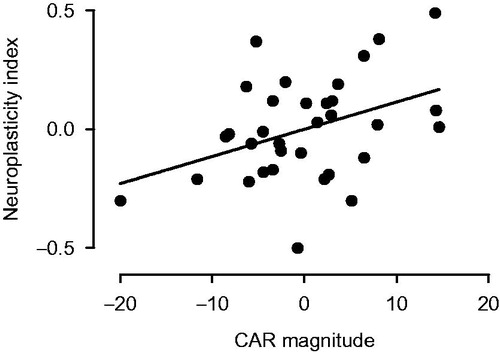
plots the relationship between morning CAR and afternoon rTMS response. Data are expressed relative to mean responses for both measures over the 4 days. The data show that if the CAR magnitude was larger than individual means, then there was a greater chance that the response to rTMS indicated greater neuroplasticity, i.e. an MEP ratio lower than the expected mean.
Figure 1. Relationship between morning CAR and afternoon mean rTMS response. Data are expressed relative to individuals’ own mean of study CAR or MEP ratio over the 4 days of sampling (i.e. zero on each axis). Because the rTMS effect is inhibitory, a small mean MEP ratio indicates a large rTMS response. The polarity of the y axis in this figure is accordingly reversed so that a more positive value indicates greater neuroplasticity.
Further modelling examined whether CAR magnitude was confounded by the covariates of awakening time and/or awakening level of cortisol. The effect was robust to such statistical control, remaining independently significant with a similar effect size. There was no association between the total amount of cortisol secreted over the post-awakening period (the AUCg) and rTMS-induced cortical plasticity the same afternoon. Sleep efficiency, determined by wrist work actigraphy, was not associated with either the CAR or rTMS-induced cortical plasticity.
Discussion
This study of temporal variation with multiple sampling across days shows a highly significant relationship between cortisol awakening response and the capacity to induce synaptic plasticity in the motor cortex on that same day. Specifically, on days when individuals’ CARs were bigger than their own individual averages there were greater changes in the size of the muscle response following the rTMS protocol, measured that afternoon, indicating greater neuroplasticity. Likewise, lower than average CARs predicted smaller changes in the size of the muscle response after rTMS.
The rTMS protocol used here (continuous theta bust, cTBS) is thought to provide a measure of the responsiveness of early long term depression (LTD)-like processes in the motor cortex (Huang et al., Citation2005, Citation2007). The measure varies considerably between individuals as it is affected by factors such as age and genetics (Hamada et al., Citation2013; Ridding & Ziemann, Citation2010). However, there are also large variations in an individual’s response measured on different days (Sale et al., Citation2007). The present results suggest for the first time that the daily magnitude of the CAR may be responsible for some daily variation in synaptic plasticity.
We have studied a specific index of plasticity in the motor cortex, but evidence suggests this measure can serve as a representative marker of effects caused by any factor that affects the whole brain (e.g. secretion of glucocorticoids). For example, administration of an NMDA antagonist in humans can be detected by reduced rTMS-induced plasticity in motor cortex (Stefan et al., Citation2002; Wolters et al., Citation2003). Similarly, the human Huntington’s disease gene in mice reduces synaptic plasticity in many cortical regions; in humans this is reflected in reduced plasticity in the motor cortex (Crupi et al., Citation2008). This TMS paradigm also has the advantage of being non-invasive and appropriate for use in healthy intact volunteers. Our hypothesis here is that the changes we see in motor cortex synaptic plasticity will reflect changes in other brain areas, all of which could be affected by the CAR. The relationship between individual day to day changes in cortical plasticity to relevant day to day differences in behaviour have yet to be examined, so the functional implications of such changes in plasticity are not yet fully described. Nevertheless, some relationship might be expected as previous experiments have shown that when such TMS protocols are used to induce changes in plasticity in the motor cortex (as here) they simultaneously affect motor function (e.g. Jung & Ziemann, Citation2009).
Aberrant patterns of the CAR have been consistently linked with indices of impaired physical and mental health (Fries et al., Citation2009; Kudielka & Kirschbaum, Citation2003). In particular, the CAR has been associated with cognitive function, an attenuated CAR has been associated with lower hippocampal volume (Buchanan et al., Citation2004; Pruessner et al., Citation2007), amnesia (Wolf et al., Citation2005), deficits in verbal memory and processing speed (Aas et al., Citation2011; Evans et al., Citation2011), and worse executive function (Evans et al., Citation2012). Furthermore, inhibition of the CAR using the cortisol synthesis inhibitor metyrapone impaired memory retrieval in healthy young participants (Rimmele et al., Citation2010).
The CAR is the most prominent and dynamic element of the circadian pattern of cortisol secretion. Despite this, its precise purpose is unknown. Evidence suggests causal pathways linking circadian cortisol disruption to sub-optimal brain function and brain disorder (Jagannath et al., Citation2013; Wulff et al., Citation2010). One putative pathway involves dysregulation of the circadian CLOCK system (Menet & Roshbash, Citation2011). In this scheme, the SCN acts as the light-activated “master” CLOCK, which synchronizes the peripheral “slave” CLOCKs through neural and humoral pathways (Nader et al., Citation2010). It has been proposed that one of these pathways may involve the CAR (Law et al., Citation2013), which is regulated by dual inputs from the SCN: via the hypothalamic pituitary adrenal axis as well as a direct neural pathway to the adrenal cortex (Clow et al., Citation2010). These dual pathways mean it can fine tune sensitivity of the adrenal cortex to adrenocorticotrophic hormone (the secretagogue for cortisol) and make it ideally suited to relay messages to the periphery from the master CLOCK. Glucocorticoids are known to affect peripheral CLOCKs in almost all organs and tissues by influencing the expression of several clock-related genes, which in turn have been shown to modify synaptic plasticity (Wang et al., Citation2009). This means that the circadian pattern of glucocorticoid secretion (and as indicated by this study the CAR in particular) can affect function in a sustained way over the day, not just by the influence of ambient levels, e.g. direct inhibition of NMDA receptor function. Although peripheral clock function was not directly examined in this study, circulating leukocytes provide an ideal tissue source for further work investigating the CAR and the circadian clock system in humans (see Kusanagi et al., Citation2008). Day differences in mRNA expression of Per1, Per2, Per3 mRNA, in particular, at a set time in the afternoon relative to morning CAR would be of particular interest.
The number of participants in this study was deliberately small and select: healthy young research staff fully able to participate over four testing days and aware of the importance of adherence to the required protocols. The focus has been to demonstrate systematic covariation over time in two biological measures. The small number of participants is however potentially relevant to issues of generalizability. While we have no theoretical reason to believe that such temporal co-variation would be restricted to the participants in this particular study, we would nevertheless recommend the usual caution in matters of of generalization until more similar studies are published.
Conclusions
This study has demonstrated for the first time significant covariation between the cortisol awakening response and rTMS-induced synaptic plasticity of the motor cortex, measured 6–7 h later the same day. These findings may indicate a pivotal role for the CAR in priming the brain for the day ahead (Clow et al., Citation2010; Fries et al., Citation2009) possibly by entrainment of peripheral CLOCKs in the brain that can influence the sensitivity of synaptic plasticity. As well as shedding light on a possible role for the CAR and informing the marked state variation in this measure of neuroplasticity it offers a plausible mechanism by which state factors which affect the CAR (such as stress) can affect brain plasticity.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the article.
References
- Aas M, Dazzan P, Mondelli V, Toulopoulou T, Reichenberg A, Di Forti M, Fisher HL, et al. (2011). Abnormal cortisol awakening response predicts worse cognitive function in patients with first-episode psychosis. Psychol Med 41:463–76
- Blackwell E, Carlos F, Mendes De Leon F, Millerm G. (2006). Applying mixed regression models to the analysis of repeated-measures data in psychosomatic medicine. Psychosom Med 68:870–8
- Buchanan TW, Kern S, Allen JS, Tranel D, Kirschbaum C. (2004). Circadian regulation of cortisol after hippocampal damage in humans. Biol Psychol 56:651–6
- Cho K, Ennaceur A, Cole JC, Suh CK. (2000). Chronic jet lag produces cognitive deficits. J Neurosci 20:RC66
- Clow A, Hucklebridge F, Stalder T, Evans P, Thorn L. (2010). The cortisol awakening response: more than a measure of HPA axis function. Neurosci Biobehav Rev 35:97–103
- Crupi D, Ghilardi MF, Mosiello C, Di Rocco A, Quartarone A, Battaglia F. (2008). Cortical and brainstem LTP-like plasticity in Huntington’s disease. Brain Res Bull 75:107–14
- Edwards S, Clow A, Evans P, Hucklebridge F. (2001). Exploration of the awakening cortisol response in relation to diurnal cortisol secretory activity. Life Sci 68:2093–103
- Evans P, Fredhoi C, Loveday C, Hucklebridge F, Aitchison E, Forte D, Clow A. (2011). The diurnal cortisol cycle and cognitive performance in the healthy old. Int J Psychophysiol 79:371–7
- Evans P, Hucklebridge F, Loveday C, Clow A. (2012). The cortisol awakening response is related to executive function in older age. Int J Psychophysiol 84:201–4
- Fries E, Dettenborn L, Kirschbaum C. (2009). The cortisol awakening response (CAR): facts and future directions. Int J Psychophysiol 72:67–73
- Gibson EM, Wang C, Tjho S, Khattar N, Kriegsfeld LJ. (2010). Experimental ‘jet lag’ inhibits adult neurogenesis and produces long-term cognitive deficits in female hamsters PLoS One 5:art. no. e15267
- Goldsworthy MR, Pitcher JB, Ridding MC. (2012). The application of spaced theta burst protocols induces long-lasting neuroplastic changes in the human motor cortex. Eur J Neurosci 35:125–34
- Hamada M, Murase N, Hasan A, Balaratnam M, Rothwell JC. (2013). The Role of interneuron networks in driving human motor cortical plasticity. Cereb Cortex 23:1593–605
- Hellhammer J, Fries E, Schweisthal OW, Schlotz W, Stone AA, Hagemann D. (2007). Several daily measurements are necessary to reliably assess the cortisol rise after awakening: state and trait components. Psychoneuroendocrinology 32:80–6
- Huang YZ, Chen RS, Rothwell JC, Wen HY. (2007). The after-effect of human theta burst stimulation is NMDA receptor dependent. Clin Neurophysiol 118:1028–32
- Huang YZ, Edwards MJ, Rounis E, Bhatia KP, Rothwell JC. (2005). Theta burst stimulation of the human motor cortex. Neuron 45:201–6
- Jagannath A, Peirson SN, Foster RG. (2013). Sleep and circadian rhythm disruption in neuropsychiatric illness. Curr Opin Neurobiol 23:888–94
- Joels M. (2008). Functional actions of corticosteroids in the hippocampus. Eur J Pharmacol 583:312–21
- Jung P, Ziemann U. (2009). Homeostatic and nonhomeostatic modulation of learning in human motor cortex. J Neurosci 29:5597–604
- Kudielka BM, Kirschbaum C. (2003). Awakening cortisol responses are influenced by health status and awakening time but not by menstrual cycle phase. Psychoneuroendocrinology 28:35–47
- Kusanagi H, Hida A, Satoh K, Echizenya M, Shimizu T, Pendergast JS, Yamazaki S, Mishima K. (2008). Expression profiles of 10 circadian clock genes in human peripheral blood mononuclear cells. Neurosci Res 61:136–42
- Law R, Hucklebridge F, Thorn L, Evans P, Clow A. (2013). State variation in the cortisol awakening response. Stress 16:483–92
- Liston C, Cichon JM, Jeanneteau F, Jia Z, Chao MV, Gan W-B. (2013). Circadian glucocorticoid oscillations promote learning dependent synapse formation and maintenance. Nat Neurosci 16:698–705
- Menet JS, Rosbash M. (2011). When brain clocks lose track of time: cause or consequence of neuropsychiatric disorders. Curr Opin Neurobiol 21:849–57
- Nader N, Chrousos GP, Kino T. (2010). Interactions of the circadian CLOCK system and the HPA axis. Trends Endocrinol Metab 21:227–86
- Perreau-Lenz S, Kalsbeek A, Garidou M-L, Wortel J, Van Der Vliet J, Van Heijningen C, Simonneaux V, et al. (2003). Suprachiasmatic control of melatonin synthesis in rats: inhibitory and stimulatory mechanisms. Eur J Neurosci 17:221–8
- Pitcher JB, Riley AM, Doeltgen SH, Kurylowicz L, Rothwell J, McAllister SM, Smith AE, et al. (2012). Physiological evidence consistent with reduced neuroplasticity in human adolescents born preterm. J Neurosci 32:1640–6
- Pruessner JC, Wolf OT, Hellhammer DH, Buske-Kirschbaum A, von Auer K, Jobst S, Kaspers F, Kirschbaum C. (1997). Free cortisol levels after awakening: A reliable biological marker for the assessment of adrenocortical activity. Life Sci 61:2539–49
- Pruessner M, Pruessner JC, Hellhammer DH, Pike GB, Lupien SJ. (2007). The associations among hippocampal volume, cortisol reactivity, and memory performance in healthy young men. Psychiatry Res Neuroimaging 155:1–10
- Ridding MC, Ziemann U. (2010). Determinants of the induction of cortical plasticity by non-invasive brain stimulation in healthy subjects. J Physiol 588:2291–304
- Rimmele U, Meier F, Lange T, Born J. (2010). Suppressing the morning rise in cortisol impairs free recall. Learn Mem 17:186–90
- Rossi S, Hallett M, Rossini PM, Pascual-Leone A. (2009). Safety, ethical considerations, and application guidelines for the use of transcranial magnetic stimulation in clinical practice and research. Clin Neurophysiol 120:2008–39
- Sale MV, Ridding MC, Nordstrom MA. (2007). Factors influencing the magnitude and reproducibility of corticomotor excitability changes induced by paired associative stimulation. Exp Brain Res 181:615–26
- Sale MV, Ridding MC, Nordstrom MA. (2008). Cortisol inhibits neuroplasticity induction in human motor cortex. J Neurosci 28:8285–93
- Sapolsky RM, Uno H, Rebert CS, Finch CE. (1990). Hippocampal damage associated with prolonged glucocorticoid exposure in primates. J Neurosci 10:2897–902
- Smyth N, Clow A, Hucklebridge F, Thorn L, Evans P. (2013). Delays of 5-15 minutes between awakening and the start of saliva sampling matter in assessment of the cortisol awakening response. Psychoneuroendocrinology 38:1476–83
- Stalder T, Hucklebridge F, Evans P, Clow A. (2009). Use of a single case study design to examine state variation in the cortisol awakening response: relationship rise after awakening: state and trait components. Psychoneuroendocrinology 32:80–6
- Stefan K, Kunesch E, Benecke R, Cohen L.G, Classen J. (2002). Mechanisms of enhancement of human motor cortex excitability induced by interventional paired associative stimulation. J Physiol 543:699–708
- Suri D, Vaidya VA. (2013). Glucorticoid regulation of brain derived neurotrophic factor: relevance to hippocampal structural and functional plasticity. Neuroscience 239:196–213
- Wang LM-C, Dragich JM, Kudo T, Odom IH, Welsh DK, Dell TJ, Colwell CS. (2009). Expression of the circadian clock gene Period2 in the hippocampus: possible implications for synaptic plasticity and learned behaviour. ASN Neuro 1(3):art. no. e00012, 139–52
- Wolf OT, Fujiwarab E, Luwinskib G, Kirschbaum C, Markowitsch HJ. (2005). No morning cortisol response in patients with severe global amnesia. Psychoneuroendocrinology 30:101–5
- Wolters A, Sandbrink F, Schlottmann A, Kunesch E, Stefan K, Cohen LG, Benecke R, Classen J. (2003). A temporally asymmetric Hebbian rule governing plasticity in the human motor cortex. J Neurophysiol 89:2339–45
- Wulff K, Gatti S, Wettstein JG, Foster RG. (2010). Sleep and circadian rhythm disruption in psychiatric and neurodegenerative disease. Nat Rev Neurosci 11:589–99