
Abstract
Restraint stress (RS) is an experimental model to study stress-related cardiovascular responses, characterized by sustained pressor and tachycardiac responses. We used pharmacologic and surgical procedures to investigate the role played by sympathetic nervous system (SNS) and parasympathetic nervous system (PSNS) in the mediation of stress-evoked cardiovascular responses. Ganglionic blockade with pentolinium significantly reduced RS-evoked pressor and tachycardiac responses. Intravenous treatment with homatropine methyl bromide did not affect the pressor response but increased tachycardia. Pretreatment with prazosin reduced the pressor and increased the tachycardiac response. Pretreatment with atenolol did not affect the pressor response but reduced tachycardia. The combined treatment with atenolol and prazosin reduced both pressor and tachycardiac responses. Adrenal demedullation reduced the pressor response without affecting tachycardia. Sinoaortic denervation increased pressor and tachycardiac responses. The results indicate that: (1) the RS-evoked cardiovascular response is mediated by the autonomic nervous system without an important involvement of humoral factors; (2) hypertension results primarily from sympathovascular and sympathoadrenal activation, without a significant involvement of the cardiac sympathetic component (CSNS); (3) the abrupt initial peak in the hypertensive response to restraint is sympathovascular-mediated, whereas the less intense but sustained hypertensive response observed throughout the remaining restraint session is mainly mediated by sympathoadrenal activation and epinephrine release; (4) tachycardia results from CSNS activation, and not from PSNS inhibition; (5) RS evokes simultaneous CSNS and PSNS activation, and heart rate changes are a vector of both influences; (6) the baroreflex is functional during restraint, and modulates both the vascular and cardiac responses to restraint.
Introduction
Exposure to environmental stress has been implicated in the etiology of several pathologies, including depression, anxiety and cardiovascular disease (Huang et al., Citation2001; Kario et al., Citation1997). Furthermore, there is strong evidence indicating an interrelation between mood disorders and cardiovascular disease, affecting homeostatic and neuroendocrine functions (Dimsdale, Citation2008; Grippo & Johnson, Citation2009; Lovallo & Gerin, Citation2003). However, the molecular and neuro-functional mechanisms involved in this association are not yet clearly elucidated. Psychological stress causes neuroendocrine changes involving dysfunction of the hypothalamic-pituitary-adrenal axis (HPA) and activation of the autonomic nervous system, evoking metabolic, immune and cardiovascular changes (McEwen, Citation1998, Citation2000). Different stressful stimuli cause differential activation of neuroendocrine and sympathetic nervous systems (Figueiredo et al., Citation2003; Graeff, Citation2007; Pacak & Palkovits, Citation2001) and both pattern and intensity of the associated cardiovascular changes may vary with the type of stress imposed on the organism. This can be observed when animals are exposed to different experimental stress models, such as air jet, open field, immobilization, fear conditioning and restraint (Kaehler et al., Citation2000; Kapusta et al., Citation2002; Kubo et al., Citation2002; Lam et al., Citation1995; Muller et al., Citation2001; Resstel et al., Citation2006; van den Buuse et al., Citation2001).
Restraint stress is an unavoidable stimulus that is widely used as an experimental model to elicit a variety of behavioral and physiological stress-related responses. These responses include: the lack of exploration of open arms, when animals are later subjected to the elevated plus maze test, defecation, ultrasonic vocalization (Mitsushima et al., Citation2006; Padovan & Guimaraes, Citation2000), alteration in body temperature and cardiovascular responses, characterized by a sustained increase in blood pressure and heart rate (Conti et al., Citation2001; Kubo et al., Citation2002; Mitsushima et al., Citation2006; Padovan & Guimaraes, Citation2000; Reis et al., Citation2011; Tavares & Correa, Citation2006; Yoshino et al., Citation2005).
The cardiovascular effects of acute stress result mainly from increased autonomic sympathetic activity and release of norepinephrine from sympathetic nerve terminals, which either line the vasculature or innervate the heart, as well as epinephrine from the adrenal medulla (Kawashima, Citation2005). Additionally, stress has been reported to affect baroreceptor reflex function (Conti et al., Citation2001; Grippo & Johnson, Citation2009; Randall et al., Citation1994; Sarenac et al., Citation2011). Although the relative contribution of these mechanisms in the generation of cardiovascular responses to stress has been the subject of previous studies (Carrive, Citation2002; van den Buuse et al., Citation2001) it has not yet been fully elucidated.
Our hypothesis is that stress-related cardiovascular responses are the resultant of an interaction between influences from the autonomic nervous system and the baroreflex. In the present study, we aimed to investigate the role played by the sympathetic nervous system (SNS) and parasympathetic nervous system (PSNS) as well that of the baroreflex in the mediation of stress-evoked hypertension and tachycardia, using the restraint stress (RS) model. To accomplish this, we used the following pharmacologic and surgical procedures: (1) treatment with the ganglion blocker pentolinium was performed to block overall autonomic outflow; (2) the muscarinic antagonist homatropine methyl bromide was used to evaluate parasympathetic involvement in RS-evoked cardiovascular responses; (3) the β1-adrenoceptor antagonist atenolol and the α1-adrenoceptor antagonist prazosin were used to evaluate sympathetic cardiac and sympathetic vasomotor activities, respectively; (4) surgical adrenal demedullation was performed to study the contribution of circulating epinephrine, and (5) surgical sinoaortic denervation was performed to study the influence of baroreflex, on restraint evoked cardiovascular responses.
Materials and methods
Subjects
Sixty-three male Wistar rats weighing approximately 250 g were used in the present experiment. Animals were housed in plastic cages in a temperature-controlled room (25 °C) in the Animal Care Unit of the Department of Pharmacology, School of Medicine of Ribeirão Preto. Animals were kept under a 12:12 h light–dark cycle (lights on between 06:00 and 18:00 h). Animals had free access to water and standard laboratory food, except during the experimental period. The rats were transported to the experiment room and remained in their own cages until experimental restraint. Experiments were performed during the morning period in order to minimize possible circadian rhythm interference. Experimental procedures were carried out following protocols approved by the Ethical Review Committee of the School of Medicine of Ribeirão Preto.
Surgical preparations
Catheterization
Animals were anesthetized with tribromoethanol (250 mg/kg, i.p.) and had a polyethylene catheter implanted into the femoral vein for drug administration. Another catheter was implanted into the femoral artery for chronic recording of arterial blood pressure (BP) and heart rate (HR). The catheters were exposed on the dorsum of the animals and attached to the skin, allowing arterial pressure recording and drug administration 24 h after surgery.
Adrenal demedullation
The adrenal glands were exposed by a longitudinal incision in the dorsal region of the animals, under tribromoethanol (250 mg/kg i.p.) anesthesia. A small incision was then performed in the adrenal cortex; the adrenal cortex was gently squeezed, and the adrenal medulla extruded. Animals were allowed to recover for 1 week. At the end of the experiments, rats were anesthetized with urethane (1.25 g/kg i.p.). Adrenal glands were removed, fixed in 10% formaldehyde, embedded in paraffin, sectioned and stained with hematoxylin and eosin for analysis by light microscopy. Control animals (“sham”) were subjected to the same surgical procedure, except removal of the adrenal medulla.
Sinoartic denervation
Bilateral sinoartic surgeries were performed according to the method reported by Krieger (Citation1964). Briefly, rats were anesthetized with ketamine (50 mg/kg, i.p.) and xylazine (5 mg/kg, i.p.), and fixed in a supine position. An extensive (3–4 cm) middle incision was made into the neck, and the bilateral sternohyoid muscle was resected, exposing the neurovascular sheath. The common carotid arteries and the vagal trunk were isolated carefully. The aortic depressive nerves (except those traveling with the recurrent laryngeal and superior laryngeal nerves) were thus severed. The fibers of the latter type were also interrupted by resection of the superior laryngeal nerves. The neck muscles were separated carefully, to widely expose the carotid bifurcation. The bifurcation and all the carotid branches were stripped of fibers and connective tissues. The sham surgery consisted of the same procedure, but no denervation was carried out.
After surgery, the animals received a poly-antibiotic (i.m., 0.27 mg/kg, Pentabiotico®, Fort Dodge, Brazil), with streptomycins and penicillins, to prevent infection, and a non-steroidal anti-inflammatory (i.m., 0.025 mg/kg, Flunixine Meglumine, Banamine®, Schering Plough, Brazil) for post-operative analgesia.
Drugs
Drugs were dissolved in sterile saline (0.9% NaCl), in a concentration that allowed their injection in 1 mL/kg. Pentolinium, prazosin, atenolol and homatropine methylbromide were purchased from Sigma-Aldrich Corporation (St Louis, MO).
Experimental procedure: acute restraint
Animals were transported to the experimental room in their own cages and a 1-h period was allowed to adapt the animals to experimental room conditions, such as sound and illumination, before starting BP and HR recording. The experimental room was acoustically isolated and an air exhauster generated a constant background noise. At least another 20 min period was allowed for baseline recording before experiments were initiated. After recording baseline values, injections of drug or vehicle were made into the femoral vein. Each animal received only one injection. Ten minutes later, the animals were subjected to restraint, which was initiated by placing animals into a small plastic cylindrical restraining tube (diameter = 6.5 cm and length = 15 cm). Restraint lasted 60 min, after which animals were returned to their cages. Each animal was subjected to one session of restraint, in order to prevent habituation. Pulsatile arterial pressure (PAP) was recorded using an amplifier (model 7754A, Hewlett Packard, Palo Alto, CA) coupled to a computerized acquisition system (MP100, Biopac, Goleta, CA). Mean arterial pressure (MAP) and HR were derived from PAP data using AcqKnowledge III software (Biopac, Goleta, CA). MAP was calculated according to the equation: diastolic pressure + (systolic-diastolic)/3. HR (beats/min; bpm) and was recorded from pulsatile arterial pressure (PAP) peak intervals integrated every 6 s. The areas under the curves (AUC) of MAP × time and HR × time were integrated using the Prism software (Graph-Pad, San Diego, CA).
Animals were divided into 11 experimental groups: (1) animals without any treatment (n = 5); (2) animals treated with vehicle (0.9% NaCl, 1 mL/kg – control group, n = 6); (3) with the ganglion blocker pentolinium (5 mg/kg, n = 6); (4) with the non-selective muscarinic receptor antagonist homatropine methylbromide (0.2 mg/kg, n = 5); (5) with β1-adrenoceptor antagonist atenolol (1 mg/kg, n = 6); (6) with α1-adrenoceptor antagonist prazosin (0.5 mg/kg, n = 6); (7) with atenolol 1 mg/kg + prazosin 0.5 mg/kg (n = 6); (8) sham-demedullated (n = 6); (9) adrenal demedullated (n = 6), (10) sham-sinoaortic denervated (n = 6), and (11) sinoaortic denervated (n = 5).
Statistical analysis
Statistical analysis was performed using the Prism software. The Student t-test was used to compare basal values of MAP and HR before and after vehicle or drug treatment. A two-way ANOVA for repeated measures was used to compare mean arterial pressure (ΔMAP) and heart rate (ΔHR) time curves generated after vehicle and drug treatments. Although PAP was recorded throughout the experimental procedure, curves for statistical analysis or illustrative figures were generated with points obtained using different data samplings. For statistical purposes, curves were generated with sampling at 0.07/min to generate five points (experiments with five subjects), or 0.08/min to generate six points (experiments with six subjects). Illustrative time-curves were generated with sampling at 0.64/min for a more accurate representation. Data are presented as mean ± S.E.M. A one-way ANOVA followed by Bonferroniás post-test for selected pairs was used to compare effects of treatments on AUC of MAP and HR RS-evoked responses. Significance was set at p < 0.05.
Results
Cardiovascular effects associated with exposure to restraint stress
Exposure to restraint stress for 60 min evoked a sustained cardiovascular response characterized by an increased arterial pressure and tachycardia, which lasted throughout the restraint session. RS-evoked cardiovascular changes were characterized by an abrupt increase in blood pressure and HR observed during the first 10 min of restraint, which was followed by a less intense but sustained pressor and tachycardiac response that lasted throughout the remaining 50 min of the restraint session. The arterial pulse remained constant throughout the restraint period and was not different from that recorded during the control period. A representative recording showing restraint evoked cardiovascular changes is presented in . Time-curves for ΔMAP; differential pressure (systolic – diastolic pressure) and ΔHR values (n = 5) are also presented in .
Figure 1. On the left – Recordings of pulsatile arterial pressure (PAP), mean arterial pressure (MAP), and heart rate (HR) of one unanesthetized rat submitted to restraint showing the cardiovascular changes observed during a restraint period of 60 min. On the right – Curves showing changes with time of differential pressure (systolic – diastolic pressure, ΔDP), mean arterial pressure (ΔMAP) and heart rate (ΔHR). The onset of restraint is at t = 0.
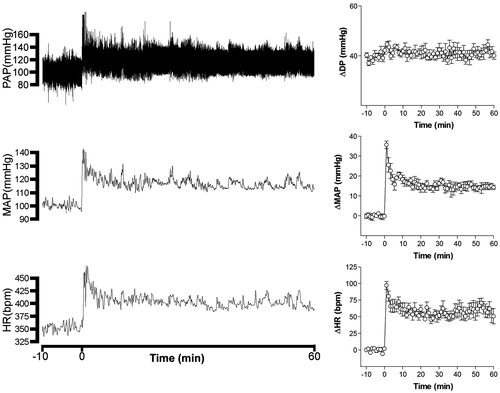
Effect of pretreatment with vehicle or the ganglion blocker pentolinium on restraint stress induced cardiovascular changes
Intravenous pretreatment with vehicle did not affect basal MAP and HR values (n = 6).
MAP was significantly reduced after i.v. treatment with 5 mg/kg of pentolinium (97 ± 4 mmHg vs. 69 ± 4 mmHg, t = 13.29, p < 0.0001, n = 6 Student’s t-test), but no significant changes were observed in HR (353 ± 13 bpm vs. 351 ± 12 bpm, t = 0.6118, p = 0.5674, n = 6 Student’s t-test).
Intravenous pretreatment with pentolinium significantly reduced RS-evoked pressor and tachycardiac responses when compared with control animals (). The two-way ANOVA indicated a significant effect of pentolinium on treatment (MAP, F(1,60) = 110.4, p < 0.0001 and HR, F(1,60) = 24.85, p < 0.0001); a significant effect on time (MAP, F(5,60) = 28.94, p < 0.0001 and HR, F(5,60) = 22.53, p < 0.0001) and a treatment-time interaction (MAP, F(5,60) = 14.76, p < 0.0001 and HR F(5,60) = 2.99, p = 0.0178) ().
Figure 2. Mean arterial pressure (ΔMAP) and heart rate (ΔHR) changes with time during restraint in the vehicle-treated control group (1 mL/kg, i.v., n = 6) and pentolinium-treated group (5 mg/kg, i.v., n = 6). Drugs were injected at t = −10 min. The onset of restraint is at t = 0. *Significantly different from control. p < 0.05; two-way ANOVA.
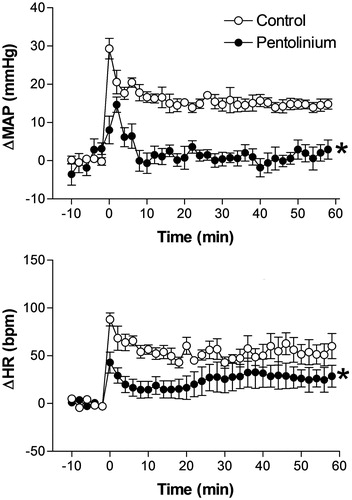
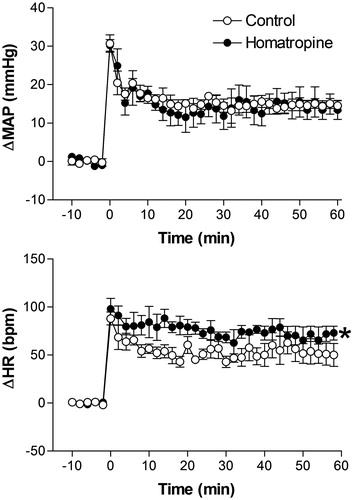
Effect of pretreatment with the non-selective muscarinic antagonist homatropine methylbromide on restraint stress induced cardiovascular changes
Intravenous pretreatment with 0.2 mg/kg of homatropine methylbromide did not affect basal MAP and HR values (98 ± 4 mmHg vs. 97 ± 6 mmHg, t = 0.8142, p = 0.7823 and 354 ± 12 bpm vs. 363 ± 11 bpm, t = 1.654, p = 0.6852, n = 5, Student’s t-test).
Treatment with homatropine methylbromide did not affect the restraint stress-induced pressor response but significantly increased the tachycardiac response, when compared with control animals (). The two-way ANOVA indicated no significant effect of homatropine methylbromide treatment on MAP (F(1,54) = 3.47, p = 0.067) but a significant effect on HR (F(1,54) = 11.38, p = 0.0014); a significant effect on time (MAP, F(5,54) = 76.39, p < 0.0001 and HR, F(5,60) = 82.45, p < 0.0001), and no treatment-time interaction (MAP, F(5,54) = 0.6498, p = 0.6628 or HR, F(5,60) = 1.446, p = 0.2228) ().
Figure 3. Mean arterial pressure (ΔMAP) and heart rate (ΔHR) changes with time during restraint in the vehicle-treated control group (1 mL/kg, i.v., n = 6), and homatropine-treated group (0.2 mg/kg, i.v., n = 5). Drugs were injected at t = −10 min. The onset of restraint is at t = 0. *Significantly different from control. p < 0.05; two-way ANOVA.
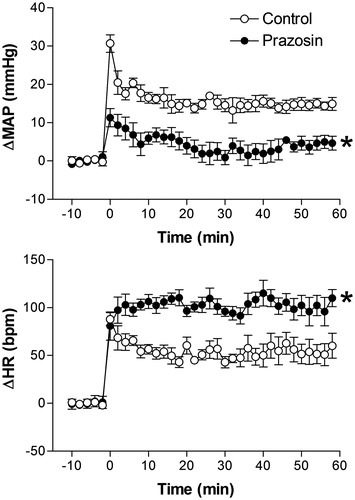
Effect of pretreatment with the α1-adrenoceptor antagonist prazosin on restraint stress-induced cardiovascular changes
There were no significant differences in basal values of MAP and HR after i.v. pretreatment with 0.5 mg/kg of prazosin (94 ± 5 mmHg vs. 95 ± 6 mmHg, t = 0.5097, p = 0.6213 and 354 ± 13 bpm vs. 361 ± 11 bpm, t = 0.5901, p = 0.5682, n = 6, Student’s t-test).
Treatment with prazosin practically blocked the RS-evoked pressor response and significantly increased the HR response, when compared with control animals (). The two-way ANOVA indicated a significant effect of prazosin on treatment (MAP, F(1,60) = 82.87, p < 0.0001 and HR, F(1,60) = 25.03, p < 0.0001); a significant effect on time (MAP, F(5,60) = 58.60, p < 0.0001 and HR, F(5,60) = 61.87, p < 0.0001), and a significant treatment-time interaction (MAP, F(5,60) = 13.2, p < 0.0001 and HR, F(5,60) = 6.94, p = 0.0178) ().
Figure 4. Mean arterial pressure (ΔMAP) and heart rate (ΔHR) changes with time during restraint in the vehicle-treated control group (1 mL/kg, i.v., n = 6), and prazosin-treated group (0.5 mg/kg, i.v., n = 6). Drugs were injected at t = −10 min. The onset of restraint is at t = 0. *Significantly different from control. p < 0.05; two-way ANOVA.
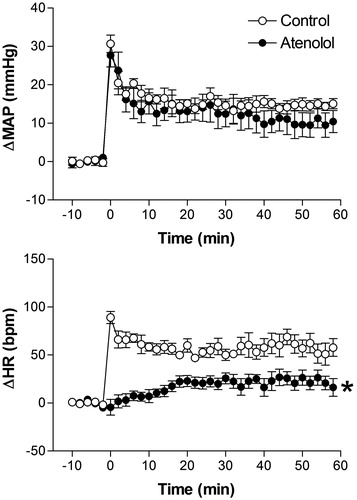
Effect of pretreatment with the β1-adrenoceptor antagonist atenolol on restraint stress-induced cardiovascular changes
There were no significant differences in basal values of MAP and HR after i.v. pretreatment with 1 mg/kg of atenolol (96 ± 3 mmHg vs. 97 ± 3 mmHg, t = 0.8102, p = 0.4547 and 352 ± 10 bpm vs. 339 ± 7 bpm, t = 1.105, p = 0.3119, n = 6, Student’s t-test).
Pretreatment with atenolol did not affect the restraint stress-induced pressor response but blocked the tachycardiac response (). The two-way ANOVA indicated no significant effect of atenolol treatment on MAP (F(1,60) =0.1056, p = 0.7464) but a significant effect on HR (F(1,60) = 107, p < 0.0001); a significant effect on time (MAP, F(5,60) = 49.89, p < 0.0001 and HR, F(5,60) = 21.54, p < 0.0001); it also indicated no treatment-time interaction for MAP (F(5,60) = 0.1994, p = 0.9615) but a significant treatment-time interaction for HR (F(5,60) = 20.94, p < 0.0001) ().
Figure 5. Mean arterial pressure (ΔMAP) and heart rate (ΔHR) changes with time during restraint in the vehicle-treated control group (1 mL/kg, i.v., n = 6), and atenolol-treated group (1 mg/kg, i.v., n = 6). Drugs were injected at t = −10 min. The onset of restraint is at t = 0. *Significantly different from control. p < 0.05; two-way ANOVA.
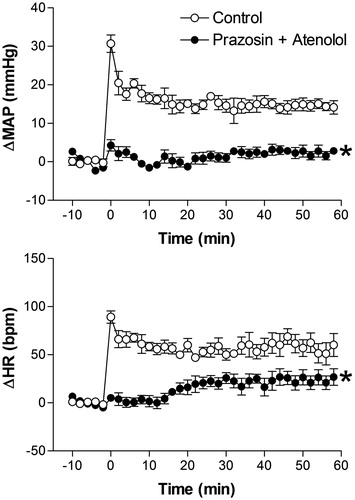
Effect of combined pretreatment with prazosin and atenolol on restraint stress-induced cardiovascular changes
Basal values of MAP were reduced after combined i.v. treatment with prazosin and atenolol (91 ± 3 mmHg vs. 85 ± 3 mmHg, t = 5.58, p = 0.0051, n = 6, Student’s t-test), but no changes were observed in basal HR values (344 ± 12 bpm vs. 342 ± 11 bpm, t = 2.064, p = 0.108, n = 6 Student’s t-test). The combined treatment with 0.5 mg/kg prazosin + 1 mg/kg atenolol blocked RS-evoked MAP and HR increases (). The two-way ANOVA indicated a significant effect of combined prazosin and atenolol on treatment (MAP, F(1,60) = 399.1, p < 0.0001 and HR, F(1,60) = 166.1, p < 0.0001); on time (MAP, F(5,60) = 102.3, p < 0.0001 and HR, F(5,60) = 41, p < 0.0001), and a significant treatment-time interaction (MAP, F(5,60) = 59.56, p < 0.0001 and HR, F(5,60) = 24.72, p < 0.0001) ().
Figure 6. Mean arterial pressure (ΔMAP) and heart rate (ΔHR) changes with time during restraint in the vehicle-treated control group (1 mL/kg, i.v., n = 6), and 0.5 mg/kg prazosin + 1 mg/kg atenolol-treated group (5 mg/kg, i.v., n = 6). Drugs were injected at t = −10 min. The onset of restraint is at t = 0. *Significantly different from control. p < 0.05; two-way ANOVA.
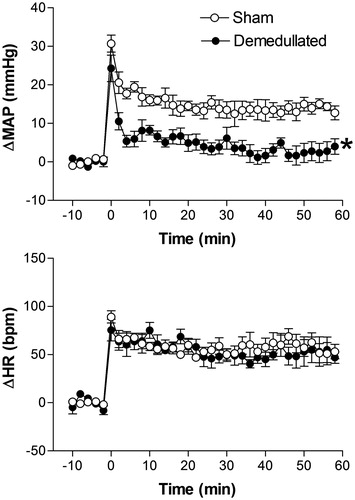
Effect of adrenal medulla surgical removal on restraint stress-induced cardiovascular changes
There were no significant differences in basal values of MAP and HR between adrenal demedullated (n = 6) and sham-operated rats (n = 6) (96 ± 3 mmHg vs. 98 ± 2 mmHg, t = 2.1, p = 0.618 and 359 ± 11 bpm vs. 356 ± 6 bpm, t = 0.38, p = 0.7084).
RS-evoked pressor responses were significantly reduced in adrenal demedullated rats, when compared with sham-operated animals (). No differences were observed between RS-evoked HR responses, when compared with control animals (). The two-way ANOVA indicated a significant effect of adrenal demedullation surgery on MAP (F(1,60) = 44.87, p < 0.0001) but not on HR (F(1,60) = 3.39, p = 0.0704); on time (MAP, F(5,60) = 119.7, p < 0.0001 and HR, F(5,60) = 67.63, p < 0.0001), and a treatment-time interaction for the MAP response (F(5,60) = 5.2, p = 0.0005) but not for the HR response (F(5,60) = 1.14, p = 0.2168) ().
Figure 7. Mean arterial pressure (ΔMAP) and heart rate (ΔHR) changes with time during restraint in the sham-operated group (n = 6) and adrenal desmedullated group (n = 6). The onset of restraint is at t = 0. *Significantly different from control. p < 0.05; two-way ANOVA.
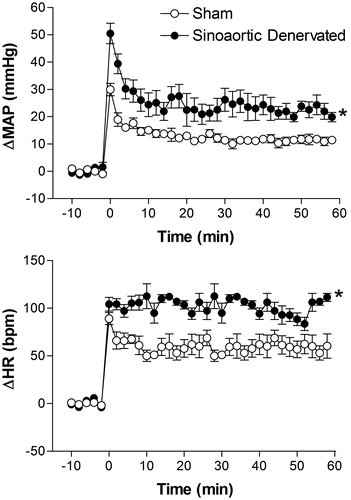
Effect of surgical sinoaortic denervation on restraint stress-induced cardiovascular changes
There were significant differences in basal values of MAP between sinoaortic denervated (n = 5) and sham-operated rats (n = 6) (103 ± 8 mmHg vs. 99 ± 3 mmHg, t = 2.1, p < 0.05). But no significant differences were observed in basal values of HR between those groups (355 ± 6 bpm vs, 359 ± 7 bpm, t = 0.38, p = 0.7084). RS-evoked pressor and tachycardiac responses were significantly increased in sinoaortic denervated rats, when compared with sham-operated animals (). The two-way ANOVA indicated a significant effect of adrenal sinoaortic denervation surgery on MAP (F(1,54) = 61.13, p < 0.0001) and HR (F(1,54) = 49.65, p < 0.0001); on time (MAP, F(5,54) = 140.2, p < 0.0001 and HR, F(5,54) = 132.4, p < 0.0001), and a treatment-time interaction for the MAP response (F(5,54) = 6.83, p < 0.0001) and HR response (F(5,54) = 8.28, p ≤ 0.0001) ().
Figure 8. Mean arterial pressure (ΔMAP) and heart rate (ΔHR) changes with time during restraint in the sham-operated group (n = 6) and sinoaortic denervated group (n = 5). The onset of restraint is at t = 0. *Significantly different from control. p < 0.05; two-way ANOVA.
Effect of different treatments on the AUC of restraint stress-induced cardiovascular changes over time
The results of treatments on integrated AUC of hypertension (ΔPAM × Time) and tachycardia (ΔHR × Time) area presented in . The one-way ANOVA indicated a significant effect of treatment on hypertension (F(9,48) =15.04) and tachycardia (F(9,48) = 23.7) evoked by restraint. The Bonferroni post-test was used to compare selected pairs of treatments for analysis. Pharmacological treatments were compared with the saline control. Prazosin was compared to prazosin + atenolol on BP, and atenolol was compared to prazosin + atenolol on HR RS-evoked response. Results from surgical procedures were compared with their respective sham-operated controls.
Table 1. Area under the curve (AUC) of restrain evoked hypertension (ΔMAP × Time) and tachycardia (ΔHR × Time) responses.
Discussion
Stress elicits a complex centrally coordinated response involving changes in mood, cognition, behavior, endocrine output and autonomic function. Cardiovascular regulation relies on feedback control systems, such as the baroreflex and the autonomic sympathetic (SNS) and parasympathetic nervous system (PSNS) as well as circulating hormones such as angiotensin II and vasopressin as their effector mechanisms (Arnold et al., Citation2013; Cowley & Liard, Citation1988; Di Rienzo et al., Citation2009; Guyenet, Citation2006). Stress-evoked autonomic changes include cardiovascular responses, such as hypertension, tachycardia and a shift in baroreflex setpoint sensitivity toward higher BP values. Under normal conditions, BP increase evokes a reflex HR decrease due to activation of the parasympathetic limb of baroreflex, but baroreflex is blunted under exercise, stress and defense reaction (Crestani et al., Citation2010; Nosaka, Citation1996).
Stimuli which act as stressors are generally divided into two categories: those with interoceptive (physical, systemic) and those with exteroceptive (processive, neurogenic, psychological) attributes (Herman & Cullinan, Citation1997; Pacak & Palkovits, Citation2001; Sawchenko et al., Citation2000). Restraint and immobilization are experimental models categorized as psychological stressors that have been widely used to study stress-evoked endocrine and autonomic responses. We have chosen the RS model to study stress-evoked cardiovascular responses because restraint is an unavoidable psychological stressor, less severe than immobilization, which evokes a hypertensive response characterized by a pressor peak observed during the first five minutes of restraint, followed by a less intense but sustained pressor respost that lasts throughout the restraint session, and a concomitant tachycardiac response (Kubo et al., Citation2002; McDougall et al., Citation2005; Tavares & Correa, Citation2006). Furthermore, RS can be categorized among the strongest psychological stressors in terms of c-fos activation in the CNS (Dayas et al., Citation2001), without the risk of being classified as a mixture of physical and psychological, as in the case of immobilization and forced swim.
In this study, we report on the relative role played by the autonomic SNS, the PSNS and the baroreflex in the mediation of RS-evoked cardiovascular responses. To achieve this, we have used pharmacologic and surgical procedures to study the systemic mechanisms involved in the mediation of RS-evoked cardiovascular responses. Effects of treatments were analyzed as a time-effect curve to evidence treatment particularities, and the area under the curve to measure their overall effect.
The resultant vectors of different treatments on RS-evoked blood pressure increase and tachycardiac responses are summarized in .
Table 2. Resultants of pharmacological and surgical procedures on blood pressure (hypertension) and heart (tachycardia) responses evoked by restraint (RS) in rats.
Pretreatment with the potent ganglion blocker pentolinium significantly reduced baseline BP values, confirming ganglion blockade effectiveness. Pentolinium reduced both the pressure response and the tachycardia evoked by RS, indicating that stress-evoked cardiovascular responses are mainly the result of autonomic nervous system stimulation. Ganglion blockade caused a 66% reduction in the AUC of hypertension and a 53% reduction in the AUC of tachycardia. Similar effects have been previously reported for fear conditioning after ganglion blockade with chlorisondamine (Carrive, Citation2002). However, treatment with pentolinium was reported to affect only the blood pressure increase, but not the tachycardia evoked by open-field stress (van den Buuse et al., Citation2001). Ganglion blockade causes less impact on sympathocardiac and parasympathetic output than on sympathoadrenal and sympathovascular autonomic outputs. Pretreatment with pentolinium reduced basal BP without affecting basal heart rate (present results (van den Buuse et al., Citation2001)), indicating a higher efficiency of cardiac autonomic output in comparison with vascular efferences. The difference between reports on ganglion blockade effect on stress-evoked tachycardia could result from stressor strength differences. Both open-field and fear conditioning models are benzodiazepine-sensitive (Resstel et al., Citation2006; van den Buuse et al., Citation2001), whereas pretreatment with diazepam did not affect RS-evoked cardiovascular responses (unpublished data). The magnitude of tachycardiac responses decreased in the following order among stressors: open-field > fear conditioning > restraint, suggesting different degrees of sympathocardiac activation as well as simultaneous coactivation of the PSNS as stressor strength increases the neuroendocrine response (Carrive, Citation2006; Paton et al., Citation2005).
Cardiovascular regulation relies on feedback control systems, such as the baroreflex and the autonomic SNS and PSNS as well as circulating hormones such as angiotensin II and vasopressin as their effector mechanisms (Arnold et al., Citation2013; Cowley & Liard, Citation1988; Di Rienzo et al., Citation2009; Guyenet, Citation2006). Results from treatment with pentolinium also indicate that RS-evoked hypertension results from sympathetic nerve activation, without a significant involvement of pressor humoral factors, such as vasopressin. In agreement with this, neither was vasopressin release reported during immobilization (Callahan et al., Citation1992; Gibbs, Citation1984), nor did intravenous pretreatment with the V1-vasopressin receptor antagonist dTyr-AVP (50 µg/kg) affects RS-evoked hypertension (unpublished data).
To examine the influence of parasympathetic cardiac activity on the cardiovascular response to RS, we treated animals with a low dose of homatropine methyl bromide that did not affect basal HR values (Tavares & Correa, Citation2006). Homatropine is a quaternary ammonium muscarinic antagonist that does not cross the blood–brain barrier (Donoso & Bacha, Citation1975), thus avoiding blockade of muscarinic receptors in the central nervous system. Inhibition of the parasympathetic outflow to the heart did not affect the pressor response to RS but caused a significant increase in the RS-evoked tachycardiac response, with a 36% increase in the AUC of tachycardia. This result reveals a parasympathetic coactivation concomitant with the predominant sympathetic activation that is observed during RS, and argues for the idea that stress-evoked tachycardia is the result of sympathocardiac activation and parasympathetic withdrawal. An even higher HR increase occurred after pretreatment with methyl-atropine in animals subjected to fear conditioning to context (Carrive, Citation2006), reinforcing the idea of a simultaneous coactivation of sympathocardiac and parasympathetic components.
Selective pharmacological protocols to block either sympathetic vasomotor tone or cardiac sympathetic activity are well established, and can be used to study the relative contribution of each one to the cardiovascular response to RS.
To test the role played by the sympathetic vasomotor tone in the cardiovascular response to RS, we treated animals with the α1-adrenergic antagonist prazosin. Given its long history of safe use, prazosin seemed to be the ideal candidate to use as a sympatholytic agent to test the effect of α1-adrenoceptor blockade on the RS-evoked cardiovascular response.
Treatment with prazosin, in a dose that did not affect baseline BP and HR values, reduced the pressor response and increased the tachycardia evoked by RS. A similar result was reported after pretreatment with phentolamine in rats subjected to conditioning fear to context (Carrive, Citation2002), The blockade of α1-adrenergic receptors reduced the peak in blood pressure observed within the first five minutes of restraint, and caused a 61% reduction in the AUC of hypertension and an 89% increase in the AUC of RS-evoked tachycardia. This result suggests that the RS-evoked pressor response is mainly mediated by activation of α1-adrenergic receptors in the vascular smooth muscle. The increase in RS-evoked tachycardia observed after α1-adrenergic blockade agrees with the results of pretreatment with homatropine methyl bromide, suggesting a concomitant activation of parasympathetic output in response to RS. Furthermore, it is indicative of functional baroreflex activity during restraint. Consequently, the tachycardiac response evoked by RS is the result of opposed sympathocardiac and parasympathetic activities.
The idea of a baroreflex influence on stress-evoked cardiovascular responses is favored by the present observation that sinoartic denervation increased pressor and tachycardiac responses to RS. Sinoartic denervation caused a 90% increase in the AUC of RS-evoked hypertension and increased 71% the AUC of tachycardia. These results are strong evidence of a functionally important negative baroreflex influence counteracting stress effects on cardiovascular system regulation. There is evidence that baroreflex is shifted toward a higher pressure set point under stress. Sinoaortic denervation has been reported to increase pressor responses evoked during escape-avoidance tests (Buchholz et al., Citation1986) and air-jet stress (Zhang et al., Citation1996). The present results are in agreement with a previous report suggesting that exposure to restraint stress affects the cardiac baroreflex response, facilitating the tachycardiac responses and shifting the set point for bradycardiac responses toward higher BP values (Crestani et al., Citation2010).
The sympathetic nervous system exerts direct chronotropic and inotropic cardio-stimulatory effects via β1-adrenergic receptors.
To block sympathocardiac output, we pretreated animals with the selective β1-adrenergic antagonist atenolol at a dose that did not affect baseline BP and HR values. Treatment with atenolol did not affect the pressor response but significantly reduced the RS-evoked tachycardiac response, resulting in a 61% inhibition of the AUC of tachycardia. Similar results were reported after β-adrenergic blockade in rats subjected to the open-field (van den Buuse et al., Citation2001) or to fear conditioning (Carrive, Citation2006). This result confirms the idea that increased cardiac sympathetic activity is the major factor responsible for RS-evoked tachycardia, and suggests a minor involvement of sympathetic cardiac output in the RS-evoked pressor response.
To block both the cardiac and vasomotor sympathetic activity, we combined treatments with prazosin and atenolol. The combined treatment with these antagonists caused a statistically significant additional reduction in RS-evoked hypertension (80% reduction in the AUC for hypertension) when compared with treatment with prazosin alone (61% reduction in the AUC for hypertension). No further reduction in the tachycardiac response was observed after combined treatment when compared with treatment with atenolol alone (63% vs. 60% reduction in the AUC for tachycardia). These data reinforce the idea that the cardiovascular response to RS is mainly mediated by activation of vascular smooth muscle α1-adrenergic receptors. However, the effect of combined treatment with prazosin and atenolol unveiled a small, although significant, involvement of β1-adrenoceptor activation in the RS-evoked hypertensive response. Since RS-evoked tachycardia was no further reduced by combined treatment with prazosin and atenolol, the effect of additional β1-adrenoceptor blockade could indicate an involvement of the circulating renin-angiotensin-system in the hypertensive response evoked by RS.
β1-adrenoceptors in juxtaglomerular cells are the target for renal sympathetic innervations and mediate renin release and consequent increases in the circulating vasopressor hormone angiotensin II (Kim et al., Citation2007). Plasma levels of renin and angiotensin II were reported to be increased after immobilization (Jindra & Kvetnansky, Citation1982) and restraint stress (Chung et al., Citation2010), respectively. Furthermore, treatment with losartan reduced the pressor response evoked by foot shock (Cierco & Israel, Citation1994; Israel & Sosa-Canache, Citation2002), or restraint (Busnardo et al., Citation2014) without affecting concomitant tachycardiac responses.
In addition to the vascular and cardiac limbs of the sympathetic nervous system, the sympathoadrenal is also extremely relevant for the functional response to stress. The adrenal medulla responds to acute or chronic stress with systemic release of catecholamines, predominantly epinephrine (Forsman & Lindblad, Citation1983; Majewski et al., Citation1986; Taylor et al., Citation1989).
Surgical adrenal gland demedullation was performed to examine the role played by the sympathoadrenal component in the cardiovascular response to RS. Adrenal demedullation significantly reduced the RS-evoked pressor response. Adrenal demedullation did not affect the peak of the pressor response observed in the first five minutes of the restraint session, but markedly attenuated the sustained pressure response that remained throughout the restraint session. Adrenal demedullation caused a 58% reduction in the AUC of hypertension when compared with that of sham-operated rats, a reduction similar to that caused by the blockade of α1-adrenoceptors with prazosin (61%), but did not affect the tachycardiac response to RS.
These results indicate that the sympathovascular component mediates the initial pressor peak of tachycardiac response, whereas a significant part of the sustained RS-evoked pressor response is mediated by epinephrine released from the adrenal medulla. Furthermore, adrenal demedullation had no effect on RS-evoked tachycardia, thus indicating that circulating epinephrine plays a marginal role in the mediation of stress evoked tachycardia that is the result of sympathocardiac activation.
Conclusion
In conclusion, the results indicate that RS-evoked cardiovascular responses are mediated by the autonomic nervous system without a significant involvement of humoral factors. They also indicate that stress-evoked hypertension results primarily from sympathovascular and sympathoadrenal activation, without the involvement of the cardiac sympathetic component (CSNS). The abrupt initial peak in the hypertensive response to restraint was shown to be sympathovascular-mediated, and the less intense but sustained hypertensive response observed throughout the remaining restraint session is mainly mediated by sympathoadrenal activation and epinephrine release, whereas stress-evoked tachycardia results from CSNS activation, and not from PSNS inhibition. RS evokes simultaneous CSNS and PSNS activation, and the resulting heart rate changes are a vector of both influences. Finally, results indicate that baroreflex is functional during restraint and modulates both the vascular and cardiac responses to restraint.
Declaration of interest
The authors report no conflicts of interest.
Daniel G. Reis is a PhD student of the Department of Pharmacology of the School of Medicine of Ribeirão Preto-USP, and recipient of a FAPESP fellowship (2011/21921-1). Eduardo Albino Trindade Fortaleza is a postdoctoral fellow in the Department of Pharmacology of the School of Medicine of Ribeirão Preto-USP, and recipient of a FAPESP fellowship (2012/18556-2). The present research was supported by grants from the CNPq and FAEPA.
Acknowledgements
We acknowledge Ms. I.A.C. Fortunato, I.I.B. Aguiar and S.S. Guilhaume for technical assistance.
References
- Arnold AC, Gallagher PE, Diz DI. (2013). Brain renin-angiotensin system in the nexus of hypertension and aging. Hypertens Res 36:5–13
- Buchholz RA, Hubbard JW, Keeton TK, Nathan MA. (1986). Cardiovascular and neuroendocrine responses to behavioral stress after central or peripheral barodenervation in rats. Brain Res 365:360–4
- Busnardo C, Tavares RF, Correa FM. (2014). Angiotensinergic neurotransmission in the paraventricular nucleus of the hypothalamus modulates the pressor response to acute restraint stress in rats. Neuroscience 270:12–19
- Callahan MF, da Rocha MJ, Morris M. (1992). Sinoaortic denervation does not increase cardiovascular/endocrine responses to stress. Neuroendocrinology 56:735–44
- Carrive P. (2002). Cardiovascular and behavioural components of conditioned fear to context after ganglionic and alpha-adrenergic blockade. Auton Neurosci 98:90–3
- Carrive P. (2006). Dual activation of cardiac sympathetic and parasympathetic components during conditioned fear to context in the rat. Clin Exp Pharmacol Physiol 33:1251–4
- Chung IM, Kim YM, Yoo MH, Shin MK, Kim CK, Suh SH. (2010). Immobilization stress induces endothelial dysfunction by oxidative stress via the activation of the angiotensin II/its type I receptor pathway. Atherosclerosis 213:109–14
- Cierco M, Israel A. (1994). Role of angiotensin AT1 receptor in the cardiovascular response to footshock. Eur J Pharmacol 251:103–6
- Conti LH, Shannon MH, Murry JD, Printz MP. (2001). Repeated restraint stress-induced increase in baroreceptor reflex sensitivity: role of corticotropin-releasing factor. Neuropeptides 35:71–81
- Cowley Jr AW, Liard JF. (1988). Vasopressin and arterial pressure regulation. Special lecture. Hypertension 11:I25–32
- Crestani CC, Tavares RF, Alves FH, Resstel LB, Correa FM. (2010). Effect of acute restraint stress on the tachycardiac and bradycardiac responses of the baroreflex in rats. Stress 13:61–72
- Dayas CV, Buller KM, Crane JW, Xu Y, Day TA. (2001). Stressor categorization: acute physical and psychological stressors elicit distinctive recruitment patterns in the amygdala and in medullary noradrenergic cell groups. Eur J Neurosci 14:1143–52
- Di Rienzo M, Parati G, Radaelli A, Castiglioni P. (2009). Baroreflex contribution to blood pressure and heart rate oscillations: time scales, time-variant characteristics and nonlinearities. Philos Trans Ser A, Math Phys Eng Sci 367:1301–18
- Dimsdale JE. (2008). Psychological stress and cardiovascular disease. J Am Coll Cardiol 51:1237–46
- Donoso AO, Bacha JC. (1975). Role of the blood-brain barrier in the anticholinergic differential effects on LH and prolactin release in proestrous rats. J Neural Trans 37:155–64
- Figueiredo HF, Bruestle A, Bodie B, Dolgas CM, Herman JP. (2003). The medial prefrontal cortex differentially regulates stress-induced c-fos expression in the forebrain depending on type of stressor. Eur J Neurosci 18:2357–64
- Forsman L, Lindblad LE. (1983). Effect of mental stress on baroreceptor-mediated changes in blood pressure and heart rate and on plasma catecholamines and subjective responses in healthy men and women. Psychosom Med 45:435–45
- Gibbs DM. (1984). Dissociation of oxytocin, vasopressin and corticotropin secretion during different types of stress. Life Sci 35:487–91
- Graeff FG. (2007). [Anxiety, panic and the hypothalamic-pituitary-adrenal axis]. Revista brasileira de psiquiatria 29(Suppl 1):S3–6
- Grippo AJ, Johnson AK. (2009). Stress, depression and cardiovascular dysregulation: a review of neurobiological mechanisms and the integration of research from preclinical disease models. Stress 12:1–21
- Guyenet PG. (2006). The sympathetic control of blood pressure. Nature Rev Neurosci 7:335–46
- Herman JP, Cullinan WE. (1997). Neurocircuitry of stress: central control of the hypothalamo-pituitary-adrenocortical axis. Trends Neurosci 20:78–84
- Huang JL, Chiou CW, Ting CT, Chen YT, Chen SA. (2001). Sudden changes in heart rate variability during the 1999 Taiwan earthquake. Am J Cardiol 87:245–8, A249
- Israel A, Sosa-Canache B. (2002). Angiotensin II supports sympathetically mediated vasopressor response to footshock-stress. J Human Hypertens 16(Suppl 1):S84–8
- Jindra Jr A, Kvetnansky R. (1982). Stress-induced activation of inactive renin. Molecular weight aspects. J Biol Chem 257:5997–9
- Kaehler ST, Sinner C, Philippu A. (2000). Release of catecholamines in the locus coeruleus of freely moving and anaesthetized normotensive and spontaneously hypertensive rats: effects of cardiovascular changes and tail pinch. Naunyn-Schmiedeberg's Arch Pharmacol 361:433–9
- Kapusta DR, Dayan LA, Kenigs VA. (2002). Nociceptin/orphanin FQ modulates the cardiovascular, but not renal, responses to stress in spontaneously hypertensive rats. Clin Exp Pharmacol Physiol 29:254–9
- Kario K, Matsuo T, Kobayashi H, Yamamoto K, Shimada K. (1997). Earthquake-induced potentiation of acute risk factors in hypertensive elderly patients: possible triggering of cardiovascular events after a major earthquake. J Am Coll Cardiol 29:926–33
- Kawashima T. (2005). The autonomic nervous system of the human heart with special reference to its origin, course, and peripheral distribution. Anatomy Embryol 209:425–38
- Kim SM, Chen L, Faulhaber-Walter R, Oppermann M, Huang Y, Mizel D, Briggs JP, Schnermann J. (2007). Regulation of renin secretion and expression in mice deficient in beta1- and beta2-adrenergic receptors. Hypertension 50:103–9
- Krieger EM. (1964). Neurogenic hypertension in the rat. Circulation Res 15:511–21
- Kubo T, Kanaya T, Numakura H, Okajima H, Hagiwara Y, Fukumori R. (2002). The lateral septal area is involved in mediation of immobilization stress-induced blood pressure increase in rats. Neurosci Lett 318:25–8
- Lam W, Louis WJ, Verberne AJ. (1995). Effect of dorsal periaqueductal grey lesion on baroreflex and cardiovascular response to air-jet stress. J Auton Nerv Syst 53:35–42
- Lovallo WR, Gerin W. (2003). Psychophysiological reactivity: mechanisms and pathways to cardiovascular disease. Psychosom Med 65:36–45
- Majewski H, Alade PI, Rand MJ. (1986). Adrenaline and stress-induced increases in blood pressure in rats. Clin Exp Pharmacol Physiol 13:283–8
- McDougall SJ, Lawrence AJ, Widdop RE. (2005). Differential cardiovascular responses to stressors in hypertensive and normotensive rats. Exp Physiol 90:141–50
- McEwen BS. (1998). Protective and damaging effects of stress mediators. The New England journal of medicine 338:171–9
- McEwen BS. (2000). Protective and damaging effects of stress mediators: central role of the brain. Progr Brain Res 122:25–34
- Mitsushima D, Yamada K, Takase K, Funabashi T, Kimura F. (2006). Sex differences in the basolateral amygdala: the extracellular levels of serotonin and dopamine, and their responses to restraint stress in rats. Eur J Neurosci 24:3245–54
- Muller JR, Le KM, Haines WR, Gan Q, Knuepfer MM. (2001). Hemodynamic response pattern predicts susceptibility to stress-induced elevation in arterial pressure in the rat. Am J Physiol Regulatory Integr Compar Physiol 281:R31–7
- Nosaka S. (1996). Modifications of arterial baroreflexes: obligatory roles in cardiovascular regulation in stress and poststress recovery. Japanese J Physiol 46:271–88
- Pacak K, Palkovits M. (2001). Stressor specificity of central neuroendocrine responses: implications for stress-related disorders. Endocrine Rev 22:502–48
- Padovan CM, Guimaraes FS. (2000). Restraint-induced hypoactivity in an elevated plus-maze. Brazil J Medical Biolog Res 33:79–83
- Paton JF, Boscan P, Pickering AE, Nalivaiko E. (2005). The yin and yang of cardiac autonomic control: vago-sympathetic interactions revisited. Brain Res Brain Res Rev 49:555–65
- Randall DC, Brown DR, Brown LV, Kilgore JM. (1994). Sympathetic nervous activity and arterial blood pressure control in conscious rat during rest and behavioral stress. Am J Physiol 267:R1241–9
- Reis DG, Scopinho AA, Guimaraes FS, Correa FM, Resstel LB. (2011). Behavioral and autonomic responses to acute restraint stress are segregated within the lateral septal area of rats. PLoS One 6:e23171
- Resstel LB, Joca SR, Guimaraes FG, Correa FM. (2006). Involvement of medial prefrontal cortex neurons in behavioral and cardiovascular responses to contextual fear conditioning. Neuroscience 143:377–85
- Sarenac O, Lozic M, Drakulic S, Bajic D, Paton JF, Murphy D, Japundzic-Zigon N. (2011). Autonomic mechanisms underpinning the stress response in borderline hypertensive rats. Exp Physiol 96:574–89
- Sawchenko PE, Li HY, Ericsson A. (2000). Circuits and mechanisms governing hypothalamic responses to stress: a tale of two paradigms. Progr Brain Res 122:61–78
- Tavares RF, Correa FM. (2006). Role of the medial prefrontal cortex in cardiovascular responses to acute restraint in rats. Neuroscience 143:231–40
- Taylor J, Harris N, Krieman M, Vogel WH. (1989). Effects of buspirone on plasma catecholamines, heart rate, and blood pressure in stressed and nonstressed rats. Pharmacol Biochem Behav 34:349–53
- van den Buuse M, Van Acker SA, Fluttert M, De Kloet ER. (2001). Blood pressure, heart rate, and behavioral responses to psychological “novelty” stress in freely moving rats. Psychophysiology 38:490–9
- Yoshino K, Hayakawa M, Niki E, Matsuoka K. (2005). Closed-loop analysis of cardiovascular variability in rats under restraint stress. Auton Neurosci 119:61–6
- Zhang ZQ, Julien C, Barres C. (1996). Baroreceptor modulation of regional haemodynamic responses to acute stress in rat. J Auton Nerv Syst 60:23–30