
Abstract
Stress and ethanol are important cardiovascular risk factors. Their vascular and blood pressure (BP) effects were evaluated alone and in combination. Adult male Wistar rats (8–10 per group) were separated into control, ethanol (ethanol 20% in drinking water for 6 weeks), stress (restraint 1 h/d 5 d/week for 6 weeks), and ethanol/stress (in combination) groups. Systolic BP was evaluated weekly. Concentration–response curves for contractile responses to angiotensin II in the absence and the presence of losartan (AT1-blocker), PD123-319 (AT2-blocker), L-NAME (nitric oxide synthase inhibitor), or indomethacin (cyclooxygenase inhibitor) were obtained in isolated intact and endothelium-denuded aortas. Effective concentration 50% (EC50) and maximum response (MR) were compared among groups using MANOVA/Tukey tests. Stress and stress plus ethanol increased BP. Ethanol and stress, alone and in combination, did not alter angiotensin responses of intact aortas. PD123-319 decreased MR to angiotensin II in intact aortas from the ethanol and ethanol/stress groups relative to control in the presence of PD123-319. Losartan increased MR to angiotensin II in intact aortas from the stress and ethanol/stress groups relative to control in the presence of losartan. None of the protocols altered angiotensin responses of denuded aortas. Neither indomethacin nor L-NAME altered angiotensin responses of intact aortas from the experimental groups. Thus ethanol and ethanol plus stress may alter endothelial signaling via AT1-receptors, without changing systemic BP. Stress and stress plus ethanol may alter endothelial signaling via AT2-receptors, and thereby increase BP. Knowledge of such vascular changes induced by stress and/or ethanol may contribute to understanding adverse cardiovascular effects of stress and ethanol consumption in humans.
Introduction
Chronic stress is an important risk factor for the development of cardiovascular pathologies such as atherosclerosis and arterial hypertension (Chung et al., Citation2010; Cordellini & Vassilieff, Citation1998; Diaconu et al., Citation2011; Ghiadoni et al., Citation2000). Moreover, the severity of the cardiovascular damage caused by stress exposure depends on the nature of the stressor, as well as on its intensity and duration (Hjemdahl, Citation2002). The literature reports that chronic stress impairs endothelial function, as evidenced by the increase of angiotensin-induced vasoconstriction in the aorta due to reduced endothelial nitric oxide (NO) buffering capacity via the angiotensin/AT2 receptor pathway (Loria et al., Citation2011). Oxidative stress, via activation of the angiotensin/AT1 receptor pathway, has also been shown to impair endothelial function (Chung et al., Citation2010).
Similarly, ethanol consumption may cause cardiocirculatory alterations that contribute to the development of cardiovascular diseases (Tirapelli et al., Citation2007, Citation2008b). In this regard, chronic ethanol consumption induces activation of the renin–angiotensin system (Wakabayashi & Hatake, Citation2001). Moreover, the arterial hypertension reported in this condition seems to be correlated with the activation of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase causing endothelial injury and impairment of the endothelial NO-generating system (Husain et al., Citation2005, Citation2008, Citation2010).
Our current understanding of the development of cardiovascular diseases recognizes the significance of multiple risk factors, which have been shown to be directly implicated in the genesis, progression, and occurrence of future cardiovascular events. Two of these cardiovascular risk factors are ethanol consumption and stress exposure. While many studies examine the effects of ethanol consumption alone, this condition usually happens in the context of various risk factors, including stress exposure able to modify the impact of ethanol consumption on the body. However, the consequences of chronic ethanol consumption in association with stress exposure on vascular function remain to be determined. Moreover, the literature has shown that although long-term ethanol consumption can affect cardiovascular functions, early damage caused by ethanol consumption on the cardiovascular system also needs to be considered (Resstel et al., Citation2006).
Thus, the purpose of the present study was to evaluate the cardiovascular risk of stress exposure and ethanol consumption, alone and in combination, through the investigation of possible changes in arterial blood pressure and vascular reactivity to angiotensin II, assessed in vitro. The second purpose was to evaluate in detail the mechanisms underlying the effects of long-term ethanol consumption and stress exposure, alone and in combination, on angiotensin-induced contraction of the aorta. This evaluation was conducted with a focus on NO, cyclooxygenase, and endothelial AT1/AT2-signaling pathways, as well as plasma antioxidant capacity.
Methods
Experimental design
Experiments were performed on adult male Wistar rats (90 to 120-d old) obtained from UNESP – Univ Estadual Paulista facilities. They were housed in individual plastic cages under a 12-h light–dark cycle (lights on/off at 07:00/19:00 h) at 23 ± 2 °C and fed with regular lab chow. The rats were separated into four groups (8–10 rats per group): control (received water ad libitum), stress (restraint for 1 h/d 5 d/week for 6 weeks), ethanol [20% (v/v) ethanol solution instead of tap water for 6 weeks (Tirapelli et al., Citation2006)], and ethanol/stress (20% ethanol solution and restraint stress for 6 weeks). Rats from the ethanol and ethanol/stress groups were adapted to ethanol consumption by gradually increasing the concentration of ethanol in their drinking water (5% in the first week, 10% in the second week, 20% in the third week, and 20% for 6 additional weeks).
Rats were restrained, but not immobilized, in a cylindrical metallic tube measuring 27.0 cm long × 6.1 cm internal diameter containing ventilation holes, individually adapted to completely restrict movements, preserving only their breathing. A slotted opening allowed for free mobility of the tail. Exposure to this stressful stimulus took place between 08:00 h and 10:00 h. Restraint is a well-validated method to induce stress (Buynitsky & Mostofsky, Citation2009). The duration of the stressor used, i.e. the length of time of exposure during each session, was based on the work of Marin et al. (Citation2007).
During the stress sessions, rats from the control and ethanol groups remained in their cages. Twenty-four hours after the last stress session, the rats were killed by decapitation, without anesthesia, and the aorta was removed for different experimental protocols.
Animal procedures were in accordance with the principles and guidelines of the National Council of Control of Animal Experimentation, and the study was approved by the Ethical Committee for Animal Experimentation (protocol number: 295-CEEA).
Measurement of blood pressure
Systolic blood pressure (mmHg) was determined weekly during the adaptation and exposure periods. Measurements were taken from conscious rats using the tail-cuff plethysmographic method (Narco Bio-Systems, Inc., Houston, TX). The rats were pre-warmed in a Bio-Heater (Insight Ltda. Ribeirão Preto, Brazil) at ∼40 °C for 5–10 min and then placed into a restrainer tube for blood pressure measurement. Three consecutive recordings (∼1 min apart) were performed, and the mean of these three measurements was recorded.
Vascular reactivity protocol
Immediately after the rat had been decapitated, the descending thoracic aorta was excised and trimmed free of adhering fat and connective tissue. Two transverse rings of the aorta, each about 4 mm in length, were cut. One ring served as a control (intact aorta), while the endothelium was mechanically removed from the other by gently rubbing the luminal surface (denuded aorta). The rings were mounted into a 2-ml organ chamber containing the Krebs–Henseleit solution, with a composition (in mM) of NaCl 130.0, KCl 4.7, CaCl2 1.6, KH2PO4 1.2, MgSO4 1.2, NaHCO3 15.0, and glucose 11.1. The Krebs–Henseleit solution was kept at pH 7.4 and 37 °C and bubbled continuously with a mixture of 95% O2 and 5% CO2. Tension was monitored continuously and recorded using a Powerlab 8/30 data-acquisition system (AD Instruments, Castle Hill, NSW, Australia). Prior to the collection of contractile responses to increasing agonist concentrations, to construct concentration–response curves, the rings were equilibrated for 60 min under a resting tension of 1.5 g, which is optimal for inducing a maximum response. All drugs were obtained from Sigma Chemical Co., St Louis, MO. They were dissolved in the Krebs–Henseleit solution and the concentrations were expressed in molarity.
The functional state of the endothelium was tested at the beginning of the concentration–response curve measurements by the ability of 10−4 M acetylcholine to elicit vasodilator responses in preparations pre-contracted by 10−5 M phenylephrine. Preparations that presented more than 80% of relaxation were considered as having an intact endothelium, whereas those that showed no relaxation were considered completely devoid of endothelium. Preparations with and without endothelium were studied in parallel.
Angiotensin II (10−10–10−6 M) was cumulatively added to the organ bath, and the evoked responses (g of tension) were plotted to obtain the cumulative concentration–response curves. At the end of obtaining these curves, a single dose of sodium nitroprusside (10−4 M) was used to test the integrity of the smooth muscle layer.
Involvement of endothelial AT1/AT2-signaling pathway
Cumulative concentration–response curves to angiotensin II were also obtained in intact and denuded aortas in the presence of losartan (10−7 M, specific AT1-receptor blocker) or PD123-319 (10−7 M, specific AT2-receptor blocker) introduced into the organ bath 20 min before starting measurement of angiotensin II responses.
Involvement of NO and cylooxygenase pathways
In another series of experiments, angiotensin II responses were obtained in intact aortas pretreated with L-NG-nitroarginine methyl ester (L-NAME; 10−4 M, non-selective nitric oxide synthase inhibitor) or indomethacin (10−5 M, non-selective cyclooxygenase inhibitor) added to the organ bath 20 min before measuring angiotensin II responses.
Plasma corticosterone concentration
Immediately after the rat had been decapitated, the trunk blood was collected in plastic tubes containing heparin solution 5000 IU/ml (10 μl/ml blood). The whole blood was centrifuged at 3000g for 20 min at 4 °C and the plasma was separated for later extraction. Plasma corticosterone concentrations, a classic index for a stressful paradigm, were determined by using the Vecsei (Citation1979) method, modified by the previous extraction of the steroid with 1 ml of ethanol. For the assay, the anti-corticosterone antibody, AB-cort-17984, was used. The antibody was sourced from rabbit, with the hormone conjugated to bovine albumin. The specificity of the antibody was tested for cross-reactivity with cortisol (1%), 17-OH-progesterone (1%), and dehydroepiandrosterone sulfate (12%). Tritium-labeled corticosterone was used as the labeled hormone and the separation of the bound and free fraction was carried out with a solution of charcoal/dextran 0.5/0.05%. The minimum detectable concentration was 0.4 µg/dl. The coefficients of intra- and inter-assay variations were 2.8% and 17.8%, respectively.
Plasma antioxidant capacity
Ferric-reducing ability of plasma (FRAP) measurements were taken using a slightly modified version of the methods outlined by Benzie & Strain (Citation1996).
The FRAP technique is based on the ability of plasma to reduce Fe+++ ions to Fe++ in the presence of 2,4,6-tripyridyl-s-triazine at low pH, forming an Fe++ complex tripyridyl-s-triazine; the blue coloring is measured as a change of absorbance at 593 nm. The working reagent was prepared by adding 25 ml of acetate buffer pH 3.6, 2.5 ml 2,4,6-tripyridyl-s-triazine, 10–40 mM HCl, and 2.5 ml of FeCl3.6H2O 20 mM. For measurements, 300 µl of the working reagent was placed in a cuvette and incubated in a water bath at 37 °C for 5 min. Next, 10 µl of plasma were added, followed by 30 µl of H2O. The solution was then incubated in a water bath at 37 °C for exactly 15 min. Subsequently, changes in the optical density were recorded in a spectrophotometer (Quimis, Madrid, Spain) against a reagent blank. The values of the antioxidant capacity of plasma were calculated by comparing the absorbance with the absorbance of Fe++ solutions of known concentrations (100–1000 mM), which were tested in parallel.
Data analysis and statistics
The concentration of angiotensin II producing a response that was 50% of the maximum (EC50) was calculated in each experiment. Data are presented as mean ± SEM or SD. The maximal responses and EC50 values were compared by the two-way ANOVA followed by Tukey’s post-test (SigmaStat 3.2, SPSS Inc., Chicago, IL). The factors in the analysis were ethanol consumption and stress exposure. p < 0.05 was considered statistically significant. If necessary, variables were log transformed prior to parametric testing.
Results
Blood pressure
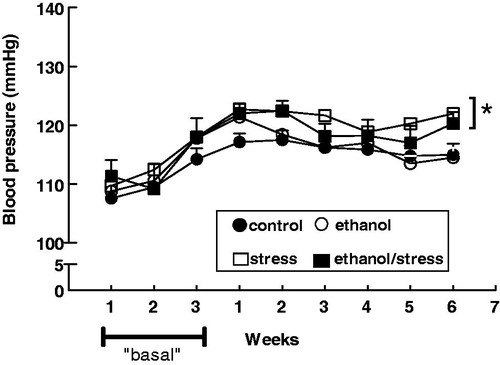
Exposure to stress, alone and in combination with ethanol, resulted in an increase in the blood pressure in the last week of exposure (sixth week) when compared with the control group. On the contrary, no change in the blood pressure was observed after chronic ethanol consumption alone ().
Figure 1. Evolution of caudal systolic blood pressure of adult male rats submitted to chronic ethanol consumption and exposed to stress, alone and in combination. Control – received water ad libitum; stress – restraint 1 h/day 5 d/week for 6 weeks; ethanol – 20% ethanol solution instead of tap water for 6 weeks; ethanol/stress – restraint stress and 20% ethanol solution for 6 weeks. “Basal” – the adaptation period for ethanol intake. Values are means ± SEM. The two-way ANOVA followed by Tukey’s post-test. (*) p < 0.05 relative to control. The number of rats per group = 8–10.
Aorta reactivity to angiotensin II
In the absence of receptor blockers, the vasoconstriction induced by angiotensin II in intact and denuded aortas was not altered by any of the experimental protocols, i.e. chronic ethanol consumption and stress exposure alone and in combination ().
Table 1. Maximum response and EC50 values to angiotensin II in the absence and presence of losartan or PD123-319 obtained in thoracic aorta rings, with and without endothelium, isolated from adult male rats submitted or not to ethanol consumption and restraint stress, alone and in combination.
Involvement of endothelial AT1/AT2-signaling pathway
In intact aortas taken from the control and ethanol groups, the presence of the AT1-receptor blocker, losartan, determined a similar decrease in the maximum response to angiotensin II compared with the same preparations in the absence of a blocker (). However, the presence of this antagonist did not cause any change in the reactivity to angiotensin II in intact aortas from rats submitted to stress – whether alone or in combination – compared with the same preparations in the absence of a blocker ().
The presence of AT2-receptor blocker, PD123-319, caused a similar decrease in the maximum response to angiotensin II in intact aortas from ethanol and ethanol/stress groups compared with the same preparations in the absence of a blocker (). However, the presence of this antagonist did not cause any change in the reactivity to angiotensin II in the intact aortas of rats from the control and stress groups compared with the same preparations in the absence of a blocker ().
In the absence of blockers, the EC50 values for angiotensin II in aortas did not differ among different experimental groups, either in the absence or in the presence of endothelium (). The presence of losartan, but not PD123-319, increased the EC50 values for angiotensin II in intact and denuded aortas, with similar values in all experimental groups ().
The removal of endothelium increased the maximum response to angiotensin II that reached similar values among all experimental groups and protocols, without altering the EC50 values ().
Involvement of NO and cyclooxygenase pathways
The presence of indomethacin did not cause any change in the reactivity to angiotensin II in intact aortas from all experimental groups (). The presence of L-NAME determined a similar increase in the reactivity to angiotensin II in intact aortas from all experimental groups ().
Table 2. Maximum response and EC50 to angiotensin II values in the presence of indomethacin or L-NAME obtained in intact aorta rings isolated from adult male rats submitted or not to ethanol consumption and restraint stress, alone and in combination.
Corticosterone concentration
The plasma concentration of corticosterone was higher in rats submitted to stress, both alone and in combination with ethanol, but was not altered by ethanol consumption alone [mean ± SEM: control 2.26 ± 0.20 µg/dl; stress 4.03 ± 0.50* µg/dl; ethanol 2.27 ± 0.27 µg/dl; ethanol/stress 4.44 ± 0.53† µg/dl; n = 8–12; *p = 0.0195 and †p = 0.0059 compared with control (the two-way ANOVA followed by Tukey’s post-test)].
Plasma antioxidant capacity
No statistically significant alteration in plasma antioxidant capacity was observed in any of the experimental groups [mean ± SEM: control 762.6 ± 37.4 µmol/l Fe+2; stress 817.6 ± 50.0 µmol/l Fe+2; ethanol 736.2 ± 31.9 µmol/l Fe+2; ethanol/stress 686.3 ± 38.0 µmol/l Fe+2; n = 10–12; p > 0.05 compared with control (the two-way ANOVA followed by Tukey’s post-test)].
Discussion
The present study demonstrated that chronic exposure to restraint stress, regardless of ethanol consumption, increased blood pressure and impaired the aortic endothelial AT2 pathway that mediates endothelium-dependent relaxation of smooth muscle to angiotensin II. Conversely, chronic ethanol consumption, regardless of exposure to stress, impaired the endothelial AT1 pathway that mediates endothelium-dependent constriction of smooth muscle to angiotensin II, but did not alter blood pressure.
Experimental and epidemiological evidence suggests that both stress and ethanol play an important role in the development of cardiovascular disease (Chung et al., Citation2010; Husain et al., Citation2010; Rozanski et al., Citation2005). However, there are discrepancies in the literature regarding the effects of ethanol consumption and stress exposure on regulatory cardiovascular responses.
Low concentrations of ethanol seem to be beneficial to endothelial function and, therefore, may decrease blood pressure (Jones et al., Citation2013; Toda & Ayajiki, Citation2010). However, high doses of ethanol or its chronic consumption can determine a loss in function and impaired viability of endothelial cells (Toda & Ayajiki, Citation2010) as well as significant changes in the heart and circulatory functions. These findings have identified high/chronic ethanol consumption as a significant risk factor in the development of cardiovascular diseases such as arterial hypertension (Husain et al., Citation2005, Citation2008; Sun & Mayhan, Citation2001; Tirapelli et al., Citation2007, Citation2008a). However, Hatton et al. (Citation1992) and Kleinhenz et al. (Citation2008) reported a decrease in blood pressure of rats subjected to chronic ethanol consumption. In the present study, however, no change in blood pressure was observed among rats submitted to chronic ethanol consumption alone.
In men, alcohol consumption has been categorized as light (0.1–9.9 g/d, or <1 drink daily), moderate (10.0–29.9 g/d, or 1–2 drinks daily), and heavier (≥30.0 g/d, or ≥3 drinks daily) (Mukamal et al., Citation2005). In the present study, the use of 20% ethanol corresponded to a daily intake of 6.0–8.0 g. Thus, the chronic ethanol consumption protocol employed in this study might be considered a light daily intake of alcohol, which could partially explain the lack of alteration in blood pressure observed after this protocol alone.
The literature is also not in agreement in regard to cardiovascular adaptive responses induced by stress exposure. Increase or decrease in blood pressure depends on the stressor and the duration and/or frequency of stress exposure (Chung et al., Citation2010; Matthews et al., Citation2001; Sparrow et al., Citation1987). In the present study, chronic exposure to restraint stress alone and in combination with ethanol consumption resulted in hypertension in the last week of exposure.
The most common responses to stress involve activation of the sympathetic nervous system, hypothalamic–pituitary–adrenal axis, and renin–angiotensin system (Hjemdahl, Citation2002; McEwen, Citation1998; Viau & Meaney, Citation1991). As a consequence, elevations in serum corticosterone and catecholamine concentrations can be seen after stress exposure (Pacák & Palkovits, Citation2001). Corroborating the literature, the chronic exposure to stress alone and in combination with ethanol consumption caused a significant increase in plasma corticosterone concentration. As the chronic stress state has been defined as persistent visceral arousal coupled with behavioral abnormalities (Ottenweller et al., Citation1992), the increase in corticosterone concentration partially meets the assumption of the chronic stress paradigm and thus validates our procedure. On the contrary, chronic ethanol consumption did not alter the plasma corticosterone concentration suggesting that this protocol did not represent a stressogenic condition.
Both stress and chronic ethanol consumption induce activation of the renin–angiotensin–system. This endocrine system has angiotensin II as one of its main effectors whose effects are experienced by interacting with the specific receptors, subtype-1 (AT1) and subtype-2 (AT2) (Leung, Citation2004; Wakabayashi & Hatake, Citation2001).
Angiotensin II induces constriction of the vascular wall by a direct action on smooth muscle cells, but this effect might be modulated by the action of angiotensin II on endothelial cells. Similar to smooth muscle, the endothelial cells present AT1 and AT2 receptors that are involved in the release of endothelium-derived relaxing and/or contracting factors (Pueyo & Michel, Citation1997). The relative predominance of one or other of the AT-receptor pathways may determine the final action of angiotensin II in these cells.
Normally, the contractile response overcomes relaxation. In several vascular beds, activation of AT1- and AT2-receptors leads to antagonistic effects, promoting a counterbalance in vasoactive responses (Fyhrquist & Saijonmaa, Citation2008). This is evident in the aorta of rats that have had the gene for the AT2-receptor deleted, where the contractile response to angiotensin II was higher compared with the control group (Akishita et al., Citation1999). Moreover, the blockade of AT1-receptors lowers while the AT2 blockade increases blood pressure in rats, indicating again a counterbalance in response to angiotensin II (Munzenmaier & Greene, Citation1996). Thus, AT2 signaling is antagonistic to AT1 signaling and results in bradykinin and NO formation (Rush & Aultman, Citation2008). However, AT1 signaling results in increased superoxide production via NADH/NADPH oxidase activation and enhanced generation of endothelium-derived constricting factors, such as endothelin-1 (MacKenzie, Citation2011; Toda et al., Citation2007). In this context, it should be considered that the AT1-receptor blockade leaves uncovered the endothelial signaling pathway through AT2-receptors that becomes a brake for the constriction caused by angiotensin II acting directly on the smooth muscle cell. Furthermore, the blockade of AT2-receptors leaves uncovered the endothelial signaling pathway through AT1-receptors that increases the vasoconstriction by angiotensin II acting directly on the smooth muscle cells. Corroborating these findings, angiotensin II-induced vasoconstriction was potentiated by the removal of endothelium and L-NAME presence, regardless of the protocols of exposure.
It is noteworthy that the increase in blood pressure of rats chronically treated with ethanol is related to increased angiotensin II concentration in plasma and aorta (Husain et al., Citation2008). Angiotensin II is also responsible for the amplification of the actions of the sympathetic nervous system (Kawasaki et al., Citation1984; Malik & Nasjletti, Citation1976). Moreover, chronic angiotensin II signaling induces vascular dysfunction; however, pharmacological management of the renin–angiotensin system can not only control blood pressure but also correct endothelial dysfunction (Rush & Aultman, Citation2008). In this context, the effects of chronic ethanol consumption and stress exposure – alone and in combination – were investigated. This investigation was conducted through analysis of vasomotor responses of aorta to angiotensin II in the absence and presence of losartan, PD123-319, indomethacin, or L-NAME.
Neither ethanol consumption nor stress exposure, alone or in combination, caused any change in aortic responses to angiotensin II. Nevertheless, previous studies from our laboratory have shown that the decrease in the vascular reserves determined by the presence of inhibitors such as L-NAME and indomethacin may unveil vascular changes that are below the level of security of vascular function (Grizzo & Cordellini, Citation2008). In this context, the final responses to angiotensin II in aortas of rats submitted to stress and/or ethanol intake may vary considering the changes in the endothelial cell output determined by the presence of AT-receptor blockers, L-NAME, or indomethacin. However, in the present study, the presence of neither inhibitors nor blockers unveiled any change in vascular reactivity to angiotensin II in denuded aortas of adult rats exposed to stress and ethanol consumption, alone and in combination.
In contrast, in intact aorta, the presence of losartan showed an impairment of AT2-receptor endothelial signaling induced by chronic exposure to stress, which was seen regardless of ethanol consumption. The impairment of this signaling was inferred from the observation that the presence of losartan resulted in similar degrees of hyperreactivity to angiotensin II in intact aortas from the stress and stress/ethanol groups when compared with the control group in the same condition, i.e. the presence of losartan. Moreover, this difference was abolished by the removal of endothelium. Conversely, the presence of PD123-319 did not show any stress-induced alteration in endothelial signaling via AT1-receptors, as the aortic reactivity to angiotensin II was similar in the control and stress groups in the presence of this blocker. These data indicate a decrease in the bioavailability of endothelial-derived relaxing factors via AT2-receptor as a result of stress exposure, alone and in combination with ethanol. Moreover, the change in the endothelial cell output could be responsible for the etiology and/or maintenance of the hypertensive state observed in the stress condition. Taken in conjunction, these findings raise concern about the higher cardiovascular vulnerability of stress-exposed individuals in the presence of associated pathologies that impair the endothelial AT2-signaling pathway.
In addition, the presence of PD123-319 showed an impairment of AT1-receptor endothelial signaling induced by chronic ethanol consumption, alone and in combination with stress. The impairment of this signaling was inferred from the observation that the presence of PD123-319 resulted in similar degrees of hyporeactivity to angiotensin II in intact aortas from the ethanol and ethanol/stress groups when compared with the control group in the same condition, i.e. the presence of PD123-319. Moreover, this difference was abolished by the removal of endothelium. Conversely, the presence of losartan did not show any ethanol-induced alteration in endothelial signaling via AT2-receptors, as the aortic reactivity to angiotensin II was similar in the control and ethanol groups in the presence of this blocker. These data indicate that ethanol consumption, alone and in combination with stress, induces a decreased bioavailability of endothelium-derived contracting factors via AT1-receptors. This beneficial effect might be partially responsible for there being no change in the blood pressure among rats submitted to chronic ethanol consumption. Thus, this finding expands on previous reports showing the beneficial cardiovascular effects of low concentrations of alcohol consumption (Jones et al., Citation2013; Toda & Ayajiki, Citation2010).
Although the results show that chronic ethanol in association with stress exposure alters endothelial signaling via AT1- and AT2-receptors in intact rat aortas, there was no sign of interaction between these factors. This conclusion was reached based on the finding that the hyperreactivity to angiotensin in the aortas of stressed rats in the presence of losartan, as well as the hyporeactivity displayed in those subjected to chronic ethanol consumption in the presence of PD123-319, of similar magnitudes to those observed in rats from the ethanol/stress group in the presence of the same antagonists. Thus, in this study, chronic consumption of ethanol and stress exposure did not constitute additional cardiovascular risk factors, reinforcing the literature reports showing that moderate alcohol consumption might reduce cardiovascular disease (Jones et al., Citation2013).
The question arises about whether changes in vascular reactivity to stress and ethanol consumption are specific for the angiotensin system. However, previous studies in our laboratory have shown that a similar protocol of stress exposure, alone and in combination with ethanol consumption, increases noradrenaline-induced contraction in rat aorta (Baptista et al., Citation2014). Moreover, Tirapelli et al. (Citation2008b) reported that a similar protocol of ethanol consumption increases phenylephrine-induced contraction, as well as decreases acetylcholine-induced relaxation, in the rat mesenteric arterial bed. It is also important to consider that the vascular reactivity alterations may be blood pressure dependent since the stress-exposed rats were hypertensive at the point of the ex vivo experiments with the vascular tissue. However, further studies are necessary to clarify this correlation.
The literature has also reported the involvement of cyclooxygenase and/or nitric oxide pathways in changes to vascular reactivity as a result of chronic ethanol consumption and stress exposure (Cordellini et al., Citation2006; Loria et al., Citation2011; Tirapelli et al., Citation2006). In order to investigate the role, these pathways may play in the alteration of aortic reactivity to angiotensin II, indomethacin and L-NAME were used. However, in our experiments, the presence of these inhibitors did not change aortic reactivity to angiotensin II in any of the experimental groups. This outcome effectively ruled out the involvement of cyclooxygenase and nitric oxide pathways in the endothelial alterations induced by ethanol consumption and stress exposure alone and in combination. These data also corroborate previous reports from our laboratory showing no change in the plasma concentration of nitrites/nitrates in rats submitted to the same protocols of exposure (Baptista et al., Citation2014). Similarly, the FRAP method did not show any change in plasma antioxidant capacity throughout all experimental groups, suggesting that the antioxidant system is not involved in the changes in bioavailability of endothelial factors induced by the ethanol and stress conditions. Taking these data into consideration, the identity of the endothelial factors involved in the aortic adaptive response brought on by stress exposure and ethanol consumption alone and in combination requires further investigation.
Conclusions
Chronic ethanol consumption alone and in combination with stress altered endothelial signaling via AT1-receptors, resulting in aortic hyporeactivity to angiotensin II in the presence of an AT2-receptor blocker. Ethanol consumption alone resulted in no change in blood pressure. Meanwhile, exposure to stress alone and in combination with ethanol altered endothelial signaling via AT2-receptors, resulting in aortic hyperreactivity to angiotensin II in the presence of an AT1-receptor blocker, as well as increased blood pressure. However, the mechanisms involved in these alterations remain to be elucidated. Thus, data presented here may contribute to the body of evidence about disordered cardiovascular function induced by chronic stress and ethanol intake.
Declaration of interest
The authors declare that they have no conflict of interest. The authors would like to thank the agency of Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES – Brasil) for their financial support.
Acknowledgements
The authors would like to thank Mr. Alisson Douglas Ventura Neves for his technical assistance and Dr. John A. Russell for his help in reviewing and editing the manuscript.
References
- Akishita M, Yamada H, Dzau VJ, Horiuchi M. (1999). Increased vasoconstrictor response of the mouse lacking angiotensin II type 2 receptor. Biochem Biophys Res Commun 261(2):345–9
- Baptista RFF, Taipeiro EF, Queiroz RHC, Chies AB, Cordellini S. (2014). Stress alone or associated with ethanol induces prostanoid release in rat aorta via α2-adrenoceptor. Arq Bras Cardiol 102(3):211–18
- Benzie IFF, Strain JJ. (1996). The ferric reducing ability of plasma (FRAP) as a measure of “antioxidant power”: the FRAP assay. Anal Biochem 239:70–6
- Buynitsky T, Mostofsky DI. (2009). Restraint stress in biobehavioral research: recent developments. Neurosci Biobehav Rev 33(7):1089–98
- Chung IM, Kim YM, Yoo MH, Shin MK, Kim CK, Suh SH. (2010). Immobilization stress induces endothelial dysfunction by oxidative stress via the activation of the angiotensin II/its type I receptor pathway. Atherosclerosis 213:109–14
- Cordellini S, Novo R, Lanza Júnior U. (2006). Exposure to stress differential vascular adaptive response in spontaneously hypertensive and Wistar rats: role of nitric oxide, and prehypertensive and hypertensive states. Life Sci 79(7):646–53
- Cordellini S, Vassilief VS. (1998). Decreased endothelium-dependent vasoconstriction to noradrenaline in acute-stressed rats is potentiated by previous chronic stress: nitric oxide involvement. Gen Pharmacol 30(1):79–83
- Diaconu C, Tartau L, Lupusoru CE. (2011). Experimental research on the influence of stress factors in an animal model of hypertension. Rev Med Chir Soc Med Nat Iasi 115(2):349–53
- Fyhrquist F, Saijonmaa O. (2008). Renin–angiotensin system revisited. J Intern Med 264(3):224–36
- Ghiadoni L, Donald AE, Cropley M, Mullen MJ, Oakley G, Taylor M, O’Connor G, et al. (2000). Mental stress induces transient endothelial dysfunction in humans. Circulation 102(20):2473–8
- Grizzo LT, Cordellini S. (2008). Perinatal lead exposure affects nitric oxide and cyclooxygenase pathways in aorta of weaned rats. Toxicol Sci 103(1):207–14
- Hatton DC, Bukoski RD, Edgar S, Mccarron DA. (1992). Chronic alcohol consumption lowers blood pressure but enhances vascular contractility in Wistar rats. J Hypertens 6:529–37
- Hjemdahl P. (2002). Stress and the metabolic syndrome: an interesting but enigmatic association. Circulation 106(21):2634–6
- Husain K, Ferder L, Ansari RA, Lalla J. (2010). Chronic ethanol ingestion induces aortic inflammation/oxidative endothelial injury and hypertension in rats. Hum Exp Toxicol 30(8):930–9
- Husain K, Mejia J, Lalla J, Kazim S. (2005). Dose response of alcohol-induced changes in BP, nitric oxide and antioxidants in rat plasma. Pharmacol Res 51:337–43
- Husain K, Vazquez M, Ansari RA, Malafa MP, Lalla J. (2008). Chronic alcohol-induced oxidative endothelial injury relates to angiotensin II levels in the rat. Mol Cell Biochem 307:51–8
- Jones A, McMillan MR, Jones RW, Kowalik GT, Steeden JA, Pruessner JC, Taylor AM, et al. (2013). Habitual alcohol consumption is associated with lower cardiovascular stress response – a novel explanation for the known cardiovascular benefits of alcohol? Stress 16(4):369–76
- Kawasaki H, Cline WH JR, Su C. (1984). Involvement of the vascular renin–angiotensin system in beta adrenergic receptor-mediated facilitation of vascular neurotransmission in spontaneously hypertensive rats. J Pharmacol Exp Ther 231(1):23–32
- Kleinhenz DJ, Sutliff RL, Polikandriotis JA, Walp ER, Dikalov SI, Guidot DM, Hart CM. (2008). Chronic ethanol ingestion increases aortic endothelial nitric oxide synthase expression and nitric oxide production in the rat. Alcohol Clin Res 32(1):148–54
- Leung PS. (2004). The peptide hormone angiotensin II: its new functions in tissues and organs. Curr Protein Pept Sci 5(4):267–73
- Loria AS, Kang KT, Pollock DM, Pollock JS. (2011). Early life stress enhances angiotensin II-mediated vasoconstriction by reduced endothelial nitric oxide buffering capacity. Hypertension 58(4):619–26
- MacKenzie A. (2011). Endothelium-derived vasoactive agents, AT1 receptors and inflammation. Pharmacol Ther 131(2):187–203
- Malik KU, Nasjletti A. (1976). Facilitation of adrenergic transmission by locally generated angiotensin II in rat mesenteric arteries. Circ Res 38(1):26–30
- Marin MT, Cruz FC, Planeta C. (2007). Chronic restraint or variable stresses differently affect the behavior, corticosterone secretion and body weight in rats. Physiol Behav 90(1):29–35
- Matthews KA, Gump BB, Owens JF. (2001). Chronic stress influences cardiovascular and neuroendocrine responses during acute stress and recovery, especially in men. Health Psychol 20(6):403–10
- McEwen BS. (1998). Protective and damaging effects of stress mediators. N Engl J Med 338(3):171–9
- Mukamal KJ, Ascherio A, Mittleman MA, Conigrave KM, Camargo CA Jr, Kawachi I, Stampfer MJ, et al. (2005). Alcohol and risk for ischemic stroke in men: the role of drinking patterns and usual beverage. Ann Intern Med 142(1):11–19
- Munzenmaier DH, Greene AS. (1996). Opposing actions of angiotensin II on microvascular growth and arterial blood pressure. Hypertension 27(3):760–5
- Ottenweller JE, Servatius RJ, Tapp WN, Drastal SD, Bergen MT, Natelson BH. (1992). A chronic stress state in rats: effects of repeated stress on basal corticosterone and behavior. Physiol Behav 51(4):689–98
- Pacák K, Palkovits M. (2001). Stressor specificity of central neuroendocrine responses: implications for stress-related disorders. Endocr Rev 22(4):502–48
- Pueyo ME, Michel J-B. (1997). Angiotensin II receptors in endothelial cells. Gen Pharmac 29(5):691–6
- Resstel LB, Tirapelli CR, Lanchote VL, Uyemura SA, de Oliveira AM, Corrêa FM. (2006). Chronic ethanol consumption alters cardiovascular functions in conscious rats. Life Sci 78(19):2179–87
- Rozanski A, Blumenthal JA, Davidson KW, Saab PG, Kubzansky L. (2005). The epidemiology, pathophysiology, and management of psychosocial risk factors in cardiac practice: the emerging field of behavioral cardiology. J Am Coll Cardiol 45(5):637–51
- Rush JW, Aultman CD. (2008). Vascular biology of angiotensin and the impact of physical activity. Appl Physiol Nutr Metab 33(1):162–72
- Sparrow MG, Roggendorf H, Vogel WH. (1987). Effect of ethanol on heart rate and blood pressure in nonstressed and stressed rats. Life Sci 40(26):2551–9
- Sun H, Mayhan WG. (2001). Temporal effect of alcohol consumption on reactivity of pial arterioles: role of oxygen radicals. Am J Physiol Heart Circ Physiol 280(3):H992–1001
- Tirapelli CR, Al-khoury J, Bkaily G, D’Orléans-Juste P, Lanchote VL, Uyemura SA, De Oliveira AM. (2006). Chronic ethanol consumption enhances phenylephrine-induced contraction in the isolated rat aorta. J Exp Pharmacol Ther 316(1):233–41
- Tirapelli CR, Fukada SY, Yogi A, Chignalia AZ, Tostes RC, Bonaventura D, Lanchote VL, et al. (2008a). Gender-specific vascular effects elicited by chronic ethanol consumption in rats: a role for inducible nitric oxide synthase. Br J Pharmacol 153:468–79
- Tirapelli CR, Leone AF, Coelho EB, Resstel LB, Corrêa FM, Lanchote VL, Uyemura SA, et al. (2007). Effect of ethanol consumption on blood pressure and rat mesenteric arterial bed, aorta and carotid responsiveness. J Pharmacol 59(7):985–93
- Tirapelli CR, Leone AF, Yogi A, Tostes RC, Lanchote VL, Uyemura SA, Resstel LB, et al. (2008b). Ethanol consumption increases blood pressure and alters the responsiveness of the mesenteric vasculature in rats. J Pharm Pharmacol 60(3):331–41
- Toda N, Ayajiki K. (2010). Vascular actions of nitric oxide as affected by exposure to alcohol. Alcohol Alcohol 45(4):347–55
- Toda N, Ayajiki K, Okamura T. (2007). Interaction of endothelial nitric oxide and angiotensin in the circulation. Pharmacol Rev 59:54–87
- Vecsei P. (1979). Glucocorticoids: cortisol, corticosterone and compounds. In: Jaffe BM, Behrmann HR, editors. Methods of hormone radioimmunoassay, vol. 39. New York: Academic Press. p 767–92
- Viau V, Meaney MJ. (1991). Variations in the hypothalamic-pituitary-adrenal response to stress during the estrous cycle in the rat. Endocrinology 129(5):2503–11
- Wakabayashi I, Hatake K. (2001). Effects of ethanol on the nervous and vascular systems: the mechanisms of alcohol-induced hypertension. Nihon Eiseigaku Zasshi 55(4):607–17