
Abstract
Individuals with post-traumatic stress disorder (PTSD) experience many debilitating symptoms, including intrusive memories, persistent anxiety and avoidance of trauma-related cues. PTSD also results in numerous physiological complications, including increased risk for cardiovascular disease (CVD). However, characterization of PTSD-induced cardiovascular alterations is lacking, especially in preclinical models of the disorder. Thus, we examined the impact of a psychosocial predator-based animal model of PTSD on myocardial sensitivity to ischemic injury. Male and female Sprague–Dawley rats were exposed to psychosocial stress or control conditions for 31 days. Stressed rats were given two cat exposures, separated by a period of 10 days, and were subjected to daily social instability throughout the paradigm. Control rats were handled daily for the duration of the experiment. Rats were tested on the elevated plus maze (EPM) on day 32, and hearts were isolated on day 33 and subjected to 20 min ischemia and 2 h reperfusion on a Langendorff isolated heart system. Stressed male and female rats gained less body weight relative to controls, but only stressed males exhibited increased anxiety on the EPM. Male, but not female, rats exposed to psychosocial stress exhibited significantly larger infarcts and attenuated post-ischemic recovery of contractile function compared to controls. Our data demonstrate that predator stress combined with daily social instability sex-dependently increases myocardial sensitivity to ischemic injury. Thus, this manipulation may be useful for studying potential mechanisms underlying cardiovascular alterations in PTSD, as well as sex differences in the cardiovascular stress response.
Introduction
Traumatized individuals who develop post-traumatic stress disorder (PTSD) are at increased risk for developing cardiovascular disease (CVD) (Boscarino, Citation2011; Buckley et al., Citation2013; Coughlin, Citation2011; Edmondson & Cohen, Citation2013; Edmondson et al., Citation2013). This association appears to be independent of comorbid depression, genetic influences and several other potentially confounding psychosocial factors (Edmondson et al., Citation2013; Vaccarino et al., Citation2013). According to Edmondson and colleagues (Citation2013), the risk for CVD in PTSD patients could be greater in females, relative to males, but the limited number of CVD studies involving female subjects has underpowered such statistical comparisons. The scientific literature suggests that the link between PTSD and CVD may be causal in nature and related to the numerous cardiovascular abnormalities observed in PTSD patients. For instance, people with PTSD exhibit lower heart rate variability (HRV), reduced baroreflex sensitivity and increased QT variability (Cohen et al., Citation1998, Citation2000; Rozanski et al., Citation2005), each of which has been linked to greater CVD incidence or cardiovascular-related mortality (Bigger et al., Citation1992; Piccirillo et al., Citation2007; Rozanski et al., Citation2005). PTSD patients also demonstrate abnormally elevated heart rate (HR), blood pressure (BP) and norepinephrine (NE) levels at baseline and in response to emotional stimuli or events (for a review, see Zoladz & Diamond, Citation2013). Coupled with reports of reduced HRV and parasympathetic nervous system (PNS) activity in PTSD patients, this exaggerated sympathetic nervous system (SNS) activity suggests that PTSD causes autonomic dysregulation.
CVD has been described as a disease of inflammation (Libby et al., Citation2009), and PTSD patients exhibit elevated levels of pro-inflammatory cytokines, such as tumor necrosis factor α and IL-1β (von Kanel et al., Citation2007, Citation2010), as well as increased incidence of autoimmune diseases (O'Donovan et al., Citation2015). These observations may be associated with baseline alterations of hypothalamus–pituitary–adrenal (HPA) axis function in PTSD, as a common finding in PTSD patients is abnormally low levels of baseline cortisol (Daskalakis et al., Citation2013; Yehuda, Citation2009; Yehuda & Seckl, Citation2011). Because corticosteroids act as major anti-inflammatory agents, insufficient HPA axis activity, coupled with exaggerated SNS activity, could promote inflammation and negative health outcomes.
One of the present authors was previously involved in developing a psychosocial predator-based animal model that has been shown to emulate several core symptoms of PTSD (Roth et al., Citation2011; Zoladz et al., Citation2008, Citation2012, Citation2013, Citation2015). In the model, rats are immobilized and placed in close proximity to a cat (i.e. predator stress) on two separate occasions. The stressed rats are also exposed to daily social instability (randomized changing of cage mates) throughout the duration of the model to increase the likelihood of producing long-lasting sequelae in the rats. Thus, the model incorporates elements, such as uncontrollability, unpredictability, a lack of social interaction and a re-experiencing of the stress that, in people, significantly increases the likelihood of PTSD onset and maintenance following trauma exposure.
Previous work has shown that exposing rats to this psychosocial stress manipulation results in a number of physiological and behavioral abnormalities that are similar to those observed in people with PTSD. Three weeks after the second predator exposure, psychosocially stressed rats exhibit a robust fear memory for the cat exposures (evidenced by greater immobility in response to the cat-associated context and cues), heightened anxiety on an EPM (evidenced by less time spent exploring open arms), an exaggerated startle response and impaired memory for newly learned information (Zoladz et al., Citation2008, Citation2012, Citation2013). The psychosocially stressed rats also display physiological changes indicative of elevated SNS activity and HPA axis abnormalities, such as greater cardiovascular and hormonal reactivity to an acute stressor, an exaggerated physiological and behavioral response to the α2-adrenergic receptor antagonist, yohimbine, abnormally low levels of baseline corticosterone and enhanced negative feedback of the HPA axis (Zoladz et al., Citation2008, Citation2012, Citation2013). Others have replicated and extended the findings from this model, revealing that the chronic psychosocial stress results in decreased serotonin, increased norepinephrine and increased measures of oxidative stress and inflammation in the brain, adrenal glands and systemic circulation (Wilson et al., Citation2013, Citation2014a). More recently, research has shown that some effects induced by the psychosocial stress manipulation persist for four months following the initiation of stress (Zoladz et al., Citation2015).
There has been limited research investigating the cardiovascular consequences of animal models of PTSD and none have assessed whether a rodent model of PTSD could influence myocardial ischemic injury. There have also been few animal models of PTSD that include females, despite the fact that females are reportedly more likely to develop the disorder (Tolin & Foa, Citation2006). Based on previous work demonstrating cardiovascular-related abnormalities and increased inflammatory markers in male rats exposed to the psychosocial predator-based animal model of PTSD, we examined whether male and female rats exposed to this psychosocial stress manipulation would exhibit greater myocardial sensitivity to ischemic injury and whether female rats would demonstrate the anxiogenic and physiological effects previously observed in males.
Methods and materials
Animals
Experimentally naïve adult male and female Sprague–Dawley rats (approximately 200–300 g at the beginning of the experiment) part of an established breeding colony (established from Sprague–Dawley rats that were obtained from Charles River laboratories) were used in the present experiment. The rats were housed on a 12-h light/dark schedule (lights on at 07:00) in standard Plexiglas cages (two per cage) with free access to food and water. All procedures were approved by the Institutional Animal Care and Use Committee at Ohio Northern University and followed those recommended by the Guide for the Care and Use of Laboratory Animals provided by the National Institute of Health.
Psychosocial stress procedure
Rats were assigned to “psychosocial stress” (males: N = 10; females: N = 14) or “no stress” (males: N = 11; females: N = 10) groups. On day 1, rats in the psychosocial stress groups were immobilized in plastic DecapiCones (Braintree Scientific; Braintree, MA) and placed in a perforated wedge-shaped Plexiglas enclosure (Braintree Scientific; Braintree, MA; 20 × 20 × 8 cm). This enclosure was then taken to a cat housing room and placed in a metal cage (61 × 53 × 51 cm) with an adult female cat for 1 h. The Plexiglas enclosure prevented any contact between the cat and rats, but the rats were still exposed to all non-tactile sensory stimuli associated with the cat. Canned cat food was smeared on top of the enclosure to direct the cat’s attention toward the rats. An hour later, the rats were returned to their home cages. Rats in the no stress groups remained in their home cages during the 1-h stress period.
Rats in the psychosocial stress groups were given two acute stress sessions, which were separated by 10 days. The first stress session took place during the light cycle (between 08:00 and 13:00), and the second stress session took place during the dark cycle (between 19:00 and 21:00). The stress sessions took place during different times of the day to add an element of unpredictability as to when the rats might re-experience the predator exposure, as per previously-employed methodology (Roth et al., Citation2011; Zoladz et al., Citation2008, Citation2012, Citation2013, Citation2015).
Daily social stress
Beginning on the day of the first stress session (day 1), rats in the psychosocial stress groups were exposed to unstable housing conditions until behavioral testing (day 32). Rats in the psychosocial stress groups were housed two per cage, but every day, their cohort pair combinations were changed. Therefore, no rats in the psychosocial stress groups had the same cage mate on two consecutive days during the 31-day stress period. Rats in the no stress groups were housed with the same cohort pair throughout the experiment; these rats were handled daily to control for handling effects on stressed animals.
Behavioral testing
Three weeks following the second stress session (day 32), rats were tested on the EPM to assess anxiety-like behavior. Rats were placed on the EPM for 5 min, and their behavior was videotaped by a JVC hard disk camera hanging above the EPM and scored offline by two separate investigators who were blind to the experimental conditions of the animals. Investigators assessed time spent in the open arms and number of entries into the open and closed arms. Rats were removed from data analysis if they fell off of the EPM during testing.
Langendorff heart preparation and measurement of cardiac function
On Day 33, rats were anesthetized with an intraperitoneal injection of pentobarbital (75 mg/kg). Hearts were rapidly removed and cannulated while bathed in ice-cold Krebs–Henseleit solution (118 mM NaCl, 4.7 mM KCl, 1.2 mM MgSO4, 25 mM NaHCO3, 1.2 mM KH2PO4, 0.5 mM Na2EDTA, 11 mM glucose, 2.5 mM CaCl2, pH 7.4). Krebs solution was perfused through the aortic cannula at a constant pressure of 80 mmHg. Contractile function of the left ventricle was measured using an intraventricular balloon connected to a pressure transducer. The balloon was inflated to an end diastolic pressure of 4–8 mmHg, and data were continually recorded using a Powerlab 4SP data acquisition system (AD Instruments, Colorado Springs, CO). Hearts were equilibrated for 25 min prior to the onset of 20 min ischemia and 2 h reperfusion. Hearts were excluded after the 25-min equilibration period if developed pressure was less than 100 mmHg, coronary flow rate was greater than 20 ml/min or if they had a persistent irregular rhythm. Ischemia was induced by closing a valve which stopped the flow of Krebs solution through the aortic cannula. Reperfusion was initiated by reopening the valve and allowing Krebs solution to resume flow through the cannula (Granville et al., Citation2004; Waterson et al., Citation2011). The heart was submerged in Krebs solution to maintain the temperature of the heart at 37.5 ± 0.5 °C throughout the experiment, and temperature was continuously monitored using a thermocouple placed on the surface of the heart. Pre-ischemic contractile function was measured immediately prior to ischemia. Post-ischemic recovery of contractile function was measured following 1 h of reperfusion.
Measurement of infarct size
Following 20 min ischemia and 2 h reperfusion, hearts were perfused with 1% triphenyltetrazolium chloride for 8 min (at a rate of 7.5 ml/min) and then submerged in 1% triphenyltetrazolium chloride and incubated at 37 °C for 15 min. Hearts were subsequently frozen at −80 °C, sliced into approximately 1 mm sections, soaked in 10% neutral buffered formalin and then photographed with a Nikon SMZ 800 microscope equipped with a Nikon DS-Fi1 digital camera. The infarcted surface area and total surface area of each slice were measured using NIH Image J software (Bethesda, MD). Infarct size was expressed as a percentage of the area at risk (AAR). The AAR was defined as the entire ventricular myocardium since hearts were exposed to global ischemia.
Body and organ weights
Rats were weighed on the days of the first and second stress sessions, as well as on the day of EPM testing. Growth rates (grams per day) were calculated for statistical analysis. In females, adrenal and thymus glands were removed and weighed to compare to previously-reported findings in psychosocially stressed males. Organ weights were expressed as mg/100 g body weight for statistical analysis.
Statistical analyses
Male and female data were analyzed separately. Most data (body weight, EPM behavior, infarct sizes, adrenal/thymus weights) were compared via independent samples t tests with treatment (psychosocial stress, no psychosocial stress) serving as the between-subjects factor. Parameters of contractile function were compared with two-way, mixed-model ANOVAs with treatment (psychosocial stress, no psychosocial stress) serving as the between-subjects factor and time (pre-ischemic, post-ischemic) serving as the within-subjects factor. Tukey post-hoc tests were employed when necessary, and alpha was set at 0.05 for all analyses. Data points more than three standard deviations beyond exclusive group means were eliminated from statistical analyses.
Results
Preliminary characterization of psychosocially stressed rats
Males
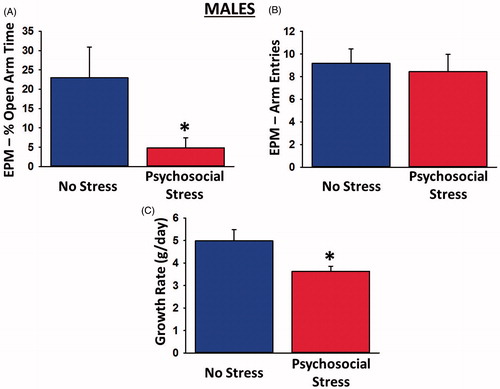
Psychosocially stressed males spent significantly less time in the open arms of the EPM than control males [t(16) = 2.17, p = 0.046; ], indicating a heightened state of anxiety. This effect was not attributable to reduced locomotor activity in the psychosocially stressed males, as both groups exhibited a statistically equivalent number of total arm entries [t(16) = 0.60, p = 0.56; ]. Psychosocial stress also led to significantly reduced growth rate in male rats over the 31-day period [t(19) = 2.39, p = 0.028; ].
Figure 1. Physiological and behavioral effects of psychosocial stress in male rats. Psychosocially stressed males spent significantly less time in the open arms of the elevated plus maze (A), despite making an equivalent numbers of overall arm entries in the maze (B). The stressed males also exhibited significantly reduced growth rates (C) relative to controls. Sample sizes were 9–11 rats per group. Data are presented as means ± SEM. *p < 0.05 versus no stress.
Females
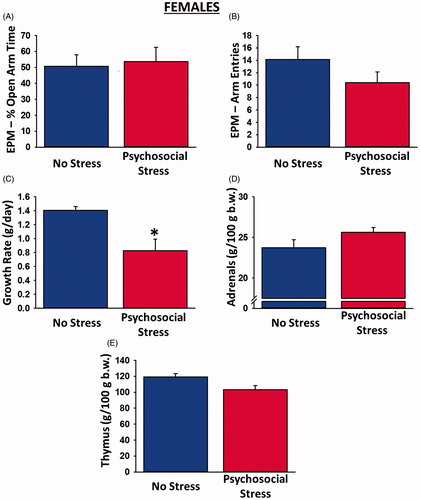
Examination of the physiological and behavioral data from females revealed inconsistent findings. Although psychosocially stressed females exhibited significantly reduced growth rate over the 31-day period [t(22) = 2.93, p = 0.008; ], they spent a statistically equivalent amount of time in the open arms of the EPM as control rats [t(20) = 0.19, p = 0.86; ], suggestive of no anxiogenic effect of the psychosocial stress manipulation. Adrenal [t(22) = 1.75, p = 0.09; ] and thymus gland [t(20) = 2.04, p = 0.055; ] comparisons were not significant.
Figure 2. Physiological and behavioral effects of psychosocial stress in female rats. Psychosocially stressed females spent a similar amount of time in the open arms of the elevated plus maze as control rats (A) and made a similar number of overall arm entries in the maze (B), despite exhibiting significantly reduced growth rates (C) relative to controls. There were no significant differences between the adrenal (D) or thymus (E) gland weights of stressed and non-stressed female rats. Sample sizes were 10–14 rats per group. Data are presented as means ± SEM. *p < 0.05 versus no stress.
Effect of psychosocial stress on cardiac function and myocardial ischemic injury
Males
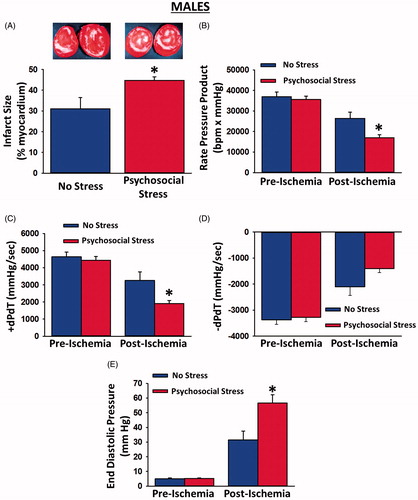
Psychosocial stress had no effect on pre-ischemic contractile function. Pre-ischemic rate pressure product, +dP/dT, −dP/dT and end diastolic pressure were similar in psychosocially stressed males and controls (). However, 20 min of ischemia induced more myocardial injury in psychosocially stressed males, relative to controls. Hearts from psychosocially stressed males exhibited significantly larger infarcts compared to controls [t(15) = 2.54, p = 0.023; ]. In addition, post-ischemic recovery of contractile function was significantly attenuated in the hearts from psychosocially stressed males. Recovery of the rate pressure product [significant Group × Time interaction: F(1, 14) = 5.38, p = 0.036; ] and +dP/dT [significant Group × Time interaction: F(1, 14) = 5.90, p = 0.029; ] was significantly reduced in psychosocially stressed males, relative to controls, indicating that recovery of systolic function was worsened by the psychosocial stress. Recovery of −dP/dT was decreased 33% in psychosocially stressed male hearts compared to control hearts, but this did not reach statistical significance [Group × Time interaction: F(1, 14) = 3.24, p = 0.094; planned comparison: t(14) = 2.08, p = 0.056; ]. Post-ischemic recovery of end diastolic pressure was significantly elevated in psychosocially stressed males [significant Group × Time interaction: F(1, 13) = 8.26, p = 0.013; ], indicating that recovery of diastolic function was also worsened by psychosocial stress. Collectively, these data indicate that chronic psychosocial stress occurring prior to the onset of ischemia increases cell death and decreases post-ischemic recovery of contractile function in male rats.
Figure 3. Myocardial infarct sizes, pre-ischemic contractile function and post-ischemic recovery of contractile function in male hearts exposed to myocardial ischemia. Hearts from psychosocially stressed males exhibited significantly larger infarcts than control hearts (A; insets depict representative myocardial slices with white areas indicative of infarction). There were no differences in pre-ischemic parameters of contractile function between hearts from psychosocially stressed and control males. Recovery of rate pressure product (B) and +dP/dT (C) were significantly attenuated in hearts from psychosocially stressed males, relative to those from controls. Recovery of −dP/dT (D) was borderline significant in hearts from psychosocially stressed males. Recovery of end diastolic pressure (E) was significantly elevated in psychosocially stressed males relative to controls. Samples sizes were 7–9 hearts per group. Data are presented as means ± SEM. *p < 0.05 relative to no stress.
Females
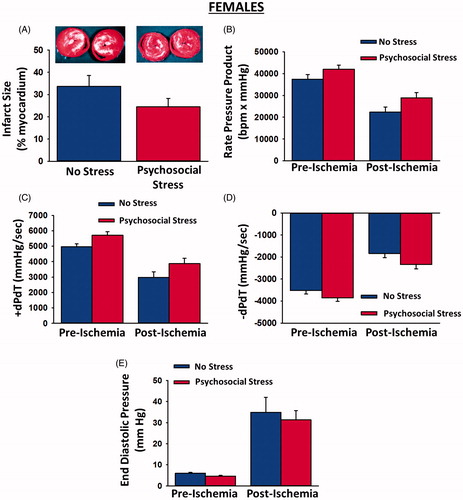
Psychosocial stress had no effect on pre-ischemic contractile function in females. In contrast to male rats, infarct size [t(22) = 1.51, p = 0.15); ] and post-ischemic recovery of rate pressure product [Group × Time interaction: F(1, 21) = 0.19, p = 0.67; ], +dP/dT [Group × Time interaction: F(1, 21) = 0.04, p = 0.84], −dP/dT [Group × Time interaction: F(1, 21) = 0.13, p = 0.73] and end diastolic pressure [Group × Time interaction: F(1, 22) = 0.07, p = 0.79] were unaffected by psychosocial stress in females.
Figure 4. Myocardial infarct sizes, pre-ischemic contractile function and post-ischemic recovery of contractile function in female hearts exposed to myocardial ischemia. Psychosocial stress had no effect on infarct sizes (A; insets depict representative myocardial slices with white areas indicative of infarction) or pre-ischemic contractile function. In addition, post-ischemic recovery of rate pressure product (B), +dp/dT (C), −dP/dT (D) and end diastolic pressure (E) were unaffected by psychosocial stress. Sample sizes were 10–14 hearts per group. Data are presented as means ± SEM.
Discussion
Despite extensive evidence of increased risk for CVD in PTSD patients, few animal models of the disorder have examined cardiovascular endpoints. In the present study, we have shown that a psychosocial stress manipulation involving predator exposure and daily social instability, one that has previously been shown to emulate several core symptoms of PTSD, increases myocardial sensitivity to ischemic injury in male, but not female, rats. Following ischemia, hearts from psychosocially stressed males exhibited significantly less recovery of contractile function and demonstrated larger infarct sizes than hearts from controls. Hearts from psychosocially stressed females, on the other hand, exhibited no differences in infarct size or recovery of contractile function, relative to controls. Moreover, psychosocially stressed females did not show increased anxiety on the EPM, despite exhibiting a reduced growth rate. Overall, our findings provide evidence of a sex-dependent increase in myocardial sensitivity to ischemic injury as a result of predator stress and chronic social instability, suggesting that this model may be used to explore mechanisms underlying cardiovascular changes that result from intense stress, at least in males.
Cardiovascular effects
PTSD promotes hypertension, atherosclerosis and the production of inflammatory markers that are detrimental to cardiovascular function (Boscarino, Citation2011; Buckley et al., Citation2013; Coughlin, Citation2011; Edmondson & Cohen, Citation2013; Edmondson et al., Citation2013; von Kanel et al., Citation2007, Citation2010). The underlying cause of CVD in PTSD patients is thought to be an acceleration of atherosclerosis and subsequent blockage of coronary arteries (Ahmadi et al., Citation2011). It is important to note, however, that myocardial ischemia in the present study was not induced by the atherosclerotic occlusion of coronary arteries but was artificially induced by termination of coronary flow. The observation that stressed males exhibited greater ischemic injury than male controls in the absence of atherosclerotic occlusion of the coronary vasculature suggests that PTSD-induced atherosclerotic blockade of coronary arteries may not be the only factor that contributes to the increased risk of myocardial infarction in patients with PTSD.
The chronic psychosocial stress manipulation utilized in the present study provides several important advantages over studying the impact of PTSD on myocardial infarction in a clinical setting. One advantage is that the psychosocial stress manipulation avoids factors that are commonly comorbid with PTSD (depression, smoking, drug and alcohol abuse, etc.) and can independently alter cardiovascular function. It also provides a mechanism to compare the extent of myocardial injury in stressed and non-stressed hearts under conditions where the ischemic insult is identical between subjects. Clinical data (death rates, hospitalization rates, rates of coronary revascularization, etc.) have been important for establishing that PTSD increases the likelihood of experiencing a myocardial infarction. However, clinical studies have been unable to establish whether or not PTSD increases the extent of myocardial injury. In addition to the common comorbid factors mentioned above, ischemic conditions in patients who are experiencing heart attacks are heterogeneous. For example, occlusion of a large coronary artery that delivers blood to a wide region of myocardial tissue induces ischemia over a much larger region of the heart than occlusion of a smaller vessel that delivers blood to a more localized area. In contrast, the psychosocial stress paradigm used here enables stressed and non-stressed hearts to be made uniformly ischemic so that the anatomical location and amount of myocardial tissue that is subjected to ischemia is consistent between individual hearts. One fundamental difference between the psychosocial stress paradigm used here and a clinical heart attack is that atherosclerotic blockage of coronary arteries develops over a long period of time and may be accompanied by the formation of collateral vessels or other myocardial compensatory changes as ischemic heart disease progresses. In contrast, the fact that the present psychosocial stress manipulation induces myocardial ischemia through a mechanism that is independent of atherosclerotic coronary occlusion represents an important distinction (and perhaps a limitation) from the clinical scenario.
Our finding that chronic psychosocial stress increases myocardial sensitivity to ischemic injury is consistent with previous work exploring the impact of chronic social, psychological or emotional stress on myocardial sensitivity to ischemic injury. For instance, Mercanoglu et al. (Citation2008) reported that consecutive exposure to a series of multiple stressors (e.g. cage crowding, isolation, changes in light/dark cycle, changes in cage mates, cage tilting, food/water restriction) increased infarct size in the rat heart. Chronic restraint stress produces a similar effect (Scheuer & Mifflin, Citation1998). In contrast, acute stress immediately prior to the onset of ischemia protects the heart from ischemic injury (Moghimian et al., Citation2012). Thus, the duration and timing of stress appear to play a critical role in how ischemia impacts the heart.
The present psychosocial stress paradigm induces multiple physiological changes that may underlie the increased sensitivity of the male heart to ischemic injury. Wilson and colleagues reported that male rats exposed to this psychosocial stress exhibit increased expression of inflammatory cytokines and other protein markers of inflammation in the brain (Wilson et al., Citation2013, Citation2014b,Citationc). These rats also have elevated concentrations of reactive oxygen species in blood, brain and adrenal glands (Wilson et al., Citation2013). In addition, male rats exposed to the psychosocial stress exhibit increased norepinephrine release in the hippocampus and prefrontal cortex (Wilson et al., Citation2014a). Elevated blood pressure and heart rate in psychosocial stressed rats are suggestive of increased SNS stimulation (Zoladz et al., Citation2008, Citation2013). Inflammation and oxidative stress play prominent roles in modulating ischemia/reperfusion injury (Frangogiannis, Citation2014; Ling et al., Citation2013; Sandanger et al., Citation2013). Chronic β-adrenergic receptor stimulation also worsens ischemic injury (Hu et al., Citation2006). Thus, pro-inflammatory conditions, increased oxidative stress and chronic β-adrenergic receptor stimulation that exist prior to the onset of ischemia may all contribute to the psychosocial stress-induced increase in myocardial sensitivity to ischemic injury.
Sex differences
Investigators have reported that traumatized females are twice as likely as traumatized males to develop PTSD (Tolin & Foa, Citation2006). However, in a recent review paper, Zoladz & Diamond (Citation2013) discussed issues with this position, noting that for several types of trauma, the incidence of PTSD is statistically equivalent across the sexes. According to Zoladz & Diamond (Citation2013), most research reporting a greater prevalence of PTSD in females, relative to males, has involved individuals whose trauma included some element of assault. Indeed, for non-assault trauma, there appears to be no difference in the prevalence of PTSD across men and women (e.g. Kessler et al., Citation1995). Moreover, the authors raise the issue of female hormone influences on emotional memory and, thus, traumatic memory formation. Basic and applied research studies have shown that the levels of progesterone and estradiol significantly influence emotional memories and their extinction (Bryant et al., Citation2011; Felmingham et al., Citation2012a,Citationb). Thus, the point at which the trauma occurs during the menstrual cycle can significantly influence a single female’s likelihood of PTSD development. To suggest that being female, per se, presents an inherent vulnerability to PTSD may be an oversimplification of a much more complicated process. It is likely that the female sex interacts with genetics, trauma type, personality characteristics and other environmental factors to affect PTSD susceptibility.
Most basic science research on stress has been conducted in males, and an established animal model of PTSD including females is non-existent. The present study included an initial attempt to extend the psychosocial, predator-based model to females. However, we observed no anxiogenic or cardiovascular effects of psychosocial stress in female rats, despite stressed females exhibiting reduced growth rate compared to non-stressed females. One possibility for the lack of anxiogenic or cardiovascular effects in stressed females is that the psychosocial stress manipulation is less “stressful” to females. Additionally, as suggested in the previous paragraph, extensive work has shown that female hormone levels significantly influence the stress response and emotional memory formation. Thus, it is possible that the random times of predator stress and physiological/behavioral testing in the present study masked any estrous-dependency of the observed effects. On the other hand, a recent meta-analysis showed that, across thousands of behavioral, morphological, physiological and molecular traits, there was no greater variability in the data obtained from female rats that were examined without estrous stage monitoring than was observed in data obtained from male rats (Prendergast et al., Citation2014). In fact, the authors of this meta-analysis observed significantly more variability in the data from male rats for certain dependent variables. Thus, it is also possible that, in the present study, estrous stage did not introduce anymore variability in the behavioral or myocardial measures than was observed in males. We did not assess the estrous cycle in females because we did not want the stress associated with estrous testing to confound our results. To conclusively determine whether females are less stressed by this psychosocial stress manipulation or if estrous stage influences female responses to the psychosocial stress, future research is necessary.
Conclusions
The psychosocial predator-based model used here provides a useful tool to study the cardiac impact of intense stress. We anticipate that future work will further our understanding of the mechanisms by which PTSD-like sequelae in rats increase myocardial ischemic injury. This could provide new therapeutic strategies to protect the hearts of PTSD patients from myocardial infarction and other ischemic heart diseases.
Declaration of interest
The authors report no conflicts of interest.
References
- Ahmadi N, Hajsadeghi F, Mirshkarlo HB, Budoff M, Yehuda R, Ebrahimi R. (2011). Post-traumatic stress disorder, coronary atherosclerosis, and mortality. Am J Cardiol 108(1):29–33
- Bigger Jr JT, Fleiss JL, Steinman RC, Rolnitzky LM, Kleiger RE, Rottman JN. (1992). Frequency domain measures of heart period variability and mortality after myocardial infarction. Circulation 85(1):164–71
- Boscarino JA. (2011). Post-traumatic stress disorder and cardiovascular disease link: time to identify specific pathways and interventions. Am J Cardiol 108(7):1052–3
- Bryant RA, Felmingham KL, Silove D, Creamer M, O'Donnell M, McFarlane AC. (2011). The association between menstrual cycle and traumatic memories. J Affect Disord 131(1–3):398–401
- Buckley T, Tofler G, Prigerson HG. (2013). Posttraumatic stress disorder as a risk factor for cardiovascular disease: a literature review and proposed mechanisms. Curr Cardiovasc Risk Rep 7:506–13
- Cohen H, Benjamin J, Geva AB, Matar MA, Kaplan Z, Kotler M. (2000). Autonomic dysregulation in panic disorder and in post-traumatic stress disorder: application of power spectrum analysis of heart rate variability at rest and in response to recollection of trauma or panic attacks. Psychiatry Res 96(1):1–13
- Cohen H, Kotler M, Matar MA, Kaplan Z, Loewenthal U, Miodownik H, Cassuto Y. (1998). Analysis of heart rate variability in posttraumatic stress disorder patients in response to a trauma-related reminder. Biol Psychiatry 44(10):1054–9
- Coughlin SS. (2011). Post-traumatic stress disorder and cardiovascular disease. Open Cardiovasc Med J 5:164–70
- Daskalakis NP, Lehrner A, Yehuda R. (2013). Endocrine aspects of post-traumatic stress disorder and implications for diagnosis and treatment. Endocrinol Metab Clin North Am 42(3):503–13
- Edmondson D, Cohen BE. (2013). Posttraumatic stress disorder and cardiovascular disease. Prog Cardiovasc Dis 55(6):548–56
- Edmondson D, Kronish IM, Shaffer JA, Falzon L, Burg MM. (2013). Posttraumatic stress disorder and risk for coronary heart disease: a meta-analytic review. Am Heart J 166(5):806–14
- Felmingham KL, Fong WC, Bryant RA. (2012a). The impact of progesterone on memory consolidation of threatening images in women. Psychoneuroendocrinology 37:1896–900
- Felmingham KL, Tran TP, Fong WC, Bryant RA. (2012b). Sex differences in emotional memory consolidation: the effect of stress-induced salivary alpha-amylase and cortisol. Biol Psychol 89:539–44
- Frangogiannis NG. (2014). The inflammatory response in myocardial injury, repair, and remodelling. Nat Rev Cardiol 11(5):255–65
- Granville DJ, Tashakkor B, Takeuchi C, Gustafsson AB, Huang C, Sayen MR, Wentworth Jr P, et al. (2004). Reduction of ischemia and reperfusion-induced myocardial damage by cytochrome P450 inhibitors. Proc Natl Acad Sci USA 101(5):1321–6
- Hu A, Jiao X, Gao E, Gao E, Koch WJ, Sharifi-Azad S, Grunwald Z, Ma XL, Sun J-Z. (2006). Chronic beta-adrenergic receptor stimulation induces cardiac apoptosis and aggravates myocardial ischemia/reperfusion injury by provoking inducible nitric-oxide synthase-mediated nitrative stress. J Pharmacol Exp Ther 318(2):469–75
- Kessler RC, Sonnega A, Bromet E, Hughes M, Nelson CB. (1995). Posttraumatic stress disorder in the National Comorbidity Survey. Arch Gen Psychiatry 52(12):1048–60
- Libby P, Ridker PM, Hansson GK. (2009). Inflammation in atherosclerosis: from pathophysiology to practice. J Am Coll Cardiol 54(23):2129–38
- Ling H, Gray CB, Zambon AC, Grimm M, Gu Y, Dalton N, Purcell NH, et al. (2013). Ca2+/Calmodulin-dependent protein kinase II delta mediates myocardial ischemia/reperfusion injury through nuclear factor-kappaB. Circ Res 112(6):935–44
- Mercanoglu G, Safran N, Uzun H, Eroglu L. (2008). Chronic emotional stress exposure increases infarct size in rats: the role of oxidative and nitrosative damage in response to sympathetic hyperactivity. Methods Find Exp Clin Pharmacol 30(10):745–52
- Moghimian M, Faghihi M, Karimian SM, Imani A. (2012). The effect of acute stress exposure on ischemia and reperfusion injury in rat heart: role of oxytocin. Stress 15(4):385–92
- O'Donovan A, Cohen BE, Seal KH, Bertenthal D, Margaretten M, Nishimi K, Neylan TC. (2015). Elevated risk for autoimmune disorders in Iraq and Afghanistan veterans with posttraumatic stress disorder. Biol Psychiatry 77(4):365–74
- Piccirillo G, Magri D, Matera S, Magnanti M, Torrini A, Pasquazzi E, Schifano E, et al. (2007). QT variability strongly predicts sudden cardiac death in asymptomatic subjects with mild or moderate left ventricular systolic dysfunction: a prospective study. Eur Heart J 28(11):1344–50
- Prendergast BJ, Onishi KG, Zucker I. (2014). Female mice liberated for inclusion in neuroscience and biomedical research. Neurosci Biobehav Rev 40:1–5
- Roth TL, Zoladz PR, Sweatt JD, Diamond DM. (2011). Epigenetic modification of hippocampal Bdnf DNA in adult rats in an animal model of post-traumatic stress disorder. J Psychiatr Res 45:919–26
- Rozanski A, Blumenthal JA, Davidson KW, Saab PG, Kubzansky L, . (2005). The epidemiology, pathophysiology, and management of psychosocial risk factors in cardiac practice: the emerging field of behavioral cardiology. J Am Coll Cardiol 45(5):637–51
- Sandanger O, Ranheim T, Vinge LE, Bliksoen M, Alfsnes K, Finsen AV, Dahl CP, et al. (2013). The NLRP3 inflammasome is up-regulated in cardiac fibroblasts and mediates myocardial ischaemia-reperfusion injury. Cardiovasc Res 99(1):164–74
- Scheuer DA, Mifflin SW. (1998). Repeated intermittent stress exacerbates myocardial ischemia–reperfusion injury. Am J Physiol 274(2 Pt 2):R470–5
- Tolin DF, Foa EB. (2006). Sex differences in trauma and posttraumatic stress disorder: a quantitative review of 25 years of research. Psychol Bull 132(6):959–92
- Vaccarino V, Goldberg J, Rooks C, Shah AJ, Veledar E, Faber TL, Votaw JR, et al. (2013). Post-traumatic stress disorder and incidence of coronary heart disease: a twin study. J Am Coll Cardiol 62(11):970–8
- von Kanel R, Begre S, Abbas CC, Saner H, Gander M-L, Schmid J-P. (2010). Inflammatory biomarkers in patients with posttraumatic stress disorder caused by myocardial infarction and the role of depressive symptoms. Neuroimmunomodulation 17(1):39–46
- von Kanel R, Hepp U, Kraemer B, Traber R, Keel M, Mica L, Schnyder U. (2007). Evidence for low-grade systemic proinflammatory activity in patients with posttraumatic stress disorder. J Psychiatr Res 41(9):744–52
- Waterson RE, Thompson CG, Mabe NW, Kaur K, Talbot JN, Neubig RR, Rorabaugh BR. (2011). Galpha(i2)-mediated protection from ischaemic injury is modulated by endogenous RGS proteins in the mouse heart. Cardiovasc Res 91(1):45–52
- Wilson CB, Ebenezer PJ, McLaughlin LD, Francis J. (2014a). Predator exposure/psychosocial stress animal model of post-traumatic stress disorder modulates neurotransmitters in the rat hippocampus and prefrontal cortex. PLoS One 9(2):e89104
- Wilson CB, McLaughlin LD, Ebenezer PJ, Nair AR, Dange R, Harre JG, Shaak TL, et al. (2014b). Differential effects of sertraline in a predator exposure animal model of post-traumatic stress disorder. Front Behav Neurosci 8:256
- Wilson CB, McLaughlin LD, Ebenezer PJ, Nair AR, Francis J. (2014c). Valproic acid effects in the hippocampus and prefrontal cortex in an animal model of post-traumatic stress disorder. Behav Brain Res 268:72–80
- Wilson CB, McLaughlin LD, Nair A, Ebenezer PJ, Dange R, Francis J, . (2013). Inflammation and oxidative stress are elevated in the brain, blood, and adrenal glands during the progression of post-traumatic stress disorder in a predator exposure animal model. PLoS One 8(10):e76146
- Yehuda R. (2009). Stress hormones and PTSD. In: Shiromani PJ, Keane TM, LeDoux JE, editors. Post-traumatic stress disorder: basic science and clinical practice. New York, NY: Humana Press. p. 257–75
- Yehuda R, Seckl J. (2011). Minireview: stress-related psychiatric disorders with low cortisol levels: a metabolic hypothesis. Endocrinology 152(12):4496–503
- Zoladz PR, Conrad CD, Fleshner M, Diamond DM. (2008). Acute episodes of predator exposure in conjunction with chronic social instability as an animal model of post-traumatic stress disorder. Stress 11(4):259–81
- Zoladz PR, Diamond DM. (2013). Current status on behavioral and biological markers of PTSD: a search for clarity in a conflicting literature. Neurosci Biobehav Rev 37(5):860–95
- Zoladz PR, Fleshner M, Diamond DM. (2013). Differential effectiveness of tianeptine, clonidine and amitriptyline in blocking traumatic memory expression, anxiety and hypertension in an animal model of PTSD. Prog Neuropsychopharmacol Biol Psychiatry 44:1–16
- Zoladz PR, Fleshner M, Diamond DM. (2012). Psychosocial animal model of PTSD produces a long-lasting traumatic memory, an increase in general anxiety and PTSD-like glucocorticoid abnormalities. Psychoneuroendocrinology 37(9):1531–45
- Zoladz PR, Park CR, Fleshner M, Diamond DM. (2015). Psychosocial predator-based animal model of PTSD produces physiological and behavioral sequelae and a traumatic memory four months following stress onset. Physiol Behav 147:183–92