Abstract
The hypothalamic–pituitary–adrenal (HPA) axis self-regulates through a glucocorticoid negative feedback mechanism that is stereotypically slow and long lasting. Rapid (seconds to minutes) glucocorticoid feedback, however, inhibits stress-induced adrenocorticotropic hormone (ACTH) secretion too quickly to result from classic transcriptional effects of the occupied glucocorticoid receptor. Cannabinoids may act as rapid intermediary messengers between glucocorticoids and HPA activation via retroactive inhibition of afferent glutamate stimulation of the corticotropin-releasing factor neurons in the paraventricular nucleus. We demonstrated fast feedback effects of GR stimulation and blockade and observed the effect of cannabinoid receptor (CB1) antagonist AM251 on HPA axis reactivity in vivo. Rats were injected intraperitoneally with varying doses of the specific GR agonist RU28362, the GR antagonist RU486, or AM251 2 min before restraint. Blood was collected at predetermined times and corticosterone and ACTH concentrations were measured. RU28362 blunted stress-induced ACTH secretion while RU486 and AM251 significantly increased stress-induced ACTH release 15 min after restraint onset. Next, we injected AM251 58 min before RU28362, 2 min before restraint, to determine if inhibition of ACTH by RU28362 was contingent on CB1 activation. Unexpectedly, CB1 blockade failed to prevent glucocorticoid negative feedback and instead enhanced it. These studies not only establish an in vivo fast feedback model but show that rapid glucococorticoid negative feedback is similarly altered by GR and CB1 blockade. Although the hormonal consequences of acute AM251 treatment were strikingly similar to those of RU486 treatment, we are unable to draw conclusions about the serial nature of the interaction between GR activation and CB release from these results.
Introduction
The cells of the medial paraventricular nucleus (mpPVN) of the hypothalamus, anterior pituitary gland, and adrenal gland collectively form the hypothalamic–pituitary–adrenal (HPA) axis. Stressful stimuli induce a cascade of events within this network that culminates in secretion of corticosterone from the cortex of the adrenal gland. Briefly, stressful stimuli cause release of corticotropin-releasing factor (CRF) from neuron terminals in the median eminence at the base of the hypothalamus. CRF then travels through the hypophyseal portal system to the anterior lobe of the pituitary gland. Upon exposure of anterior pituitary corticotroph cells to CRF, adrenocorticotropic hormone (ACTH) is released into the general circulation, ultimately causing secretion of corticosterone from the adrenal cortex, which then distributes through the circulation and acts to regulate energy stores. The HPA axis is self-regulatory, utilizing its end-product, glucocorticoids (corticosterone in rats, cortisol in humans), to normalize its own activation, function, and responsiveness through negative feedback mechanisms. This self-regulatory mechanism is referred to widely as glucocorticoid negative feedback. Regulation of the HPA axis via glucocorticoid receptors depends on many dynamic factors including, but not limited to, previous phasic increases in glucocorticoid concentrations, the hormonal milieu prior to the stressful stimulus including other hormones, and the timing, duration, and magnitude of the applied stressor (Keller-Wood and Dallman Citation1984; Dallman et al. Citation1987a,Citationb). Adding still further complexity, glucocorticoid negative feedback acts through multiple molecular mechanisms via glucocorticoid receptors at numerous cell populations both intrinsic (Keller-Wood and Dallman Citation1984) and extrinsic to the HPA axis (Herman and Cullinan Citation1997; Herman et al. Citation2002).
Unlike the higher affinity type I glucocorticoid receptor (MR), the type II receptor (GR) is not fully occupied under basal conditions, therefore the strength of GR signaling varies in response to phasic increases in endogenous hormone secretion (de Kloet et al. Citation1998). Consequently, GR is the main receptor of interest for regulation of the HPA axis above basal circulating corticosteroid levels. Furthermore, GR is the only glucocorticoid receptor expressed in the mpPVN, the major integratory hub for stress input and negative feedback signals (Herman and Cullinan Citation1997). Although the multiple actions of GR make definitive characterization of glucocorticoid negative feedback very complex (Losel et al. Citation2003), regulatory glucocorticoid actions on HPA responsivity can be putatively parceled into two temporally defined phases: a fast mechanism and a slow mechanism (Jones et al. Citation1979). Most of the consequences of the slower “classic” GR action as a nuclear transcription factor are not detectable until hours or even days after exposure. More rapidly, bound GR have nontranscriptional effects through protein–protein interactions (Reichardt and Schutz Citation1998; Falkenstein et al. Citation2000a). The faster inhibition (seconds or minutes, up to 1 h) has been demonstrated convincingly by a number of researchers (Keller-Wood and Dallman Citation1984; Liberzon et al. Citation1997; Thrivikraman et al. Citation2000), but the underlying mechanism and dynamics have not been definitively characterized in vivo.
The rapid, primary HPA response to phasic increases in glucocorticoid concentration is of great importance to understanding the dynamics of glucocorticoid negative feedback. Fast glucocorticoid feedback, although brief and small in magnitude plays an important role in the overall balance and responsiveness of the HPA axis and can also affect sexual, aggressive, and ingestive behaviors (Dallman Citation2005). Many of the seminal in vivo studies of rapid glucocorticoid actions were conducted in the 1960s and 1970s (Yates et al. Citation1961; Dallman and Yates Citation1969; Jones and Hillhouse Citation1976; Jones et al. Citation1979). At that time, not only were the molecular processes of HPA regulation yet to be well understood, but specific assays for hormones such as ACTH and corticosterone were not yet fully developed. Since then, as details of nuclear cytosolic steroid receptor actions have become known, most attention has been paid to the classic, slowly acting feedback effects of glucocorticoid. In recent years, however, rapid effects of systemically administered glucocorticoid have been demonstrated across species from rats and humans to migratory birds (Breuner and Wingfield Citation2000; Falkenstein et al. Citation2000b; Dallman Citation2005).
In 2003, Di et al. (Citation2003) presented compelling in vitro evidence that endocannabinoids (eCBs) act as fast-acting intermediaries between glucocorticoid and neuronal activation in the mpPVN. In the model proposed by Tasker's group, glucocorticoids activate a putative post-synaptic G-protein-coupled membrane receptor that leads to the release of post-synaptic eCBs from CRF neurons as rapidly as 3–5 min after glucocorticoid exposure (Di et al. Citation2003). Released by postsynaptic neurons, eCBs then act as retrograde messengers binding to presynaptic eCB receptors (CB1) to modulate the release of excitatory and inhibitory neurotransmitters (Schlicker and Kathmann Citation2001). In this case, eCBs act at presynaptic glutamatergic terminals to inhibit glutamate release onto the CRF neurons, thereby altering synaptic drive to these cells. CB1 are expressed in the hypothalamus and pituitary of rats and on the anterior lobe of the human pituitary gland (Breivogel et al. Citation1997), suggesting some involvement with stress networks. There have been a few studies observing the effects of cannabinoids on HPA axis function in vivo (Navarro et al. Citation1997; Patel et al. Citation2004a,Citationb; Wade et al. Citation2006). None of these, however, have been conducted in the appropriate timeframe to observe the rapid actions of eCBs on HPA responsivity that could result from the rapid glucocorticoid action proposed by the above model.
In previous experiments, we have shown that the specific GR agonist RU28362 injected 15 min or more before the onset of restraint significantly reduced stress-induced secretion of ACTH and corticosterone (Ginsberg et al. Citation2003a, Citation2006). In order to follow up on these experiments, and to establish an in vivo fast feedback paradigm, we have conducted a dose response experiment observing the effect of RU28362, and a GR antagonist, RU486, administered intraperitoneally (i.p.) 2 min before the onset of restraint on subsequent HPA responsivity. Next, we examined whether eCBs rapidly alter HPA responsivity in vivo by conducting a dose response experiment similar in design to those for RU28362 and RU486 using the CB1 receptor antagonist AM251. Lastly, we attempted to establish a connection between the observed rapid effects of glucocorticoid and eCB, with a polypharmacological study in which we injected the CB1 antagonist AM251 58 min before the GR agonist RU28362 with the onset of restraint 2 min later. Based on evidence that glucocorticoids act via eCB to prevent glutamate release from stress-responsive afferents (Di et al. Citation2003, Citation2005; Tasker et al. Citation2006), we hypothesized that blockade of CB1 would prevent glucocorticoid negative feedback by preventing the action of increased extracellular eCB concentrations caused by rapid actions of bound GR.
Methods
Animals
Naïve, 60 day-old male Sprague–Dawley rats (300–375 g) from Charles River, North Carolina were used. Rats were housed separately in hanging cages, with chow and water available ad libitum. Colony lighting schedule was a 12 h:12 h light:dark cycle, lights on at 0700 h and off at 1900 h. Rats were allowed a 2-week acclimatization period during which they were handled and weighed daily. All experiments took place within 6 h after lights on during the HPA axis circadian nadir. The dependent variables in these experiments were extremely sensitive to stressful stimuli of any kind, therefore care was be taken to maintain a noise-controlled environment with minimal physical disturbances. Permission for the experimental protocols and animal use was obtained from the UCSF Institutional Animal Care and Use Committee (IACUC).
Drugs
The doses of RU28362 were chosen based on a preliminary dose-response experiment (Ginsberg et al. Citation2003b). RU28362 (Roussel Uclaf, France, a gift from Dr Robert Spencer, University of Colorado at Boulder) and RU486 were dissolved in 0.9% saline +40% 2-hydroxypropyl-beta-cyclodextrin (HBC, Sigma, St Louis, MO, USA). AM251 (Tocris International, St Louis, MO, USA) was dissolved in a solution consisting of 0.9% saline (97%), Tween (1%), and dimethyl sulfoxide solution (2%). The doses chosen for the AM251 dose response experiment were based on previously established anti-cannabinoid effects on feeding and HPA activation. Both drugs and their corresponding vehicles were delivered i.p. at a designated time before the initiation of restraint.
Restraint procedure
Rats were placed in a clear Plexiglas tube (23.5 cm in length and 7 cm in diameter) with tails protruding. The size of the tube restricted both lateral and forward/backward movement but did not interfere with breathing. Rats remained in the restrainer for 120 min. Blood samples (∼300 μl) were collected via tailnick at 0, 15, 30, 60, and 120 min following the initiation of restraint and kept on ice. The tip (∼1 mm) of the tail was removed with a scalpel for the 0 time sample; thereafter the formed clot was wiped away and blood collected from the initial wound. Owing to the sensitive nature of dependent variables, no anesthetic was used prior to restraint or tailnick. “Time zero” samples were collected within a minute after the rat was placed in the restrainer.
Experimental design
Experiment 1
RU28362 dose response. Rats (n = 5–10) were injected i.p. with 37.5, 75, or 150 μg/kg RU28362 in HBC or vehicle 2 min before being placed in restrainers.
Experiment 2
RU486 dose response. Rats (n = 5–8) were injected i.p. with 3.33, 10, or 30 mg/kg RU486 in HBC or vehicle 2 min before being placed in restrainers.
Experiments 3 and 4
AM251 dose responses. Rats (n = 5) were injected i.p. with 0, 1, 2, or 4 mg/kg AM251 2 min (experiment 3) or 60 min (experiment 4) before being placed in a restrainer. In experiment 4, a 15-min blood sample was not collected, and the restraint period lasted for 120 min.
Experiment 5
AM251/RU28362 combination. Rats (n = 17–20) received an i.p. injection of vehicle or 2 mg/kg AM251 1 h before the initiation of restraint. The dose of 2 mg/kg was selected because it was the only dose in experiment 4 that injected 60 min before the onset of restraint, caused an increase in stress-induced ACTH and corticosterone concentration. Fifty-eight minutes following the first injection, rats received an injection of vehicle or 150 μg/kg RU28362 and were placed in restrainers 2 min later. Rats remained in restraint for 120 min. A 15-min blood sample was not collected.
Radioimmunoassay
Blood samples were collected into chilled tubes containing 0.3 M disodium EDTA (100 μl/tube) and centrifuged at 3000 rpm and 4°C. Separated plasma was stored at − 20°C prior to radioimmunoassay. Plasma corticosterone and ACTH were measured using an Immuchem double-antibody corticosterone assay kit for rat (ICN Biochemicals, Costa Mesa, CA, USA) and ACTH kit for rat (Diasorin Inc., Stillwater, MN, USA). Protocol and assay variability have been described previously (La Fleur et al. Citation2004).
Statistical analysis
Analyses were conducted using the statistical analysis program SPSS. For dose response experiments, an initial two-way univariate ANOVA (dose X time) was performed to assess the overall effects of dose across time. Next, the data were analysed by one-way ANOVA discretely at each timepoint, and relevant planned comparisons were then conducted using Fisher's LSD tests. For the final experiment, a multivariate 2 × 2 × 5 ANOVA (AM251 × RU28362 × time) was used followed by planned comparisons. Outliers more than 2.5 standard deviations from the mean were excluded from all analyses. Error bars in the figures represent ± SEM. For all comparisons, the criterion for significance was p < 0.05.
Results
Experiment 1
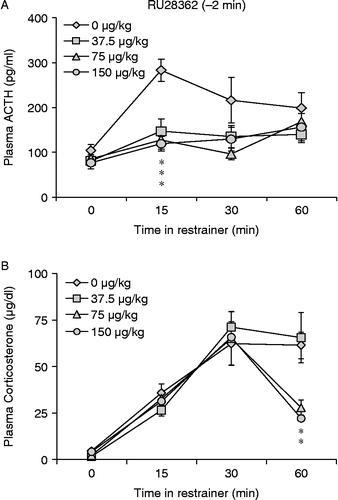
RU28362 treatment significantly blunted hormonal stress responses when administered 2 min before the onset of restraint (n = 5–10, ). ACTH: As a whole, the data showed a main effect of both drug [F(3,106) = 5.058, p < 0.01)] and time [F(3,106) = 5.888, p < 0.01)] on plasma ACTH concentrations following the onset of restraint. When the data were parceled into groups by time point, a significant overall effect of RU28362 on plasma ACTH concentration [F(3,30) = 2.895, p = 0.05) was found 15 min following the onset of restraint. Planned comparisons show that all three doses of RU28362 significantly inhibited the ACTH response to restraint compared to vehicle treated controls x (). Corticosterone: Overall, there was a significant increase in corticosterone concentration during the restraint period [F(3,109) = 34.01, p < 0.001] and this depended on the dose of RU28362 administered [interaction, F(9,103) = 1.969, p = 0.05]. There was no difference in plasma corticosterone concentration among treatment groups at any individual time during restraint. Plasma corticosterone concentration of rats treated with 75 and 150 μg/kg RU28362 returned to baseline values 60 min into the restraint period [F(3,19) = 7.708, p < 0.01] while in rats treated with vehicle or 37.5 μg/kg RU28362 corticosterone remained significantly elevated compared to baseline at 60 min ().
Figure 1. RU28362 injected i.p. 2 min before the onset of restraint blunted the hormonal responses to restraint stress. All three doses blunted ACTH 15 min following the onset of restraint (A). There was no difference in corticosterone secretion until 60 min into the restraint period, when plasma corticosterone values in rats that received 75 and 150 μg/kg RU28362 returned to baseline levels (B). Values are group mean + SEM; n = 5–10 per group. *p < 0.05 vs. other group (A) or groups (B).
Experiment 2
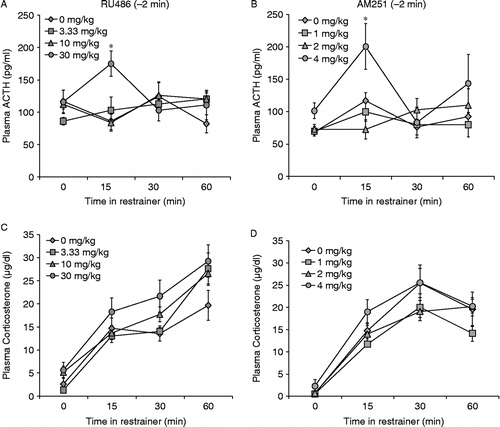
ACTH: GR antagonist RU486 administered 2 min before restraint increased ACTH secretion (n = 5–8, ). Overall, stress-induced changes in plasma ACTH concentration depended on the dose of RU486 administered to the rat [interaction, F(9,109) = 2.273, p < 0.05]. At 15 min, RU486 caused a significant increase plasma ACTH concentration overall [F(3,28) = 3.459, p < 0.05]. Subsequent comparisons revealed that only 30 mg/kg RU486 produced an increase in ACTH concentration that was different from vehicle treated controls (). At 30 min, ACTH values of all groups were at baseline. Corticosterone: Overall, plasma corticosterone increased with restraint [F(3,88) = 40.30, p < 0.001], and this was not changed by RU486 treatment. There were no differences in plasma corticosterone secretion among treatment groups at any time ().
Figure 2. RU486 and AM251 acted rapidly to increase the ACTH response to restraint stress. Fifteen minutes after the onset of restraint, rats that received RU486 i.p. (30 mg/kg) 2 min before the onset of restraint had significantly higher plasma ACTH values than those treated with 0, 3.33, or 10 mg/kg (A). There was no significant effect of RU486 on stress-induced corticosterone secretion (C). 4 mg/kg AM251 administered i.p. 2 min before the onset of restraint stress caused increased ACTH concentration at the initiation of restraint (2 min after injection) and 15 min into the restraint period (B), but did not alter stress-induced corticosterone secretion (D). Values are group mean + SEM; n = 5–8 per group. *p < 0.05 vs. other groups.
Experiment 3
ACTH: AM251 increased ACTH secretion when given 2 min before restraint (n = 5, .) ACTH: There was an overall effect of time [F(3,75) = 5.163, p < 0.01] and drug [F(3,75) = 4.3, p < 0.01] on plasma ACTH concentration. The effect of restraint on plasma ACTH values depended somewhat on both the time examined and the dose of AM251 the rat received, but not significantly (p = 0.08). There was a significant main effect of AM251 on ACTH concentration [F(3,17) = 3.785, p < 0.05]. Rats that received 4 mg/kg AM251 had significantly higher baseline ACTH values than groups treated with 0,1, or 2 mg/kg AM251 (). By 30 min of restraint, ACTH values of all treatment groups had returned to baseline. Corticosterone: Overall, plasma corticosterone concentrations increased with time in the restrainer [F(3,77) = 55.130, p < 0.001]. There were no significant differences among treatment groups at any single time ().
Experiment 4
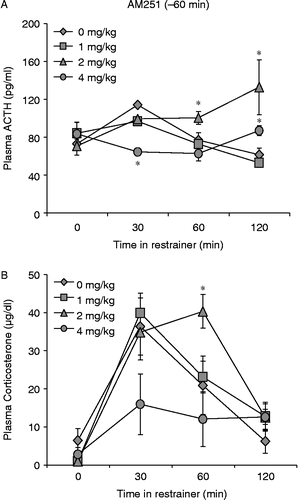
ACTH: AM251 had dose-dependent effects on stress- induced ACTH secretion when administered 1 h before the onset of restraint (n = 4–5, ). There was a significant overall effect of AM251 administered 60 min before the onset of restraint on stress-induced ACTH secretion [F(3,77) = 3.136, p < 0.05], and this effect varied across time [interaction, F(9,71) = 3.314, p < 0.005]. There were no significant differences among treatment groups until 30 min after the onset of restraint [F(3,17) = 3.237, p = 0.05] when rats treated with the highest dose of AM251, 4 mg/kg, had significantly lower plasma ACTH values than vehicle treated controls (p < 0.05). Sixty minutes into the restraint period, plasma ACTH concentrations in rats that received vehicle and 1 mg/kg AM251 decreased to levels comparable to those of rats treated with 4 mg/kg AM251, but rats treated with 2 mg/kg AM251 had significantly higher plasma ACTH concentrations compared to the other three groups (p < 0.05 for all comparisons). Rats that received 2 mg/kg AM251 continued to have higher plasma ACTH values compared to vehicle and 1 mg/kg AM251 treated rats 120 min into the restraint period [F(3,17) = 3.335, p < 0.05 for both comparisons). At this time, however, ACTH values of animals treated with 4 mg/kg AM251 increased slightly to a level that was significantly higher than vehicle and 1 mg/kg treated rats (). Corticosterone: Restraint generally caused an increase in plasma corticosterone concentrations [F(3,73) = 18.59, p < 0.001] which varied with increasing doses of AM251 [F(9,67) = 2.431, p < 0.05] (). Rats that received 4 mg/kg AM251 showed the same trend observed previously for ACTH. Plasma corticosterone values for this group were in general lower than those of other treatment groups throughout the restraint period. The differences observed here with 4 mg/kg AM251 were only trends (p < 0.1), unlike the results with ACTH. Furthermore, rats that received 4 mg/kg AM251 were the only treatment group that did not have a significant corticosterone peak 30 min after the initiation of restraint. However, the 2 mg/kg dose of AM251 produced a higher peak in corticosterone than the three other doses at 60 min after the onset of restraint ().
Figure 3. AM251 injected 1 h before the onset of restraint stress alters hormonal responses. Four mg/kg AM251 injected 1 h before restraint initially caused a blunted ACTH response to restraint at 30 min. After 120 min rats that received 4 mg/kg AM251 had increased levels of plasma ACTH, significantly higher than vehicle treated controls (A). Two mg/kg AM251 augmented stress-induced ACTH release at 60 and 120 min after the initiation of restraint (A), and produced significantly higher corticosterone levels compared to vehicle treated controls 60 min following the onset of restraint (B). Values are group mean + SEM; n = 5 per group. *p < 0.05 vs. other groups.
Experiment 5
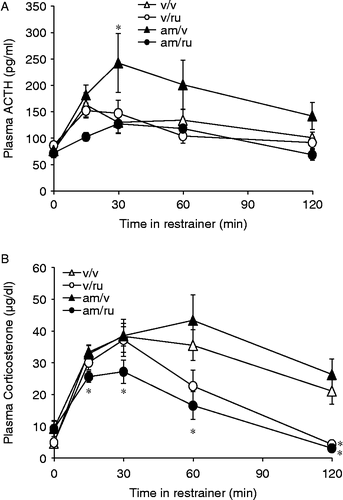
AM251 administered 58 min before RU28362 and 60 min before the onset of restraint blunted the HPA response to restraint (n = 17–20, ). ACTH: rats that received vehicle treatments at − 60 and − 2 min before restraint had a small but significant peak in stress-induced ACTH concentration 15 min after the onset of restraint [t(19) = − 2.103, p < 0.05] which returned to baseline levels 45 min later (). Vehicle at − 60 min and 150 μg/kg RU28362 treatment at − 2 min failed to alter ACTH compared with vehicle at any time. Two mg/kg AM251 58 min before vehicle at − 2 min produced effects similar to those seen in experiment 3; there was a significant augmentation of restraint-induced ACTH secretion by 30 min compared to vehicle treated controls [F(1,49) = 3.972, p = 0.05]. Unexpectedly, the rats treated with AM251 58 min before RU28362 at − 2 min showed a significantly blunted ACTH response to restraint, first at 15 min after the onset of restraint stress (not significant, p = 0.059) and again at 120 min [F(1,26) = 7.737, p < 0.05] (). Corticosterone: Overall, time in the restraint tube had a significant effect on plasma corticosterone concentration (F[4,45] = 3.986, p < 0.01], and this effect varied with RU28362 treatment (interaction, F[4,45] = 18.903, p < 0.001] (). Corticosterone concentrations in rats treated with AM251 alone did not differ from vehicle treated rats at any time during the restraint period while rats treated with RU28362 alone exhibit delayed glucocorticoid negative feedback 120 min following the onset of restraint [F(1,26) = 9.488, p < 0.01]. Rats given the AM251 –RU28362 combination, in accord with the ACTH values in the same rats, had consistently low corticosterone values throughout the restraint period showing lower corticosterone values starting at 15 min [F(1,26) = 3.658, p < 0.05] and continuing until the termination of restraint 30 min [F(1,26) = 11.058 p < 0.01], 60 min [F(1,26) = 4.537, p < 0.05], and 120 min [F(1,26) = 29.31, p < 0.001] ().
Figure 4. Two mg/kg AM251 injected 60 min before 150 μg/kg RU28362, 2 min before the onset of restraint, produced blunted hormonal responses to restraint stress. Rats that received the combination of AM251/RU28362 showed lower plasma ACTH values at 15 min following the onset of restraint and significantly lower values at 120 min (A). AM251 administered 1 h before RU28362 administration and 62 min before the onset of restraint produced consistently lower stress-induced plasma corticosterone concentrations from 15 to 120 min into the restraint period. RU28362 alone produced significantly blunted corticosterone levels only 120 min following the onset of restraint (B). Values are group mean + SEM; n = 17–20 per group. *p < 0.05 vs. other groups.
Discussion
These experiments demonstrate that: (1) systemic administration of the GR agonist RU28362 2 min before the onset of acute restraint rapidly suppresses ACTH secretion, when not preceded by any other manipulation; (2) systemic administration of either the GR antagonist RU486 or the CB1 antagonist AM251 augments stress-induced ACTH secretion within the same timeframe; and (3) cannabinoids and glucocorticoids interact in some way to regulate HPA activity. We originally based our hypothesis on the work of Di et al. and sought to test in vivo whether eCBS serve as a mechanism through which glucocorticoids exert fast negative control over HPA activation. Although some results generally corroborated this hypothesis others, using long temporal separation between the cannabinoid antagonist and GC agonist, refuted the hypothesis.
After a single injection of the GR agonist, RU28362, 2 min before restraint, there was clear and dose-related fast feedback inhibition of ACTH and corticosterone secretion. It is this rapid effect of glucocorticoids, normally secreted after a stressor, that critically limits both the magnitude and duration of HPA responses to stressors (Dallman and Jones Citation1973; Keller-Wood et al. Citation1984). Despite the uniform reduction in ACTH at 15 min after all doses of RU28362, only the higher doses of the agonist decreased plasma corticosterone concentrations, and then, only at 60 min. This result suggests that in all groups ACTH secretion did occur at some time during restraint, but that both higher doses of the steroid were effective in limiting the magnitude and/or duration of ACTH secretion, thus reducing drive to the adrenal cortex. The adrenals have a “memory” for the total amount of ACTH have been exposed to, and when ACTH is above the adrenal maximum for an immediate, proportional glucocorticoid response, the glands respond to higher ACTH with longer duration of secretion; i.e. the adrenals integrate ACTH signals over time (Keller-Wood et al. Citation1983; Keith et al. Citation1986). Moreover, the maximal adrenal response to ACTH saturates in the lower quarter of the normal ACTH stress-response range (Keller-Wood et al. 1984); thus, prolongation of the corticosteroid response is usually observed after stressor-induced ACTH secretion has subsided.
The inhibition of HPA responses observed when the feedback signal was applied systemically 2 min before restraint strongly suggests that the effect was not mediated through translocation of the GR-glucocorticoid complex to the nucleus, but rather by membrane effects. The rapid hormonal effects are too fast to be compatible with the classic genomic actions of GR (Dallman et al. Citation1987a; de Kloet et al. Citation1993). Typically, GR are considered to act through a complex, multistep process beginning with binding of the ligand to the cytosolic steroid receptor complex and continuing with dissociation of a heatshock protein from the DNA binding domain, translocation into the nucleus, dimerization, binding to a glucocorticoid response element on DNA, and subsequent alteration of target gene transcription (Falkenstein et al. Citation2000a). Translocation of the bound receptor into the nucleus alone takes up to 30 min (Robertson et al. Citation1993; Makara and Haller Citation2001; Nishi and Kawata Citation2007; Nishi et al. Citation2007), twice as long as the time it takes for the glucocorticoid effects observed in our experiments, suggesting that the GR-dependent actions observed within the timeframe of our paradigm were independent of steroid-induced transcriptional alterations. It is important to consider that preliminary stages of DNA-dependent glucocorticoid actions occur simultaneously with the rapid glucocorticoid effects. The consequences of the slower, glucocorticoid-mediated actions are generally not detectable at the level of ACTH secretion within the time constraints of our experiment.
Use of the GR antagonist, RU486, given 2 min prior to restraint was expected to increase ACTH responses during the restraint period by blocking the endogenous fast feedback of stress-induced corticosterone secretion. This was observed with the highest dose of RU486 in the ACTH response 15 min after the onset of restraint. Although corticosterone concentrations were higher than the control at all times examined after the two higher doses of RU486, these results were not significantly different from the control or low dose groups, possibly because the adrenal response was near its maximum. Again, as in the agonist experiment, it appears, because of the uniform increase in corticosterone in all groups, as though ACTH was secreted in response to restraint.
The test of a role for eCBS on fast GC feedback, through the use of the CB1-antagonist AM251 injected 2 min before restraint, strikingly mirrored the effects of injecting the GR-antagonist RU486, and suggested that eCBS were involved in the ACTH response to glucocorticoids. The highest dose of the CB-1 antagonist significantly increased the ACTH response to restraint at 15 min, as predicted by the hypothesis. Again, corticosterone responses were non-significantly increased compared to vehicle-injected rats. As with the RU486 results, these results suggest that the CB-1 antagonist augmented ACTH responses that occurred during the period of restraint. The result suggests strongly that endogenous cannabinoids may, indeed mediate the fast feedback actions of GCs on restraint stress in vivo, as they do on glutamatergic input in vitro (Di et al. Citation2003).
Our ultimate goal in these experiments was to test whether combined treatments with the GR-agonist and the CB-1-antagonist would reveal that fast feedback induced by RU28362 was reduced by ongoing blockade of eCB receptors. The two compounds could not be given in a single injection; however, accurate timing was essential for the prestress administration of the steroid. Therefore, we first tested the effect on restraint-induced ACTH and corticosterone responses of giving AM251 60 min before restraint. Treatment of rats with AM251 an hour before restraint provided a confusingly different spectrum of HPA responses from those observed when it was given at − 2 min. The middle dose of AM251 augmented the late ACTH response at 60 and 120 min after the onset of restraint. The highest dose of AM251 significantly inhibited ACTH at 30 min and augmented it 120 min after the onset of restraint. In general, except for at 120 min, the corticosterone responses reflected the ACTH responses.
Double treatment with AM251 at − 60 min and RU28362 treatment at − 2 min before restraint at 0 min had similarly unanticipated effects. The middle dose of AM251 was used in this experiment and resulted, in combination with vehicle injection at − 2 min, in stimulation of ACTH secretion at 30, rather than 60 or 120 min as was found in the single injection experiment. However, when vehicle was injected at − 60 min and RU28362 at − 2 min, the GR-agonist did not inhibit secretion of ACTH during the first 30 min, compared to control, and only inhibited corticosterone at 120 min. This may have been a consequence of the known facilitatory effects of a prior stressor (in this case, injection; Dallman Citation1993). When the CB-1 antagonist was given at − 60 min, and RU28362 was given at − 2 min, there was diminished mean ACTH concentration, and consistently decreased corticosterone responses across all times examined. The results of these studies do not support the hypothesis that fast glucocorticoid feedback is mediated by CB-1 receptors.
It is clear from these experiments that the effects of AM251 on the HPA response to restraint are both time and dose dependent. When given 2 min before restraint, the dose of 4 mg/kg produced an (anticipated) increase in stress-induced ACTH secretion. Given 1 h before the onset of restraint, 4 mg/kg AM251 suppressed restraint-induced ACTH secretion. This is consistent with previous studies in which cannabinoids have been shown to have biphasic effects (Murphy et al. Citation1998; Manzanares et al. Citation1999; Patel et al. Citation2004b). Because in the final experiment, the CB-1 antagonist was injected a long time before restraint stress, and because eCBS act on sites throughout the brain (Freund et al. Citation2003; Piomelli Citation2003; Patel et al. Citation2004a), it is likely that the results were a consequence of blocking normal action of CB-1 receptor occupancy at other sites in addition to the hypothalamus. Furthermore, CB1 agonists and antagonists have been shown to act in vivo inconsistently in studies of HPA regulation (Weidenfeld et al. Citation1994; Barna et al. Citation2004). However, Patel and Hillard showed not only that cannabinoids inhibit stress-induced corticosterone release, but that blockade of CB1 receptors potentiated the HPA response to restraint after 1 h of exposure. Antagonists have been shown to stimulate the HPA axis when given systemically (Patel et al. Citation2004b; Cota Citation2008) or intracerebroventricularly (Manzanares et al. Citation1999) although in some cases they have reduced plasma ACTH production (Murphy et al. Citation1998). Alternatively, CB1 agonists can either stimulate (Puder et al. Citation1982; Patel et al. Citation2004b) or inhibit ACTH and corticosterone secretion as well as secretion of CRH (Di et al. Citation2003; Patel et al. Citation2004b) from the hypothalamus.
Recently, eCBS in the basolateral amygdala have been shown to mediate glucocorticoid effects on learning and memory (Campolongo et al. Citation2009), and it seems quite possible that this rapid interacting response system may exist at other sites in brain as well (Hill and McEwen Citation2009).
Limitations of this study
Because it seems highly likely, as indicated by the consistent corticosterone responses, that we missed early ACTH responses that occurred in most treatment groups, we should have sampled ACTH more frequently early after the onset of restraint. Further, because there were no unrestrained controls treated with the drugs, we can only guess that ACTH would not have changed appreciably after the injections. We worked in the morning, when ACTH concentrations are low, and when ACTH secretion appears to be constitutive rather than driven by CRF (Dallman et al. Citation1987b), therefore, it seems unlikely that the patterns of ACTH observed after RU28362 would have been markedly affected by the drug. However, there should have been unrestrained controls for the groups treated with RU486 and AM251, since these drugs could have stimulated ACTH by themselves.
Finally, it is abundantly clear that a better design for testing the interactions between corticosterone and eCBS on inhibition of ACTH secretion would have been to provide the CB1 antagonist directly into the hypothalamic paraventricular nuclei 2 min before restraint while giving the glucocorticoid agonist systemically at the same time. We could, with that design, expect to see both acute- and site-dependent effects of manipulating the eCB receptors. Prior systemic application of CB1 blockers may have activated other brain pathways that normally modulate HPA responses to stress, as well as activating facilitatory stress networks as a consequence of prior manipulation. More selective studies are called for at this point.
Acknowledgements
This work was supported by NIH grants DK28172, DA16944, and DK074319. Disclosure statement: The authors have nothing to declare.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Barna I, Zelena D, Arszovski AC, Ledent C. 2004. The role of endogenous cannabinoids in the hypothalamo-pituitary-adrenal axis regulation: In vivo and in vitro studies in CB1 receptor knockout mice. Life Sci. 75:2959–2970.
- Breivogel CS, Sim LJ, Childers SR. 1997. Regional differences in cannabinoid receptor/G-protein coupling in rat brain. J Pharmacol Exp Ther. 282:1632–1642.
- Breuner CW, Wingfield JC. 2000. Rapid behavioral response to corticosterone varies with photoperiod and dose. Horm Behav. 37:23–30.
- Campolongo P, Roozendaal B, Trezza V, Hauer D, Schelling G, McGaugh JL, Cuomo V. 2009. Endocannabinoids in the rat basolateral amygdala enhance memory consolidation and enable glucocorticoid modulation of memory. Proc Natl Acad Sci USA. 106:4888–4893.
- Cota D. 2008. The role of the endocannabinoid system in the regulation of hypothalamic–pituitary–adrenal axis activity. J Neuroendocrinol. 20:35–38.
- Dallman MF. 1993. Stress update. Adaptations of the hypothalamic–pituitary–adrenal axis to chronic stress. Trends Endocrinol Metab. 4:62–69.
- Dallman MF. 2005. Fast glucocorticoid actions on brain: Back to the future. Front Neuroendocrinol. 26:103–108.
- Dallman MF, Jones MT. 1973. Corticosteroid feedback control of ACTH secretion: Effect of stress-induced corticosterone secretion on subsequent stress responses in the rat. Endocrinology. 92:1367–1375.
- Dallman MF, Yates FE. 1969. Dynamic asymmetries in the corticosteroid feedback path and distribution-metabolism-binding elements of the adrenocortical system. Ann N Y Acad Sci. 156:696–821.
- Dallman MF, Akana SF, Cascio CS, Darlington DN, Jacobson L, Levin N. Regulation of ACTH secretion: Variations on a theme of B. Rec Prog Horm Res. 1987a; 43:113–173.
- Dallman MF, Akana SF, Jacobson L, Levin N, Cascio CS, Shinsako J. Characterization of corticosterone feedback regulation of ACTH secretion. Ann N Y Acad Sci. 1987b; 512:402–414.
- de Kloet ER, Oitzl MS, Joels M. 1993. Functional implications of brain corticosteroid receptor diversity. Cell Mol Neurobiol. 13:433–455.
- de Kloet ER, Vereugdenhil E, Oitzl MS, Joels M. 1998. Brain corticosteroid receptor balance in health and disease. Endocr Rev. 19:269–301.
- Di S, Malcher-Lopes R, Halmos KC, Tasker JG. 2003. Nongenomic glucocorticoid inhibition via endocannabinoid release in the hypothalamus: A fast feedback mechanism. J Neurosci. 23:4850–4857.
- Di S, Boudaba C, Popescu IR, Weng FJ, Harris C, Marcheselli VL, Bazan NG, Tasker JG. 2005. Activity-dependent release and actions of endocannabinoids in the rat hypothalamic supraoptic nucleus. J Physiol. 569:751–760.
- Falkenstein E, Christ M, Fuering M, Wehling M. Specific nongenomic actions of aldosterone. Kidney Int. 2000a; 57:1390–1394.
- Falkenstein E, Tillmann HC, Christ M, Fuering M, Wehling M. Multiple actions of steroid hormones-a focus on rapid, nongenomic effects. Pharm Rev. 2000b; 52:513–555.
- Freund TF, Katona I, Piomelli D. 2003. Role of endogenous cannabinoids in synaptic signaling. Physiol Rev. 83:1017–1066.
- Ginsberg AB, Campeau S, Day HE, Spencer RL. Acute glucocorticoid pretreatment suppresses stress-induced hypothalamic–pituitary–adrenal axis hormone secretion and expression of corticotropin-releasing hormone hnRNA but does not affect c-fos mRNA or fos protein expression in the paraventricular nucleus of the hypothalamus. J Neuroendocrinol. 2003a; 15:1075–1083.
- Ginsberg AB, O'Connor KA, Spencer RL. 2003b. Negative feedback effects of glucocortioid receptor agonist RU28362 on stress-induced gene expression in the PVN and pituitary, Society for Neuroscience Meeting 2003, Washington DC, Program No. 282.17
- Ginsberg AB, Frank MG, Francis AB, Rubin BA, O'Connor KA, Spencer RL. Specific and time-dependent effects of glucocorticoid receptor agonist RU28362 on stress-induced pro-opiomelanocortin hnRNA, c-fos mRNA and zif268 mRNA in the pituitary. J Neuroendocrinol. 18:129–138.
- Herman J, Cullinan W. 1997. Neurocircuitry of stress: Central control of the hypothalamo-pituitary-adrenocortical axis. Trends Neurosci. 20:78–84.
- Herman JP, Tasker JG, Ziegler DR, Cullinan WE. 2002. Local circuit regulation of paraventricular nucleus stress integration. Glutamate–GABA connections. Pharm Biochem Behav. 71:457–468.
- Hill MN, McEwen BS. 2009. Endocannabinoids: The silent partner of glucocorticoids in the synapse. Proc Natl Acad Sci USA. 106:4579–4580.
- Jones MT, Hillhouse EW. 1976. Structure–activity relationship and the mode of action of corticosteroid feedback on the secretion of corticotrophin-releasing factor (corticoliberin). J Steroid Biochem. 7:1189–1202.
- Jones MT, Gillham B, Mahmoud S, Holmes MC. 1979. In: Jones MT, Gillham B, Dallman MF, Chattopadhyay S, editors. The characteristics and mechanism of actions of corticosteroid negative feedback at the hypothalamus and anterior pituitary: Interaction within the brain–pituitary adrenocortical system. New York: Academic Press Inc. 163–180.
- Keith LD, Tam B, Greer MA. 1986. Stimulation of corticosterone secretion in vitro by brief ACTH exposure. Am J Physiol. 250:E629–E633.
- Keller-Wood ME, Dallman MF. 1984. Corticosteroid inhibition of ACTH secretion. Endocr Rev. 5:1–24.
- Keller-Wood ME, Shinsako J, Dallman MF. 1983. Integral as well as proportional adrenal responses to ACTH. Am J Physiol. 245:R53–R59.
- Keller-Wood M, Shinsako J, Dallman MF. 1984. Interaction between stimulus intensity and corticosteroid feedback in control of ACTH. Am J Physiol. 247:E489–E494.
- la Fleur SE, Akana SF, Manalo SL, Dallman MF. 2004. Interaction between corticosterone and insulin in obesity: Regulation of lard intake and fat stores. Endocrinology. 145:2174–2185.
- Liberzon I, Krstov M, Young EA. 1997. Stress–restress: Effects on ACTH and fast feedback. Psychoneuroendocrinology. 22:443–453.
- Losel RM, Falkenstein E, Feuring M, Schultz A, Tillmann HC, Rossol-Haseroth K, Wehling M. 2003. Nongenomic steroid action: Controversies, questions and answers. Physiol Rev. 83:965–1016.
- Makara GB, Haller J. 2001. Non-genomic effects of glucocorticoids in the neural system. Evidence, mechanisms and implications. Prog Neurobiol. 65:367–390.
- Manzanares J, Corchero J, Romero J, Fernandex-Ruiz JJ, Ramos JA, Fuentes JA. 1999. Pharmacological and biochemical interactions between opioids and endocannabinoids. Trends Pharmacol Sci. 20:287–294.
- Murphy LL, Munoz RM, Adrian BA, Villanua MA. 1998. Function of endocannabinoid receptors in the neuroendocrine regulation of hormone secretion. Neurobiol Dis. 5:432–446.
- Navarro M, Hernandez E, Munoz RM, del Arco I, Villanua MA, Carrera MR, Rodriquez de Fonseca F. 1997. Acute administration of the CB1 cannabinoid receptor antagonist SR 141716A induces anxiety-like responses in the rat. Neuroreport. 8:491–496.
- Nishi M, Kawata M. 2007. Dynamics of glucocorticoid receptor and mineralocorticoid receptor: Implications from live cell imaging studies. Neuroendocrinology. 85:186–192.
- Nishi M, Usuku T, Itose M, Fujikawa K, Hosokawa K, Matsuda KI, Kawata M. 2007. Direct visualization of glucocorticoid receptor positive cells in the hippocampal regions using green fluorescent protein transgenic mice. Neuroscience. 146:1555–1560.
- Patel S, Cravatt BF, Hillard CJ. Synergistic Interactions between cannabinoids and environmental stress in the activation of the central amygdala. Neuropsychopharmacology. 2004a; 30:497–507.
- Patel S, Roelke CT, Rademacher DJ, Cullinan WE, Hillard CJ. Endocannabinoid signaling negatively modulates stress-induced activation of the hypothalamic–pituitary–adrenal axis. Endocrinology. 2004b; 145:5431–5438.
- Piomelli D. 2003. The molecular logic of endocannabinoid signalling. Nat Rev Neurosci. 4:873–884.
- Puder M, Weidenfeld J, Chowers I, Nir I, Conforti N, Siegel RA. 1982. Corticotrophin and corticosterone secretion following delta 1-tetrahydrocannabinol, in intact and hypothalamic deafferented male rats. Exp Brain Res. 46:85–88.
- Reichardt HM, Schutz G. 1998. Glucocorticoid signalling—multiple variations of a common theme. Mol Cell Endocrinol. 146:1–6.
- Robertson N, Schulman G, Karnik S, Alnemri E, Litwack G. 1993. Demonstration of nuclear translocation of the mineralocorticoid receptor (MR) using an anti-MR antibody and confocal laser scanning microscopy. Mol Endocrinol. 7:1226–1239.
- Schlicker E, Kathmann M. 2001. Modulation of transmitter release via presynaptic cannabinoid receptors. Trends Pharmacol Sci. 22:565–572.
- Tasker JG, Di S, Malcher-Lopes R. 2006. Minireview: Rapid glucocorticoid signaling via membrane-associated receptors. Endocrinology. 147:5549–5556.
- Thrivikraman KV, Nemeroff CB, Plotsky PM. 2000. Sensitivity to glucocorticoid-mediated fast-feedback regulation of the hypothalamic–pituitary–adrenal axis is dependent upon stressor specific neurocircuitry. Brain Res. 870:87–101.
- Wade MR, Degroot A, Nomikos GG. 2006. Cannabinoid CB1 receptor antagonism modulates plasma corticosterone in rodents. Eur J Pharmacol. 551:162–167.
- Weidenfeld J, Feldman S, Mechoulam R. 1994. Effect of the brain constituent anandamide, a cannabinoid receptor agonist, on the hypothalamo-pituitary-adrenal axis in the rat. Neuroendocinology. 59:110–112.
- Yates FE, Leeman SE, Glenister DW, Dallman MF. 1961. Interaction between plasma corticosterone concentration and adrenocorticotropin-releasing stimuli in the rat. Endocrinology. 69:67–80.