Abstract
Chronic exposure to high concentrations of hexavalent chromium (Cr(VI)) in drinking water causes intestinal adenomas and carcinomas in mice, but not in rats. Cr(VI) causes damage to intestinal villi and crypt hyperplasia in mice after only one week of exposure. After two years of exposure, intestinal damage and crypt hyperplasia are evident in mice (but not rats), as are intestinal tumors. Although Cr(VI) has genotoxic properties, these findings suggest that intestinal tumors in mice arise as a result of chronic mucosal injury. To better understand the mode of action (MOA) of Cr(VI) in the intestine, a 90-day drinking water study was conducted to collect histological, biochemical, toxicogenomic and pharmacokinetic data in intestinal tissues. Using MOA analyses and human relevance frameworks proposed by national and international regulatory agencies, the weight of evidence supports a cytotoxic MOA with the following key events: (a) absorption of Cr(VI) from the intestinal lumen, (b) toxicity to intestinal villi, (c) crypt regenerative hyperplasia and (d) clonal expansion of mutations within the crypt stem cells, resulting in late onset tumorigenesis. This article summarizes the data supporting each key event in the MOA, as well as data that argue against a mutagenic MOA for Cr(VI)-induced intestinal tumors.
Introduction
Chromium is a naturally occurring element that primarily exists in two oxidation states: hexavalent chromium (Cr(VI)) and trivalent chromium (Cr(III)). Inhalation and intratracheal instillation of Cr(VI) increases lung tumors in rodents (Glaser et al., Citation1986; Steinhoff et al., Citation1986), and occupational exposure to high concentrations of Cr(VI) is associated with increased risk of lung cancer among workers of certain industries (IARC, Citation1990; Luippold et al., Citation2003; Machle & Gregorius, Citation1948; Park et al., Citation2004). The International Agency for Research on Cancer lists Cr(VI) as “carcinogenic to humans (Group I)” (IARC, Citation1990). In contrast, Cr(III) induces only limited acute and chronic toxicity, is not carcinogenic, and is equivocally considered a micronutrient (Anderson, Citation2000; Di Bona et al., Citation2011; IARC, Citation1990; NTP, Citation2008a; Stout et al., Citation2009b). Differences in toxicity between Cr(III) and Cr(VI) are primarily due to the lower cell permeability of Cr(III). The toxicity of Cr(VI) is mitigated by reduction to Cr(III) in bodily fluids, especially in the acidic reducing conditions of the stomach (De Flora et al., Citation1987, Citation1997; Febel et al., Citation2001). Recognizing differences in kinetics, biochemistry and toxicology of Cr(III) and Cr(VI), health risk assessment of chromium compounds has historically been both route- and valence-specific (US EPA, Citation1998).
In 2008, the National Toxicology Program (NTP) reported findings from 2-year cancer bioassays for both Cr(III) and Cr(VI) in feed and drinking water, respectively (NTP, Citation2008a,Citationb). Consistent with its low bioavailability, Cr(III) (administered as chromium picolinate) demonstrated “no evidence of carcinogenicity in mice or female rats”, and only “equivocal evidence for preputial gland adenomas in male rats” (NTP, Citation2008a; Stout et al., Citation2009b). In contrast, Cr(VI) (administered as sodium dichromate dihydrate; SDD) produced a dose-related increase in tumors in the small intestine of mice and oral mucosa of rats (NTP, Citation2008b; Stout et al., Citation2009a). However, because the drinking water concentrations in the NTP study were orders of magnitude greater than typical environmental levels, there has been debate about whether the high Cr(VI) concentrations depleted the reductive capacity of the mouse stomach and intestinal lumen (Collins et al., Citation2010; De Flora et al., Citation2008; Proctor et al., Citation2011; Stern, Citation2010). Recently published studies on species-specific investigations of Cr(VI) toxicokinetics suggest that the carcinogenic concentrations in the 2-year bioassay depleted gastric reductive capacity thereby leading to higher concentrations of Cr(VI) entering the intestinal lumen (Kirman et al., Citation2012; Proctor et al., Citation2012).
Cr(VI) can be genotoxic, especially in in vitro systems (Chiu et al., Citation2010; Holmes et al., Citation2008; Nickens et al., Citation2010; Zhitkovich, Citation2011). It has therefore been argued that the tumors observed following chronic oral exposure to Cr(VI) arose by a mutagenic mode of action (MOA) involving DNA mutation as an early key event in the carcinogenic process (McCarroll et al., Citation2010; US EPA, Citation2010; Zhitkovich, Citation2011). However, the NTP study authors described the non-neoplastic lesions in the mouse intestine (namely diffuse hyperplasia) as secondary to previous epithelial injury (NTP, Citation2008b). In contrast to mice, diffuse hyperplasia and tumors were not observed in rats (NTP, Citation2008b). These disparate observations in the two species suggested that the intestinal tumors in mice likely occurred via a cytotoxic MOA; however, there was insufficient information to determine the MOA due to a dearth of data describing the dose–response and sequence of key events in the target tissue. Therefore, we conducted an MOA analysis using the MOA framework outlined in the US EPA Guidelines for Carcinogen Risk Assessment (US EPA, Citation2005a) to: (a) hypothesize a plausible MOA for the mouse intestinal tumors, (b) identify data gaps and (c) design and conduct a 90-day drinking water study in the same strains of mice and rats in order to acquire critical data to fill these gaps (Thompson et al., Citation2011a). The study design, described in detail in Thompson et al. (Citation2011b, Citation2012c), employed the same drinking water concentrations as the NTP 2-year bioassay (NTP, Citation2008b) as well as two lower drinking water concentrations; one of which was 100 μg/l, the federal maximum contaminant level (MCL) for total chromium (US EPA, Citation1991). Histopathological, biochemical, toxicogenomic and pharmacokinetic data were collected in the target tissues after 7 and 90 days of exposure, and many of the study results have been published (Kirman et al., Citation2012; Kopec et al., Citation2012a,Citationb; Proctor et al., Citation2012; Thompson et al., Citation2011b, Citation2012a,Citationb,Citationc).
The purpose of this article is to present a synthesis of the MOA data, as well as other available data, and to characterize a plausible MOA for mouse intestinal tumors using a weight-of-evidence (WOE) approach. An assessment of the oral mucosal tumors observed in rats will be presented in a separate publication.
Brief review of Cr(VI) genotoxicity and carcinogenicity
The term genotoxicity is broadly used to describe genetic damage that can arise from both mutagenic and nonmutagenic processes. These latter two terms, mutagenic and nonmutagenic, have various meanings in different contexts; and what exactly constitutes a mutagenic MOA is an ongoing debate. As an example, the 2007 US EPA draft report Framework for Detecting a Mutagenic Mode of Action for Carcinogenicity remains unfinalized (US EPA, Citation2007). As an alternative to a mutagenic MOA, many scientists prefer to use terminology such as DNA-reactive (Boobis et al., Citation2009; Preston and Williams, Citation2005; Williams, Citation2008). However, it is important to realize that DNA reactivity (e.g. DNA adducts) does not necessarily correlate with mutagenicity, and chemicals that can react with DNA may not necessarily induce tumors solely as a result of direct interaction with DNA (Swenberg, et al., Citation2008). Although the US EPA framework remains unfinished, it nevertheless contains insightful approaches for determining whether a chemical has a mutagenic MOA. The framework asks the critical question, “Is mutation an early key event in the chemical’s induction of cancer?” (US EPA, Citation2007). In the balance of this review, it is important to keep this critical question in mind, because it has important implications for how risk assessors derive safe values for exposure to Cr(VI).
The genotoxicity of Cr(VI) outside of the small intestine has been the subject of several recent reviews (Chiu et al., Citation2010; Nickens et al., Citation2010; O’Brien et al., Citation2003; Zhitkovich, Citation2005, Citation2011) and will therefore be summarized only briefly herein. In vitro evidence of genotoxicity is summarized in the section “Assessment of Cr(VI) genotoxicity in vitro”, and in vivo genotoxicity is summarized in the section “Assessment of Cr(VI) genotoxicity in vivo” – with particular attention given to the doses and route of administration in such studies. Although the in vitro findings are informative and demonstrate that Cr(VI) can be genotoxic, the in vivo studies, arguably more relevant in the context of human health risk assessment, are far less convincing with respect to the ability of Cr(VI) to induce genotoxicity under environmentally relevant exposure scenarios. The section “Cr(VI) carcinogenicity in the lung” briefly reviews data on Cr(VI)-induced lung tumors, which appear to involve nonmutagenic mechanisms. The section “Summary of the alimentary canal lesions in the NTP 2-year CR(VI) bioassays” and the sections thereafter summarize recent findings that allow for development of a highly plausible MOA for Cr(VI)-induced tumors in the small intestine. Importantly, a plausible MOA does not require in-depth mechanistic understanding, but rather is a high level description of major events that are required to result in tumor formation in a specific tissue (Sonich-Mullin et al., Citation2001; US EPA, Citation2005a).
Assessment of Cr(VI) genotoxicity in vitro
Cr(VI) primarily exists as a chromate anion under most physiological conditions; this anion is structurally similar to sulfate
and phosphate
, and therefore Cr(VI) uptake occurs through anion transporters (Alexander & Aaseth, Citation1995; Buttner & Beyersmann, Citation1985; Collins et al., Citation2010; Markovich, Citation2001; Salnikow & Zhitkovich, Citation2008; Zhitkovich, Citation2005). In fact, resistance to Cr(VI) toxicity can be conferred by changes in anion transporter expression or function (Campbell et al., Citation1981; Lu & Yang, Citation1995). In contrast, Cr(III) is likely absorbed via pinocytosis or endocytosis thereby resulting in far less uptake into cells than Cr(VI) (Sedman et al., Citation2006; Vincent, Citation2000). Extracellular reduction of Cr(VI) to Cr(III) therefore effectively limits chromium absorption. Reduction of Cr(VI) to Cr(III) is mediated primarily by binding to low molecular weight thiols (e.g. GSH), antioxidants (e.g. ascorbate) and macromolecules (e.g. proteins) (De Flora et al., Citation1985; O’Brien et al., Citation2003; Zhitkovich, Citation2005). This reduction can lead to binary [Cr(III)–ligand] and ternary [ligand–Cr(III)–ligand] complexes [e.g. GSH–Cr(III)–GSH]. Reduction of Cr(VI) can also lead to the formation of less stable intermediates such as Cr(V) and Cr(IV), as well as thiol radicals (O’Brien et al., Citation2003; Yao et al., Citation2008). Molecular oxygen can also reduce Cr(VI) to Cr(V) thereby generating reactive oxygen species (ROS) (Liu & Shi, Citation2001), which in turn can lead to the formation of hydrogen peroxide.
Noncellular in vitro studies indicate that Cr(VI) does not bind directly to DNA, whereas Cr(III) can bind directly to DNA (De Flora et al., Citation1990; Jennette, Citation1979). Cr(VI) that is reduced to binary Cr(III)–ligands can also bind DNA to form ternary [e.g. GSH-Cr(III)-DNA] DNA adducts (Zhitkovich, Citation2005). Simple bacterial systems are frequently used as an initial assessment of the genotoxicity and mutagenicity of chemicals (Ames et al., Citation1975; McCann et al., Citation1975; OECD, Citation1997). Cr(VI) has been shown to increase reversions (i.e. mutations) in Salmonella typhimurium and Escherichia coli; however, mutation formation is mitigated by the addition of S9 (De Flora, Citation1978; NTP, Citation2008b; Petrilli & De Flora, Citation1978a,Citationb). This mitigation of Cr(VI) mutagenicity is consistent with extracellular reduction of Cr(VI) to Cr(III) by constituents present in the microsomal S9 mix (Petrilli & De Flora, Citation1978a,b). This is supported by the findings that Cr(III) itself does not induce revertants in S. typhimurium (Petrilli & De Flora, Citation1978b). In 1982, the S. typhimurium strain TA102 was developed, which better detected mutations from oxidants than typical S. typhimurium tester strains such as TA100 (Levin et al., Citation1982). Subsequently, the TA102 strain was shown to be more sensitive to Cr(VI) than eight other S. typhimurium strains (Bennicelli et al., Citation1983). Similar conclusions can be drawn from the more recent NTP study (NTP, Citation2007), where the number of Cr(VI)-induced revertants in TA100 were much lower than the positive control sodium azide (Supplementary Figure 1) – indicating the relative weakness of Cr(VI) mutagenicity. In contrast, the number of Cr(VI)-induced mutants in E. coli strain WP2uvrA (equivalent to TA102) was similar to the positive control methyl methanosulfonate (Supplementary Figure 1). In fact, Cr(VI) has been characterized as having only modest mutagenic activity in bacterial systems (Petrilli & De Flora, Citation1982). Consistent with the chemistry of Cr(VI), mutations in TA102 and WP2uvrA indicate involvement of oxidative DNA damage as well as direct DNA reactivity. It is important to recall that bacterial DNA is not sequestered by a nuclear envelope and thus Cr(III) and Cr(III)–ligands have equal access to DNA, proteins and other cellular constituents. In contrast, Cr(VI) that enters eukaryotic cells through anion transporters is more likely to react with cytoplasmic components (e.g. proteins, RNA, GSH, ascorbate) than with nuclear DNA. De Flora and colleagues have long posited that Cr(VI)-induced mutagenesis (and carcinogenesis) is likely limited by extracellular reduction to Cr(III), as well as cytoplasmic trapping in ternary complexes when Cr(VI) is reduced intracellularly. Only when the intracellular Cr(VI) concentration is sufficient to escape cytoplasmic reduction will Cr(VI) enter the nucleus and be reduced to DNA-reactive Cr(III) (De Flora, Citation2000; De Flora et al., Citation1984; Petrilli & De Flora, Citation1982). Cytoplasmic and nuclear reduction of Cr(VI) can also generate ROS that might damage DNA.
The genotoxicity of Cr(VI) in mammalian cells has been the subject of several recent reviews (Chiu et al., Citation2010; Holmes et al., Citation2008; Nickens et al., Citation2010; Zhitkovich, Citation2011); the reader is referred to these reviews and the articles therein for detailed discussion. Genotoxic lesions associated with Cr(VI) exposure include DNA adducts, DNA strand breaks, DNA protein cross-links, inter- and intra-strand DNA cross-links, oxidative DNA lesions, mutations and epigenetic alterations () (Holmes et al., Citation2008; Nickens et al., Citation2010; O’Brien et al., Citation2003; Zhitkovich, Citation2011). Based on the known chemistry of Cr(VI) reduction, oxidative stress is a very likely mechanism. However, as concluded by Patierno and colleagues, “although there are many studies reporting on the relationship between oxidative stress and Cr(VI)-mediated cytotoxicity, genotoxicity, and potential carcinogenesis, due to nonuniform treatment protocols, conflicting data and controversial methods used for detection of Cr(VI)-induced oxidative stress, this relationship remains unclear” (Nickens et al., Citation2010). Nevertheless, recent in vitro studies continue to implicate oxidative stress in Cr(VI) genotoxicity. For example, several studies have reported that continuous passage (4–24 weeks) of human bronchial epithelial Beas-2B cells in relatively low concentrations of Cr(VI) (e.g. 0.25–5 µM) can result in transformation (Azad et al., Citation2010; Sun et al., Citation2011; Wang et al., Citation2011), and one study demonstrated that such transformation could be ameliorated by transfection of plasmids containing superoxide dismutase, catalase or RNA inhibitors of NADPH oxidase (Wang et al., Citation2011). As will be described in greater detail in the section “In vitro intestinal Caco-2 cell model”, Cr(VI) was shown to induce 8-hydroxy-2′-deoxyguanosine (8-OHdG) DNA damage at lower concentrations than that which induced DNA double strand breaks (DSBs) in Caco-2 cells (Thompson et al., Citation2012a).
Table 1. Summary of Cr-induced DNA lesions.
As discussed above, intracellular Cr(VI) reduction leads to binary and ternary Cr(III) adducts. The abundance of cytoplasmic macromolecules suggests that Cr(III) ternary ligands likely represent inactive chromium species (Petrilli & De Flora, Citation1982). It has been suggested that Cr(III) might be oxidized intracellularly to Cr(VI) thereby creating a cycle of Cr(VI) formation and reduction (Levina & Lay, Citation2008); however, it appears that very strong nonbiological oxidants are required to convert Cr(III) to Cr(VI) (Petrilli & De Flora, Citation1978b). Binary Cr-DNA adducts are estimated to represent more than 75% of all Cr-DNA adducts (i.e. binary and ternary), but are also thought to have weaker mutagenic potential than ternary adducts (Zhitkovich, Citation2011). Ternary Cr-DNA adducts appear to be more mutagenic, as these lesions can lead to DNA DSBs (Nickens et al., Citation2010; Zhitkovich, Citation2011). Cr(VI)-induced DNA lesions are thought to be repaired by base excision repair (BER), nucleotide excision repair (NER) and mismatch repair (MMR) pathways (Nickens et al., Citation2010; O’Brien et al., Citation2009).
Cr(VI) is also reported to induce epigenetic (e.g. microRNA, histone modification and DNA methylation) changes in cells. For example, treatment of transgenic gpt+ Chinese hamster V79 fibroblasts with soluble and insoluble chromate both induced mutant colonies (Klein et al., Citation2002). However, whereas the revertants from cells treated with insoluble chromate arose from gpt deletions, the revertants from cells treated with soluble chromate arose from gpt silencing due to hypermethylation of the gpt promoter region (Klein et al., Citation2002). Similarly, treatment of A549 cells with Cr(VI) was shown to increase cellular methylation of histone H3 lysine 9 (H3K9), as well as decrease mRNA levels of MLH1 (discussed further in the section “Cr(VI) Carcinogenicity in the lung”) (Sun et al., Citation2009). Another study reported that cells deficient in MMR were resistant to Cr(VI)-induced toxicity (Peterson-Roth et al., Citation2005), and it has been posited that Cr(VI) might “select” for cells deficient in MMR, which could increase genomic instability (Salnikow & Zhitkovich, Citation2008). Other studies, however, have shown that transformation of Beas-2B cells by repeated passage in low levels of Cr(VI) results in different phenotypes (Rodrigues et al., Citation2009). These different outcomes in different cell models underscore the complexities of interpreting in vitro findings and applying them to in vivo conditions.
One widely used method for assessing genotoxicity is the micronucleus (MN) assay, which can detect DNA damage that arises through either clastogenic or aneugenic mechanisms (Fenech et al., Citation2011; Kirsch-Volders et al., Citation1997, Citation2011; OECD, Citation2010; Parry et al., Citation2002). Studies in human diploid fibroblasts (MRC-5) have indicated that Cr(VI) induces MN through aneuploidic mechanisms (Seoane & Dulout, Citation2001; Seoane et al., Citation2002). Recent studies in CHO-K1 and A549 cells indicate that Cr(VI) induces MN only at cytotoxic concentrations (Thompson et al., Citation2012a). An earlier study reported MN in CHO cells; however, there was no dose-dependent response or statistical analyses presented (Howard et al., Citation1992), and as such these results are highly uncertain. The strongest evidence for induction of MN in vitro comes for studies that first supplement cells with dehydroascorbic acid (DHA) prior to Cr(VI) exposure (Reynolds et al., Citation2007, Citation2012). Pretreatment with DHA elevates cellular ascorbate to levels reported in tissues and freshly isolated cells (Chen et al., Citation2005; Martensson and Meister, Citation1992; Vissers et al., Citation2011). DHA is used because supplementation of culture media with ascorbate generates toxic levels of peroxide through interactions with cell culture constituents (Azzolini et al., Citation2011; Chen et al., Citation2005; Long and Halliwell, Citation2012), and may potentiate the genotoxicity of some compounds (Crott & Fenech, Citation1999; Krishnaja & Sharma, Citation2003). Reynolds and colleagues reported that preloading cells with DHA caused significant decreases in cellular chromium levels, which they posited were due to ascorbate leakage from cells. However, such leakage also implies that ascorbate might have generated peroxide through reaction with media constituents. It is also unknown whether artificially loading cells with DHA/ascorbate prior to Cr(VI) treatment better recapitulates the in vivo cell because, for example, DHA/ascorbate reduction is mediated nonenzymatically by GSH as well as enzymatically through GSH-dependent and NADPH-dependent reactions (Lister & Van Schaftingen, Citation2007; Martensson & Meister, Citation1992). This suggests that DHA loading may alter cellular redox status. Without pretreatment with DHA, little to no genotoxicity was observed (Reynolds et al., Citation2007, Citation2012).
Despite evidence of Cr(VI) genotoxicity in vitro, recent studies suggest that at lower Cr(VI) concentrations, oxidative stress and oxidative DNA damage may be the primary drivers of genotoxicity (Thompson et al., Citation2012a; Wang et al., Citation2011), not direct DNA reactivity. As will be seen in the following section, the evidence for in vivo genotoxicity is relatively weak, and most of the positive findings are based on nonphysiological doses and exposure routes, and may be the result of oxidative mechanisms.
Assessment of Cr(VI) genotoxicity in vivo
Several of the available in vivo genotoxicity studies for Cr(VI) are summarized in . Like in vitro MN formation, in vivo MN formation can arise through aneugenic and clastogenic mechanisms, and is not necessarily indicative of direct DNA reactivity. Examination of shows a clear pattern in MN studies in mice where positive results are primarily found in studies where Cr(VI) was administered by intraperitoneal (i.p.) injection, and negative results are primarily found in studies where Cr(VI) was administered orally (via drinking water or gavage). Considering that i.p. administration bypasses normal routes of exposure and results in rapid delivery into the blood stream, these results are of questionable relevance to the oral toxicity of Cr(VI). Moreover, many of the doses administered i.p. exceed the mg/kg doses that were carcinogenic to the mouse small intestine by the drinking water exposure route (NTP, Citation2008b). A few studies have directly compared MN formation of Cr(VI) when administered orally or i.p. (De Flora et al., Citation2006; Shindo et al., Citation1989). In each case, positive results were observed only after i.p. exposure.
Table 2. In vivo genotoxicity studies with Cr(VI)*.
In vivo MN assays in rodents exposed to Cr(VI) via gavage or drinking water are generally negative (De Flora et al., Citation2006; Harris et al., Citation2012; Mirsalis et al., Citation1996; NTP, Citation2007; Shindo et al., Citation1989). The NTP (Citation2007) conducted four in vivo MN tests in peripheral blood erythrocytes of three strains of mice exposed to Cr(VI) for three months. Two studies were conducted in B6C3F1 mice exposed to ≤1000 mg/l SDD: one of which was judged to be negative in both male and female mice, and the other equivocal (p value not significant) by the NTP study authors. A third study in male BALB/C mice was negative; and a fourth study in male transgenic am3-C57BL/6 mice was positive. Interestingly, the latter strain contains a transgene for detecting forward and reverse mutations; however, to our knowledge a mutation analysis has never been reported (NTP, Citation2007, Citation2008b). As will be discussed in greater detail later in this review, exposure to 0.1–182 mg/l Cr(VI) in drinking water for 7 or 90 days did not increase MN formation in the mouse small intestine (Harris et al., Citation2012).
Two studies have reported DNA mutations in mice following i.p. administration of 10–40 mg/kg chromium compounds (Itoh & Shimada, Citation1998; Knudsen, Citation1980). The findings reported in Knudsen (Citation1980) appear equivocal as significant effects were reported following three days of i.p. exposure to 10 mg/kg potassium chromate, but not 20 mg/kg. For the reasons cited above, the relevance of gene mutations in these i.p. studies for oral exposure to Cr(VI) is uncertain. These mutations might have resulted from oxidative stress, as we have shown that doses of ≥10 mg/kg Cr(VI) achieved by oral exposure in drinking water for 90 days results in significant decreases in the plasma GSH/GSSG ratio – indicating oxidative stress in the blood (Thompson et al., Citation2011b). Thus, i.p. administration of such doses is likely to result in acute oxidative stress. In this regard, Bagchi et al. (2002) have reported that gavage doses of ≥19 mg/kg sodium dichromate increase DNA fragmentation in liver and brain samples by a mechanism that the authors concluded involved oxidative DNA damage. Similar conclusions were drawn from gavage studies in rats () (Bagchi et al., Citation1995a,Citationb, Citation1997).
Other studies have reported DNA damage following oral exposure to Cr(VI) (Coogan et al., Citation1991; Dana Devi et al., Citation2001; Sarkar et al., Citation1993). Coogan et al. (Citation1991) reported that exposure to 100 and 200 ppm (i.e. 100–200 mg/l) potassium chromate in drinking water for three weeks increased DNA-protein cross-links (DPX) in liver tissue but not splenic lymphocytes. The DPX was not quantified, but rather demonstrated qualitatively by electrophoresis. Notably, Coogan et al. (Citation1991) were unable to demonstrate DPX in the liver using the more standard alkaline elution method, but did report increases in liver DPX by this method following i.p. administration of 50 mg/kg potassium chromate, which is consistent with previous studies (Cupo & Wetterhahn, Citation1985; Tsapakos et al., Citation1981). Although i.p. injections of Cr(VI) indicate a potential for chromium to bind DNA in vivo, the exposure route bypasses normal pharmacokinetic barriers to Cr(VI) exposure. Notably, the 100–200 ppm potassium chromate exposure employed by Coogan et al. far exceeds the 0.005 ppm Cr(VI) concentrations in typical US drinking water. It should also be pointed out that exposure up to 182 ppm Cr(VI) for two years did not result in liver tumors in mice or rats (NTP, Citation2008b), which calls into question the relevance of the DPX reported by Coogan et al. following oral exposure.
Sarkar et al. (Citation1993) administered 20 mg/kg chromium trioxide (CrO3) to mice by gavage and reported a significant increase in chromosomal aberrations (CAs) in bone marrow cells. It should be noted that the LD50 for hexavalent chromium compounds is estimated at 50 mg/kg (Katz & Salem, Citation1993). It is also notable that a conservative estimate of a 0.25 ml gavage administration to a 25 g mouse implies that the concentration administered was ∼2000 ppm. Given the exposure scenario in this study, it is not clear how informative these findings are to the toxicity of Cr(VI) at environmentally relevant concentrations.
Danadevi et al. (Citation2001) exposed Swiss albino mice to 0.59–76 mg/kg Cr(VI) via gavage. Blood samples were collected 24 h to 2 weeks post exposure. DNA damage was then assessed by Comet assay. Significant increases in tail moment (DNA damage) were noted at all concentrations. While these results undoubtedly indicate potential genotoxicity from Cr(VI) exposure, these results could be due to either direct DNA damage or oxidative damage (as noted by Danadevi and colleagues). In addition, the toxicokinetics of Cr(VI) must be considered. While 0.59 mg/kg is a seemingly low dose, the stated 50 µl gavage volume implies a concentration of 265 mg/l.Footnote1 Considering that 35% of this dose is Cr(VI), the concentration is ∼93 mg/l Cr(VI), a concentration clearly shown to induce oxidative stress and cytotoxicity in the mouse small intestine (NTP, Citation2007; Proctor et al., Citation2012; Thompson et al., Citation2011b). Thus, oxidative DNA damage might have been responsible for the observed change in tail moment.
To date, very few studies have examined the genotoxicity of Cr(VI) in the small intestine, where tumors were observed in mice following chronic exposure to Cr(VI) (NTP, Citation2008b). De Flora et al. (Citation2008) exposed mice to ≤20 mg/l Cr(VI) for nine months and then measured DPX and 8-OHdG in the forestomach, glandular stomach and small intestine. Neither marker was increased in vivo; however, ex vivo treatment of intestinal tissues (from unexposed animals) with Cr(VI) resulted in significant levels of both markers. Similarly, Thompson et al. (Citation2011b) did not detect any increases in oxidative DNA damage following three months of exposure to ≤182 mg/l Cr(VI). Given that significant decreases in intestinal GSH/GSSG ratio and significant induction of genes related to oxidative stress following Cr(VI) ingestion, the absence of 8-OHdG in De Flora et al. (Citation2008) and Thompson et al. (Citation2011b) might be explained by adaptation after such long exposure durations (see the section “Application of the mode of action framework”). As will be described in greater detail in the section “Key event 2: Villous cytotoxicity”, in vivo MN analyses in the small intestine were negative (Harris et al., Citation2012).
Cr(VI) Carcinogenicity in the lung
Although this review focuses on oral exposure to Cr(VI), it is useful to briefly discuss what is known about the carcinogenicity of Cr(VI) following inhalation exposure. Occupational exposure to Cr(VI) has long been recognized to increase the risk of lung cancer among historical chromate production workers (IARC, Citation1990; Luippold et al., Citation2003; Machle & Gregorius, Citation1948; Park et al., Citation2004). Lung tumor biopsies from chromium and nonchromium workers indicate fewer p53 point mutations in chromate workers (20% versus ∼50%), but greater signs of genomic instability (79% versus 15%) (Hirose et al., Citation2002; Kondo et al., Citation1997). Tumors from chromate workers also exhibit hypermethylation and reduced expression of MLH1 (Takahashi et al., Citation2005), a phenotype that is associated with genomic instability. In addition, among workers with elevated lung cancer risk, respiratory tissue irritation and inflammation were common clinical findings (Gibb et al., Citation2000; Miller, Citation1953), suggesting that cytotoxicity and inflammation contribute to carcinogenesis. Several authors have concluded that these findings, together with the generally weak mutagenic properties of Cr(VI), suggest that Cr(VI)-induced lung tumors likely arise through nonmutagenic mechanisms as opposed to direct DNA reactivity (Holmes et al., Citation2008; Nickens et al., Citation2010).
Animal studies also indicate a nonmutagenic MOA in the formation of lung tumors. Steinhoff et al. (Citation1986) exposed rats to Cr(VI) via intratracheal instillation for up to 30 months. Some rats received up to 0.25 mg/kg five times per week, and other rats were administered a single weekly dose of 1.25 mg/kg. In rats exposed to a single weekly dose of 1.25 mg/kg, there were signs of chronic inflammation including the presence of alveolar macrophages, proliferation of bronchiolar epithelium and chronic inflammatory thickening of alveolar septa. These lesions were much milder in rats exposed to the same weekly dose but in five installments of 0.25 mg/kg, as well as rats receiving five installments of 0.05 or 0.01 mg/kg, or single installments of 0.5 or 0.05 mg/kg/week for 30 months. The rats that received ≤0.25 mg/kg five times per week did not exhibit an increase in lung tumors. In contrast, 17.5% of the rats that received a single dose of 1.25 mg/kg exhibited tumors in the lung. Steinhoff et al. (Citation1986) concluded that concentration and irritancy/inflammation were more important in tumor formation than overall cumulative weekly dose. Similar effects were associated with lung tumors induced by calcium chromate (Steinhoff et al., Citation1986).
Glaser et al. (Citation1986) conducted an inhalation study with Cr(VI) as sodium dichromate, and a chromium oxide mixture of 3Cr(VI):2Cr(III) (Cr5O12) for 18 months followed by a 12-month observation period. In rats exposed to 25, 50 and 100 µg/m3 dichromate, lung tumors were observed only in the 100 µg/m3 dichromate group (16% of animals). Exposure to Cr5O12 at 100 µg/m3 increased lung tumor incidence from 0% to 6% (1/18). Glaser and colleagues (Citation1986) characterized these findings as indicating weak carcinogenicity of Cr(VI). At study termination (12 months after the last exposure), Glaser et al. (Citation1986) reported similar lesions as Steinhoff et al. (Citation1986) in the rats exposed to Cr5O12, namely accumulation of macrophages in lungs, eosinophilic substances inside the alveolar lumens, focal thickened septa and fibrosis. These lesions were not observed following exposure to dichromate. However, Glaser et al. (Citation1986) showed that the chromium lung burden in Cr5O12-treated rats was 10-fold higher than in rats exposed to 100 µg/m3 dichromate, indicating that the less soluble Cr5O12 was more slowly cleared from the lung than dichromate over the 12-month recovery period. This likely explains why Glaser et al. (Citation1986) did not observe similar non-neoplastic histological lesions (i.e. inflammation) as Steinhoff et al. (Citation1986) following the 12-month recovery period.
To date, a single study has examined mutations in lung tissue following exposure to Cr(VI), specifically in Big Blue mice following intratracheal administration (Cheng et al., Citation2000). As was discussed for the i.p. and oral studies, it is important to consider dose when drawing conclusions from a study. Prior to determining the dose for the mutation assay, Cheng et al. (Citation2000) found that instillation of a single dose of 6.75, 9 and 22.5 mg/kg potassium chromate was lethal in 0/3, 1/3 and 3/3 mice, respectively. Therefore, they used 6.75 mg/kg potassium chromate for the mutation assay, which is ∼2.4 mg/kg Cr(VI). Although this dose was not lethal, it was likely toxic given that 9 mg/kg potassium chromate was lethal in 33% of mice. Moreover, the concentration of Cr(VI) administered was extremely high. Based on the instillation volume of 25 µl, Cheng et al. (Citation2000) instilled Cr(VI) at a concentration of 2400 mg/l (2.4 mg/kg × 0.025 kg ÷ 0.000025 l). This concentration is ∼46 mM, which is several orders of magnitude greater than concentrations that are cytotoxic to cells in vitro. Intratracheal administration of 6.75 mg/kg increased the mutation frequency (MF) in lung by ∼4-fold. Cheng et al. (Citation2000) also reported increases in lung MF at lower concentration (∼3.3. mg/kg); however, fewer details were provided and 23 mM is also several orders of magnitude greater than cytotoxic concentrations in vitro. Another issue with this study is that the MF in chromium-treated animals 4 weeks after exposure was compared to the MF in saline-treated controls at only 1 week post exposure. The lack of time-matched controls assumes that the background MF would be unaltered by the intratracheal surgery. Interestingly, Cheng et al. (Citation2000) reported that sequencing of mutants from chromium-treated mice revealed a large increase in GC to TA transversions. Such transversions are commonly induced by reactive oxygen and nitrosative species (Klaunig & Kamendulis, Citation2008; Klaunig et al., Citation2010; Marnett, Citation2000) – suggesting, perhaps, that the increased MF was a result of oxidative and inflammatory processes induced by tissue damage expected from such high concentrations of Cr(VI). Given the high concentrations of Cr(VI) utilized in this study, it is difficult to conclude that any observed mutations in lung tissue were the result of a direct DNA-reactive mutagenic MOA.
Recent studies have implicated inflammation as a possible mechanism for lung tumor development. Mice exposed to 0.6 mg/ml zinc chromate via intranasal instillation either once or repeatedly (every 14 days for 64 days) exhibited clear signs of peribronchiolar, alveolar and interstitial inflammation, as well as elevated and aberrant cell proliferation in the airway lining (Beaver et al., Citation2009). These effects are similar to those reported above in the cancer assays (Glaser et al., Citation1986; Steinhoff et al., Citation1986). Beaver et al. (Citation2009) concluded that this Cr(VI)-induced inflammation contributes to the initiation and promotion of neoplastic growth in the lung. It is well established that inflammation can induce oxidative stress and that chronic inflammation and cell proliferation both contribute to carcinogenesis. Thus, the MOA for Cr(VI)-induced lung cancer may very well be nonmutagenic.
Taken together, the in vivo lung studies indicate that Cr(VI) induces inflammatory responses in the lung, which likely contribute to the carcinogenic response. Recently, scientists at the Texas Commission on Environmental Quality developed an inhalation reference value for Cr(VI) using nonlinear/threshold modeling approaches based, in part, on evidence for biologically based thresholds (e.g. extracellular Cr(VI) reduction) in the MOA for Cr(VI)-induced lung tumors (Haney et al., Citation2012).
As described in this section, there is ample evidence that Cr(VI) causes genotoxicity and can react with DNA under certain circumstances – primarily in in vitro systems and in in vivo systems under extreme exposure scenarios (e.g. nonphysiological routes and environmentally irrelevant concentrations). Although the relevance of Cr(VI) mutagenicity is debated, the carcinogenicity of Cr(VI) is not in dispute. Animals studies clearly establish that Cr(VI) can cause tumors at the portal of entry (e.g. alimentary canal and lung) (Glaser et al., Citation1986; NTP, Citation2008b; Steinhoff et al., Citation1986). Notably, inhalation exposure to Cr(VI) in the US is, for the most part, a historical problem as exposure mitigation efforts in occupational settings have greatly limited inhalation exposure to Cr(VI) (Luippold et al., Citation2005; Pastides et al., Citation1994). Moreover, oral exposure to Cr(VI) is unlikely to pose a risk for lung cancer, as oral exposure to Cr(VI) has not been reported to increase chromium lung burdens (Coogan et al., Citation1991; Kargacin et al., Citation1992). However, low levels of Cr(VI) in both ambient air and drinking water exist due to natural and anthropogenic sources and is therefore of public health interest. For these reasons, the remainder of this review focuses on the MOA for oral exposure to Cr(VI) – specifically the MOA underlying intestinal tumors in the mouse small intestine and their relevance to humans.
Summary of the alimentary canal lesions in the NTP 2-year CR(VI) bioassays
NTP conducted 13-week and lifetime bioassays for Cr(VI) in F344 rats and B6C3F1 mice (NTP, Citation2007, 2008b). In rats, the only treatment-related tumors observed were squamous cell carcinomas of the oral mucosa (buccal and tongue regions). Tumors were only significantly elevated relative to concurrent controls at the highest treatment concentration (516 mg/l SDD), and no preneoplastic lesions were observed in any rats in the 90-day or 2-year bioassays (NTP, Citation2007, Citation2008b; Stout et al., Citation2009a). The study authors noted that of 21 chemicals previously shown to induce oral neoplasms, only one produced such tumors in mice (Stout et al., Citation2009a) – indicating a potential species susceptibility to these tumors. It should also be noted that the body weights of rats in the 516 mg/l group decreased more than 10% relative to controls, and that water intake significantly decreased ≥15% at the two highest doses (172 and 516 mg/l). We previously reported significant changes in the redox status in the oral mucosa of rats following 90 days of exposure, as well as clear signs of iron deficiency (Thompson et al., Citation2012c). These various factors suggest diminished general and oral health in the rats; an investigation into the MOA and human relevance of these tumors is ongoing and will be the subject of a future article.
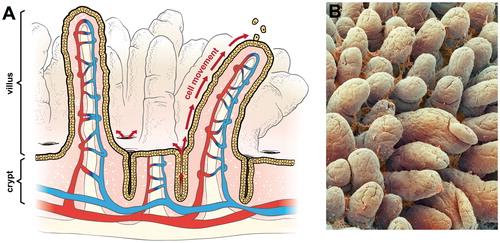
In contrast to rats, mice in the NTP studies developed treatment-related and dose-dependent tumors in the small intestine. The small intestine is comprised (proximal to distal) of the duodenum, jejunum and ileum. Each segment is lined with a single layer of epithelial cells that is renewed every few days –making the intestinal mucosa one of the most proliferative tissues in the body (Berlanga-Acosta et al., Citation2001). The small intestinal epithelium is extensively folded to maximize the absorptive surface area. Anatomically, the small intestinal epithelium is comprised of crypt and villus regions. The villi are multicellular finger-like projections that extend into the intestinal lumen (); inside each villus is a capillary and lymphatic vessels (). The crypts (also called glands of Lieberkühn) are invaginations of the epithelial surface at the base of the villi. Inside the crypts are actively dividing progenitor cells that differentiate into secretory (goblet cells), absorptive cells (enterocytes) and other cell types (e.g. Paneth cells) (Neal et al., Citation2011; Potten et al., Citation1997). Cell proliferation begins in the base of the intestinal crypts within a population of progenitor cells. Epithelial cells differentiate (mature) as they migrate toward the tips of the villi (). The cell cycle for intestinal enterocytes is 10–17 h in rodents and ∼24 h in humans, and there is complete replacement of the entire epithelium within 2–3 days in rodents and 3–6 days in humans (Greaves, Citation2007). Upon migration from the crypt, cells differentiate and express proteins involved in nutrient absorption – which occurs primarily in the duodenum and proximal jejunum (DeSesso and Jacobson, Citation2001). At the tips of the villi, cells slough into the lumen and/or undergo apoptosis. Importantly, the absorptive villous enterocytes do not have proliferative potential, and thus it is unlikely that tumors originate in these cells (Barker et al., Citation2009; Potten & Loeffler, Citation1990).
Figure 1. Intestinal structure: (A) diagram of small intestine showing crypts, villi, and vasculature. The arrows show the migration of enterocytes from the crypts below the luminal surface to the tips of villi where enterocytes are sloughed into the intestinal lumen and (B) scanning electron micrograph of small intestine showing villi and mucus (© Unlisted Images / Fotosearch.com).
Findings from the NTP 2-year bioassay relevant to the small intestine are summarized in . Similar effects were observed in the small intestines of both male and female mice (Supplementary Figure 2); therefore, for brevity only the results for females are summarized herein. In female mice, the incidence of diffuse hyperplasia was significantly increased in all treatment groups in a dose-dependent manner and was characterized by short, broad and blunt villi and by elongated crypts that contained increased numbers of epithelial cells and mitotic figures (NTP, Citation2008b). These lesions were not observed in female or male rats. The NTP study authors considered the lesions in mice to be “consistent with regenerative hyperplasia secondary to previous epithelial cell injury” (NTP, Citation2008b).
Table 3. Incidence of intestinal lesions in female rodents of NTP (Citation2008b) bioassay†.
The incidence of adenomas and carcinomas was statistically elevated relative to concurrent controls in the duodenum of female mice at ≥172 mg/l and 516 mg/l SDD, respectively. In the female mouse jejunum, diffuse hyperplasia and adenomas were significantly elevated relative to concurrent controls at 516 mg/l SDD. The overall combined incidence of intestinal tumors in female mice was significantly elevated relative to concurrent controls at ≥172 mg/l SDD (). Adenomas and carcinomas were not elevated in the small intestine in either male or female rats (NTP, Citation2008b).
Although the onset of occult tumors is not readily observable, such tumors would likely have caused significant problems (e.g. intestinal blockage) if they had developed early in life, and thus might be expected to have increased mortality. Early tumors might also have metastasized to other organs and thus increased mortality. With the exception of a dose-dependent decrease in water intake that was attributed to unpalatability, the NTP authors characterized the exposure to Cr(VI) as well tolerated (NTP, Citation2008b); thus gastrointestinal problems such as bleeding or malnourishment were not observed and suggest late onset tumor development. The first intestinal neoplasm was observed at day 451; and although nine mice died prior to day 451, none of the seven that were in treatment groups were found to have intestinal tumors (NTP, Citation2008b). Thus, the relatively late tumor onset, absence of tumors beyond the portal of entry and absence of mortality are seemingly inconsistent with a highly mutagenic compound.
Application of the mode of action framework
US EPA (2005a) defines MOA as “a sequence of key events and processes, starting with interaction of an agent with a cell, proceeding through operational and anatomical changes, and resulting in cancer formation”. EPA defines a key event as “an empirically observable and quantifiable precursor step that is itself a necessary element of the mode of action or is a biologically based marker for such an element”. The EPA MOA Framework, like those proposed by the other scientists and organizations (Boobis et al., Citation2006, Citation2008; Meek & Klaunig, Citation2010; Sonich-Mullin et al., Citation2001), stresses that the MOA for each tumor site should be evaluated separately. Moreover, toxicology data should ideally be evaluated (or generated if absent) in the target tissues of interest. Therefore, the MOA described below focuses on data obtained from the duodenal and jejunal intestinal mucosae of mice.
Summary of the key events in the MOA for intestinal tumors
The histological, biochemical, toxicogenomic and pharmacokinetic data collected as part of our MOA studies were evaluated along with other relevant data in the published literature using the MOA and human relevance frameworks developed by the US EPA and other organizations. Based on this assessment, we conclude that the overall WOE supports a cytotoxic nonmutagenic MOA with the key events shown in : Key event 1: Absorption of Cr(VI) from the intestinal lumen; Key event 2: Villous cytotoxicity; Key event 3: Sustained compensatory crypt hyperplasia to repair/replace the damaged intestinal mucosa; and Key event 4: Mutagenesis within crypt cells, ultimately leading to tumorigenesis. Each of these key events, and supporting data, are summarized in the following sections.
Figure 2. Proposed MOA for Cr(VI) carcinogenesis in the small intestine.

Key event 1: Absorption from the lumen
In most physiological conditions, Cr(VI) exists primarily as chromate anions that are structurally similar to sulfate
and phosphate
, and therefore, enter cells through anion transporters (De Flora, Citation2000; Markovich, Citation2001; Salnikow & Zhitkovich, Citation2008; Zhitkovich, Citation2005). Cr(III) is not structurally similar to sulfate and phosphate and thus enters cells more passively. Extracellular reduction of Cr(VI) to Cr(III) prevents cellular absorption through these transporters and is a well-recognized kinetic process that limits toxicity (De Flora, Citation2000; De Flora et al., Citation1997; Donaldson & Barreras, Citation1966; Febel et al., Citation2001; Kerger et al., Citation1996; US EPA, Citation1998). The stoichiometry of Cr(VI) reduction to Cr(III) in the lumen of the stomach and small intestine is not fully known, but reduction capacity varies depending on stomach conditions including pH and whether in a fed or fasted state (De Flora et al. Citation1987; Proctor et al. Citation2012). Cr(VI) that escapes reduction in the stomach will be transported to the small intestine and taken up into intestinal enterocytes, or transited along the lumen of the gastrointestinal tract where it can be reduced by microbiota (Shrivastava et al., Citation2003) and intestinal secretions such as cysteine (Dahm & Jones, Citation2000), and excreted in feces. Following Cr(VI) administration in drinking water, pharmacokinetic data indicate low chromium tissue burdens in the ileum, higher levels in the jejunum and still higher levels in the duodenum (), likely demonstrating that Cr(VI) absorption decreases distally through the small intestine due to (1) reduction of Cr(VI) to Cr(III) in the intestinal lumen, and/or (2) differences in the relative absorption of chromium in the intestinal segments (Kirman et al., Citation2012; Thompson et al., Citation2011b). Consistent with these data, tissue damage and tumor formation are increased mostly in the duodenum, only slightly in the jejunum and not increased in the ileum or large intestine (NTP, Citation2007, Citation2008b).
Figure 3. Total chromium (Cr) tissue concentration in alimentary canal: (A) total Cr levels in mice at day 91; data plotted are mean ± SEM. Total Cr was significantly (p < 0.01) elevated in all four tissues at ≥60 mg/l SDD by Shirley’s test, and in the duodenum at ≥14 mg/l SDD by Shirley’s test and (B) comparison of duodenal total Cr levels in mice (same data as in A) and the predicted exceedance of Cr(VI) reductive capacity by mouse gastric fluid in ex vivo experiments (Proctor et al., Citation2012). Bars represent the percentage of reductive capacity (left axis) and red line represents mean ± SEM duodenal Cr (right axis). (A) is plotted in terms of mg/l SDD, whereas (B) is plotted in terms of mg/l Cr(VI). *Significantly (p < 0.01) elevated Cr in duodenum by Shirley’s test.
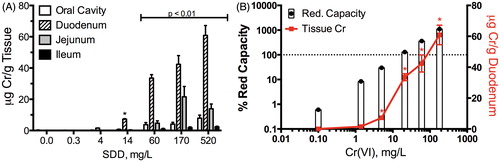
In vivo and ex vivo pharmacokinetic data collected in the alimentary tract of mice provide evidence that the carcinogenic doses in the NTP study deplete the reductive capacity of the mouse gastric contents (Proctor et al., Citation2012; Thompson et al., Citation2011b). Cr(VI) cannot be speciated in tissues; therefore, only the total chromium concentration in tissues can be measured. However, because Cr(VI) is much better absorbed than Cr(III), a significant increase in tissue total chromium concentrations suggests that extracellular reducing capacity was exceeded and that higher concentrations of Cr(VI) are available for absorption. After 90 days of exposure to Cr(VI) in drinking water, duodenal chromium levels were not significantly increased at the two lowest treatment doses (). At these doses, the physiologically based pharmacokinetic (PBPK) model developed for chromium in mice (Kirman et al., Citation2012) predicts that the amount of reducing equivalents (lumped concentration of reducing agents) in the gastrointestinal lumen of mice is relatively unchanged compared to controls (). In contrast, exposure to the three highest doses of Cr(VI) in drinking water, which were carcinogenic in the NTP 2-year bioassay (NTP, Citation2008b), resulted in a significant increase in duodenal total Cr tissue concentrations from <0.02 μg/g (controls) to 34, 42 and 61 μg/g, respectively (). At the three highest doses administered to mice, the PBPK model (Kirman et al., Citation2012) predicts that Cr(VI) exposure produces a substantial depletion in the reducing equivalents present in the gastrointestinal lumen ().
Many researchers have advocated the use of toxicogenomic data for application in risk assessment (Thomas et al., Citation2007; US EPA, Citation2009; Wilson et al., Citation2011). One of the key assumptions is that omics changes will provide more sensitive end points for detecting toxicity. We have previously reported that the total number of gene changes observed in each treatment group at day 91 correlated well with tissue dose. Specifically, the number of transcripts significantly altered ±1.5-fold at ≤14 mg/l SDD were each ≤76 genes, whereas at ≥60 mg/l more than 1800 genes were altered in each group (Kopec et al., Citation2012a). Although the total number of transcript changes represents only a cursory assessment of the effect of Cr(VI) on the transcriptome, the changes may serve as a biomarker of both exposure and effect. In this regard, the total number of gene changes generally corresponded with pharmacokinetic evidence of tissue chromium and histopathological evidence of lesions, mostly at ≥60 mg/l Cr(VI). Notably, the median EC50 for significant gene changes in the duodenum at day 91 was 39 mg/l SDD (Kopec et al., Citation2012a). These findings are consistent with other reports demonstrating that changes in the transcriptome are only slightly more sensitive than frank adverse effects (Thomas et al., Citation2011).
As discussed in the following section, histopathological lesions in our 90-day study were only observed in the mouse duodenum at doses that depleted reducing equivalents. Because these lesions are thought to be key events that precede tumor formation in the MOA, small amounts of Cr(VI) that enter intestinal villi at low concentrations (i.e. below those producing cytotoxicity) are likely inconsequential. A PBPK model capable of accounting for the competing kinetic rates in humans has been developed (Kirman et al., Citationaccepted) and will be useful for estimating the concentration of Cr(VI) that would be safe for humans.
Key event 2: Villous cytotoxicity
The non-neoplastic lesions observed in the mouse small intestine following Cr(VI) exposure were characterized by the NTP study authors as being secondary to previous epithelial cell injury (NTP, Citation2008b). The villi of treated animals were short, broad and blunt relative to unexposed animals (NTP, Citation2007, 2008b). Such effects are a nonspecific response to various types of injuries, and can be associated with either hyperproliferation or hypoproliferation of crypt enterocytes (Greaves, Citation2007; Serra & Jani, Citation2006). Chemicals that are cytotoxic to cells of the villi can stimulate crypt proliferation to repair the damaged mucosa. Alternatively, chemicals that are toxic to crypt cells can also cause blunting of villi due to hypoproliferation in the crypt and inability to replenish the mucosa. Because cytotoxicity can result from genotoxic and nongenotoxic effects, and the intestinal stem cells reside in the crypt, it is critical to determine (a) whether toxicity is originating in the crypt or villus, (b) whether there is any evidence of cytogenetic damage in the crypts and (c) whether there is evidence for mechanisms associated with intestinal carcinogenesis such as oxidative stress and inflammation. Each of these issues are discussed in the following subsections, and as a whole, indicate that Cr(VI) induces toxicity in the nonproliferating, nonpluripotent cells of the intestinal villus.
Histological evidence of cytotoxicity
The term “diffuse hyperplasia” used in the NTP studies includes the blunting effects on the villi, as well as elongation of the crypts (see Plate 21 in NTP, Citation2008b). This terminology was not used in Thompson et al. (Citation2011b); instead, villus damage and crypt hyperplasia were noted separately, which provides more resolution with respect to the location of intestinal lesions. As shown in , cytoplasmic vacuolization in the duodenal villi was the most sensitive end point –occurring at 170 and 60 mg/l SDD at days 8 and 91, respectively; while atrophy of the villi and crypt hyperplasia were first evident at higher concentrations (i.e. 520 and 170 mg/l on days 8 and 91, respectively). Examples of duodenal tissue damage are shown in . Vacuolization can be indicative of many cellular effects such as altered lipid metabolism, sequestration of absorbed material, autophagy, endoplasmic reticulum (ER) stress or proteasome dysfunction (Franco & Cidlowski, Citation2009; Henics & Wheatley, Citation1999; Mimnaugh et al., Citation2006). Although the exact cause of the SDD-induced vacuolization is unclear, vacuolization is often regarded as a sign of cytotoxicity (Henics & Wheatley, Citation1999; Mimnaugh et al., Citation2006).
Figure 4. Quantitative assessment of intestinal structure in mice. (A) Duodenum of mouse exposed to 520 mg/l SDD for 90 days: with villous atrophy, blunting and fusion (arrow) and crypt epithelial hyperplasia, (B) duodenum of mouse exposed to 520 mg/l SDD for 90 days: with histiocytic cellular infiltration of the villous lamina propria (arrows) and cytoplasmic vacuolization of the villous epithelium, (C) representative image of crypt area analysis in (D)–(E), (D) measures of crypt area in three contiguous tissue sections at day 91 (*p < 0.05 by ANOVA/Dunn’s), (E) villus/crypt area ratios at days 8 and 91 (*p < 0.05 by ANOVA/Dunnett’s), (F) number of enterocytes per crypt at day 91 (10 full crypts assessed per animal; p < 0.01 by ANOVA/Dunnett’s), (G) representative image of crypt enterocyte analysis in (F) and , and (H) fold change in Ki67 expression at days 8 and 91 of exposure (dotted line represents 1.5-fold).
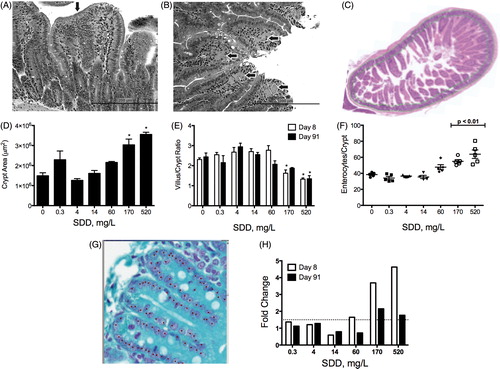
Table 4. Summary of intestinal histopathology in B6C3F1 mice.
The presence of cytoplasmic vacuolization in villi at lower concentrations than crypt hyperplasia suggests a mechanism whereby toxicity originates at the point of contact (i.e. villi) and subsequently triggers compensatory cell proliferation of crypt enterocytes. Area measurements () of the crypts of the mouse duodenum at day 8 increased ∼45% at ≥170 mg/l SDD. At day 91, the crypt area increased ∼45% at 60 mg/l, and significantly increased ≥2-fold of control at ≥ 170 mg/l (). Consistent with the increase in crypt area, the ratio of the villus to crypt area decreased at ≥170 mg/l SDD (). If SDD were toxic to crypt enterocytes, then one would expect a dose-dependent decrease in crypt area under a chronic exposure scenario. In fact, the number of enterocytes per crypt increased significantly in mice exposed to ≥170 mg/l SDD (). Despite the increase in the number of crypt enterocytes, the intestinal villi were damaged and blunted – implying that the increased supply of crypt enterocytes was not sufficient to maintain the normal length and structure of healthy duodenal villi. Hence, the alterations to the villi were not the result of toxicity to crypt enterocytes.
As further evidence that the cytotoxicity of SDD is limited to the intestinal villi, the number of mitotic and apoptotic cells were counted in fully intact crypts in order to compute both a mitotic index (MI) and apoptotic index (AI). As shown in , there were no significant or dose-dependent effects on either end points – suggesting that Cr(VI) did not cause cell cycle arrest or increases in the percentage of crypt enterocytes undergoing programmed cell death (Harris et al., Citation2012). MI results are further discussed in the section “Key event 3: Crypt cell proliferation” as they relate to cell proliferation.
Table 5. Total number of aberrant nuclei in fully intact duodenal crypts.
Assessment of cytogenetic damage in crypts
Data summarized in the previous section indicate that Cr(VI)-induced damage to the intestinal mucosa originates in the villus. However, because Cr(VI) can be genotoxic (see the section “Brief review of CR(VI) genotoxicity and carcinogenicity”), crypt enterocytes were also examined for aberrant nuclei such as karyorrhectic nuclei and MN. The former indicates a type of cell death that can be either a result of programmed death or necrosis (Kumar et al., Citation2005), while the latter can arise from either DNA breakage or chromosomal disjunction (Fenech et al., Citation2011; Vanhauwaert et al., Citation2001). There were no treatment-related effects on the number of karyorrhectic nuclei or MN in duodenal crypts of exposed mice (). MN were also counted in three entire tissue sections, regardless of whether the crypts were fully intact. This analysis has the advantage of increasing the overall amount of crypt tissue that was analyzed for aberrant nuclei. There were no treatment-related increases in MN or karyorrhectic nuclei in crypts across any of the tissue sections examined in animals exposed to Cr(VI) for 7 or 90 days (). These findings are consistent with previous data that Cr(VI) did not affect the levels of DPX in mouse duodenum (De Flora et al., Citation2008).
Table 6. Total number of aberrant nuclei in duodenal mucosal sections.
In contrast to crypts, both forms of aberrant nuclei were observed in villi (). Specifically, karyorrhectic nuclei were significantly increased in duodenal villi at ≥60 mg/l SDD at day 91 and primarily occurred in the tips, whereas MN were statistically increased in villi at ≥170 mg/l (). The concentrations where aberrant nuclei were observed in duodenal villi correspond to the concentrations where villous damage was observed (). Importantly, we considered whether it was possible that the MN present in the villi were the result of MN formation within the crypt. Given that our analyses revealed no MN within the proliferative crypt compartment, it seems highly implausible that MN present in the nonproliferating villous compartment originated within the crypts – especially under the chronic exposure scenario. Instead, these nuclear aberrations are likely the result of cytotoxic mechanisms. Notably, we found no previous studies in the small intestine that have examined villi for MN. We also note that several in vivo MN studies have been conducted in the colon (Morita et al., Citation2011), which has crypts but no villus counterpart. These studies report on MN in the crypts, but not in the mature differentiated epithelium (Blakey et al., Citation1985; Celik et al., Citation2005; Goldberg et al., Citation1983; Le Hegarat et al., Citation2006; Percy et al., Citation1989; Ronen & Heddle, Citation1984; Vanhauwaert et al., Citation2001). Given that the average cell turnover time is similar in the small and large intestines (Rizk & Barker, Citation2012), there is no reason to believe that the villus is necessary to assess chemical-induced genotoxicity in the crypt. Also, we conducted two different analyses, an intact crypt analysis and an analysis of three entire slides per animal. This latter method greatly increases the number of crypt enterocytes analyzed, but no Cr(VI)-induced MN were observed. Finally, we note that it has been previously demonstrated that nuclear aberrations (e.g. MN) can be induced in colonic crypts following γ-irradiation and treatment with the mitotic inhibitor colchicine (Duncan et al., Citation1985). Under the conditions in their study, colchicine increased the MI greatly and aberrant nuclei slightly – both due to spindle poisoning. However, colchicine treatment following irradiation greatly increased the number of aberrant nuclei observed (relative to expected), leading Duncan et al. (Citation1985) to conclude that mitosis is not required for all crypt nuclear anomalies and further suggested that some putative crypt MN were more likely “manifestations of the apoptotic process”. Taken together, the absence of crypt MN and presence of a small number of villous MN at the highest Cr(VI) treatment concentrations imply that the latter were most likely a manifestation of a dose-dependent increase in villous cytotoxicity. Overall, the data support that toxicity to the villi caused the regenerative response in the crypt in the absence of any direct genotoxicity or cytotoxicity in the crypt compartment (see the section “Key event 3: Crypt cell proliferation”).
Oxidative stress
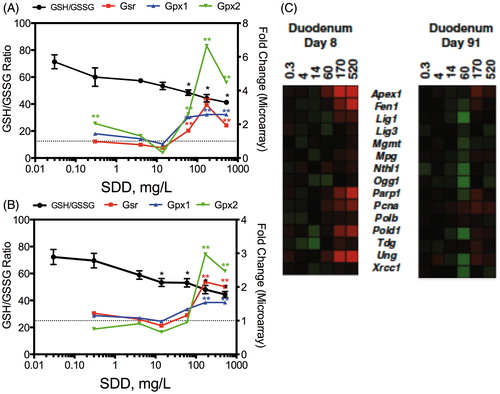
Cr(VI) that is not reduced in the intestinal lumen can be absorbed by villous enterocytes and reduced to Cr(III) intracellularly. As discussed in the section “Brief review of Cr(VI) genotoxicity and carcinogenicity”, the chemistry and biochemistry of Cr(VI) reduction imply that Cr(VI) can alter the redox status of enterocytes through the generation of ROS and/or through cytotoxicity. Cr(VI) significantly decreased the GSH/GSSG ratio, a key indicator of cellular redox status (Meister & Anderson, Citation1983; Moriarty-Craige & Jones, Citation2004; Schafer & Buettner, Citation2001), in the mouse small intestine in a time-dependent and concentration-dependent manner (; (Thompson et al., Citation2011b). Despite the change in redox status, the duodenum appeared to synthesize GSH as evidenced by dose-dependent increases in duodenal GSH levels (Thompson et al., Citation2011b). The increase in GSH was accompanied by significant increases in expression of genes involved in GSH synthesis. As described in Kopec et al. (Citation2012a), Nrf2 oxidative stress signaling and GSH metabolism were among the top five enriched canonical pathways at the lowest treatment concentrations at day 8. Transcription factor analysis in IPA also indicated activation of Nrf2 signaling pathways at ≥170 mg/l SDD day 91 (Kopec et al., Citation2012b). As shown in , Gsr, Gpx1 and Gpx2 were increased significantly in the higher treatment groups. These data suggest that oxidative stress in response to Cr(VI) was initiated early (by day 8) and continued to increase by day 90 – implying that the mice exposed to Cr(VI) in the 2-year NTP bioassay (NTP, Citation2008b) experienced increased intestinal oxidative stress nearly their entire lifespan.
Figure 5. Redox changes in the intestine at day 8 (A) and day 91 (B). The left axis represents the GSH/GSSG ratio and the right axis shows the fold-change in gene expression. Gsr, glutathione reductase; Gpx, glutathione peroxidase. *Statistically significant decrease in GSH/GSSG ratio by Shirley’s test; **statistically significant increase (>1.5-fold, P1(t) > 0.999) relative to control by empirical Bayes’ method for posterior probabilities. (C) Heat map of DNA repair genes in mouse duodenum at days 8 and 91. Adapted from Kopec et al. (Citation2012a).
Intestinal mucosal health is influenced by redox status (Circu & Aw, Citation2011; John et al., Citation2011), and there is evidence that the intestinal mucosa regulates luminal redox status by secretion of thiols such as cysteine (Dahm & Jones, Citation2000; Hagen et al., Citation1990). Chemical-mediated inhibition of GSH synthesis has been shown to cause a decrease in intestinal epithelial cell height, desquamation of microvilli, swelling of mitochondria and vacuolization in the tips of jejunal villi – all of which could be mitigated by GSH supplementation (Martensson et al., Citation1990). These lesions are similar to those induced by Cr(VI). Although Cr(VI) caused a dose-dependent increase in duodenal GSH levels after 7 and 90 days of exposure, the GSH/GSSG ratio was nonetheless significantly decreased (Thompson et al., Citation2011b). It seems highly likely that Cr(VI)-induced changes in oxidative status contribute to cytotoxicity in the intestinal villi.
Despite clear signs of oxidative stress, we did not detect significant changes in 8-OHdG in the duodena of mice exposed to Cr(VI) for up to 90 days (Thompson et al., Citation2011b). These findings are consistent with those of De Flora et al. (Citation2008) who exposed mice to 20 mg/l Cr(VI) for up to 9 months. The apparent lack of 8-OHdG induction in both studies might be explained by upregulation of DNA repair genes or general oxidative stress responses. Cr(VI) increased the expression of genes involved in BER at day 8 (and less so at day 91) (). Significant enrichment of canonical pathways related to DNA repair was not observed at day 91; however, there was enrichment for pathways related to BRCA1 and MMR at 520 mg/l SDD at day 8, as well as enrichment for pathways related to NER at ≥170 mg/l (Kopec et al., Citation2012a). Both BER and NER pathways repair oxidative DNA damage (Go & Jones, Citation2010; Klaunig et al., Citation2010; Sedelnikova et al., Citation2010).
Inflammation
Oxidative stress and inflammation are typically linked physiologically. For example, oxidative stress leads to activation of nuclear factor-κB (NF-κB) and subsequent downstream pathways resulting in the release of cytokines; this is well documented in the scientific literature for airway inflammation, intestinal inflammation and certain cancers (Kruidenier & Verspaget, Citation2002; Rahman & MacNee, Citation2000; Roberts et al., Citation2009). Increases in histiocytic infiltration of macrophages in the duodena of rats and mice were observed in the 90-day and 2-year NTP studies (NTP, Citation2007, Citation2008b), as well as in subsequent 90-day studies (Thompson et al., Citation2011b, Citation2012c); however, clear signs of chronic inflammation were not apparent in these studies. Although histiocytic infiltration can be associated with mild/chronic inflammation, its meaning is unclear. In fact, the NTP study authors concluded that the biological significance of histiocytic infiltration was uncertain (NTP, Citation2008b).
Thompson et al. (Citation2011b, Citation2012c) measured changes in ∼20 cytokines and chemokines in both serum and duodenal mucosa of both mice and rats following 90 days of exposure to Cr(VI). In the serum, there were no clear treatment-related effects in mice, whereas IP-10 and IL-12(p70) were decreased in rats (Supplementary Table 1). In the duodenum, several cytokines were significantly decreased in treated mice relative to untreated mice. TNFα and IL-1β both showed clear dose-dependent decreases following 90 days of exposure (Thompson et al., Citation2011b). These decreases were supported by toxicogenomic analyses, which showed decreased transcript levels for TNFα and IL-1β at ≥ 60 mg/l SDD (Kopec et al., Citation2012a). Other cytokines reduced were IL-7, 9, 12, 13, 17, IFNγ, and RANTES (Supplementary Table 1). In rats, only IL-1α was clearly altered by SDD, and was increased at ≥60 mg/l.
Broadly, the cytokine data support a pro-inflammatory response in rats and a decrease in cytokines and chemokine production in mice. Inflammatory responses intentionally increase ROS production in order to fight infections. Nrf2 signaling can inhibit inflammatory responses (Jung & Kwak, Citation2010; Kim et al., Citation2010), perhaps as feedback control to limit oxidative damage to the host. For example, in a dextran sulfate sodium (DSS) model for induced colitis, Nrf2 null mice demonstrated increased inflammation (viz. TNFα and IL-1β), increased oxidative stress and increased colonic mucosal injury – suggesting an important role for Nrf2 in protecting against inflammation-induced tumorigenesis (Osburn et al., Citation2007). Indeed, Nrf2 regulation of inflammation is mediated by increasing heme oxygenase-1 (HO-1) expression (Jung & Kwak, Citation2010; Kim et al., Citation2010), and HO-1 transcript levels were increased following Cr(VI) exposure (Kopec et al., Citation2012a). HO-1 catabolizes heme to iron, biliverdin and bilirubin – the latter two of which have anti-inflammatory properties (Whittle & Varga, Citation2010; Zhu et al., Citation2011). In vitro studies have also shown that Cr(VI) increases HO-1 mRNA levels, as well as inhibits NF-κB mediated cytokine signaling (Joseph et al., Citation2008; Shumilla et al., Citation1999). Thus, the activation of Nrf2 signaling in response to Cr(VI)-induced oxidative stress might also inhibit inflammatory signaling due to suppression of TNFα and IL-1β.
Key event 3: Crypt cell proliferation
Chronic cell proliferation is a well-known risk factor for carcinogenesis (Ames et al., Citation1993; Boobis et al., Citation2009; Cohen, Citation2010; Gaylor, Citation2005). Diffuse hyperplasia occurred in the duodenum of mice at all SDD concentrations examined in the 2-year bioassay, while there was no evidence of diffuse hyperplasia in the rat duodenum at any dose level (NTP, Citation2008b). The same pattern was observed in the NTP 90-day study (NTP, Citation2007). The two 90-day drinking water studies conducted by Thompson et al. (Citation2011b, Citation2012c) included evaluations at both day 8 and day 91 of exposure. At day 8, duodenal crypt cell hyperplasia was present in 3 of 5 mice exposed to 520 mg/l SDD (; Thompson et al. Citation2011b). Consistent with the increase in crypt proliferation, Myc expression was increased greater than 4-fold at 520 mg/l at day 8 (Kopec et al., Citation2012a). By day 91, crypt hyperplasia was present in almost all animals at ≥170 mg/l SDD (). Although Myc itself was not significantly increased, transcription factor analysis indicated increased MYC signaling (based on expression patterns of Myc downstream targets) at ≥60 mg/l SDD (Kopec et al., Citation2012b). In addition, transcript levels of Ki67, a common marker of crypt cell proliferation (Itzkovitz et al., Citation2012; Potten et al., Citation1997), were significantly elevated at ≥170 mg/l SDD at both day 8 and day 91 (). Together, these data suggest that Cr(VI) increased crypt hyperplasia as early as one week of exposure to very high concentrations in drinking water.
Other authors have suggested a sequence of key events whereby Cr(VI)-induced hyperplasia occurs subsequent to DNA mutation (McCarroll et al., Citation2010). This alternative MOA will be addressed in the section “Alternative MOAs”; however, a strict interpretation of MOA analysis would imply that under the MOA proposed by McCarroll et al. (Citation2010) preventing DNA mutation would prevent cell proliferation. In this regard, it has long been recognized that inactivating mutations in adenomatous polyposis coli (Apc) results in uncontrolled cell proliferation and intestinal adenoma formation through constitutive activation of Wnt/β-catenin signaling pathway (Rizk & Barker, Citation2012; Takahashi-Yanaga & Sasaguri, Citation2007). Given the clear evidence of crypt hyperplasia as early as day 8 of exposure, and the high incidence of hyperplasia by day 90, alterations in Apc might be expected. However, the toxicogenomic data collected in mice exposed to Cr(VI) for up to 90 days did not indicate changes in Apc expression that might result from genetic or epigenetic silencing of the Apc gene (Kopec et al., Citation2012a,Citationb). Likewise, there was no increase in β-catenin (Ctnnb1) expression level or indication of Ctnnb1 activation by transcription factor analysis that is expected by loss of APC; nor was there functional enrichment of Wnt/β-catenin signaling pathways. Importantly, animal models have shown that loss of APC leads to rapid onset of adenomas in both the small and large intestines of mice (Barker et al., Citation2009; Luongo et al., Citation1994; Rizk & Barker, Citation2012; Shibata et al., Citation1997; Su et al., Citation1992). Thus, the absence of Wnt-signaling/β-catenin activity or preneoplastic lesions in the small intestine of mice exposed to Cr(VI) in drinking water indicate that crypt hyperplasia is not likely the result of early genetic or epigenetic changes related to APC. These findings do not preclude the eventual involvement of APC loss and consequent increase in Wnt/β-catenin signaling in the development of Cr(VI)-induced intestinal tumors that might result from gradual accumulation of mutations as a consequence of chronic intestinal injury.
As noted previously, Cr(VI) had no effect on the MI in the crypt cells. A decrease in MI can indicate cytotoxicity or perhaps cytostasis. An increase in MI can indicate a decrease in cell cycle time, or increase in mitosis time (perhaps indicating toxicity). As was discussed above, the total number of crypt cells increased without a change in the MI. These findings along with the absence of change in AI in crypts suggest that the normal balance between cell birth and death was unaltered. This increase in crypt proliferation without change in MI is not unique (Snow & Altmann, Citation1983; Sunter et al., Citation1978). As posited by Snow & Altmann (Citation1983), an increase in crypt cells without change in MI can be explained by either an increase in the number of crypt basal stem cells or an increase in the number of divisions an upper crypt cell (sometimes referred to as a transit-amplifying cell) undergoes prior to differentiation. One recent study has reported that the number of intestinal stem cells (in the crypt base) can increase through self-renewal following calorie restriction (Yilmaz et al., Citation2012).Footnote2 Apparently, the surrounding Paneth cells, unlike the stem cells, sense caloric restriction and stimulate proliferation in nearby stem cells. Yilmaz et al. (Citation2012) posited that the preference for stem cell renewal over more differentiated cells may provide for more rapid mucosal regeneration when food becomes available. These findings imply that damage to crypt cells is not a prerequisite for stem cell renewal. In theory, chronic wounding to villi might also increase signals that stimulate basal stem cell renewal.
Interestingly, Lgr5 has received much attention as a putative biomarker for the intestinal basal stem cell (Barker et al., Citation2009; Rizk & Barker, Citation2012; Yan et al., Citation2012). One recent study showed that γ-irradiation significantly ablated Lgr5-positive basal cells in the small intestine, but increased a population of quiescent Bmi1-positive stem cells (Yan et al., Citation2012). We did not detect any significant (≥1.5-fold) decreases in Lgr5 at day 8, but did detect a significant 1.8-fold increase at 520 mg/l SDD (the only concentrations associated with crypt hyperplasia at day 8). At day 91, we did not detect any significant ±1.5-fold changes in Lgr5; nor did we detect any significant changes in Bmi1 at day 8 or day 91. Whether whole genome microarray analysis is sensitive enough to reliably detect these changes in intestinal tissue remains to be determined, but the results suggest that Cr(VI) did not ablate Lgr5-positive basal stem cells or stimulate an increase in Bmi1-positive stem cells. Importantly, γ-irradiation studies also induce MN in crypt enterocytes (Duncan et al., Citation1985; Goldberg et al., Citation1983; Ronen & Heddle, Citation1984). As discussed previously, the absence of MN in the crypts of mice exposed to Cr(VI) suggests that the proliferative response is not initiated by direct damage to crypt stem cells.
Finally, in addition to diffuse hyperplasia, NTP also reported the incidence of focal hyperplasia, which they considered a preneoplastic lesion (NTP, Citation2008b). Focal hyperplasia was observed in the duodena of a few mice, although the incidence was not statistically different from controls and was not observed in any female mice in the 520 mg/l SDD treatment group (). Notably, focal hyperplasia was not observed in any animals in the 90-day drinking water studies (NTP, Citation2007; Thompson et al., Citation2011b, Citation2012c); altogether, 260 rodents were exposed to ≥60 mg/l SDD, yet focal hyperplasia (or other preneoplastic lesions) was not observed. The presence of diffuse hyperplasia without focal hyperplasia is consistent with proliferation that is secondary to mucosal injury.
Key event 4: Crypt cell mutagenesis
According to US EPA’s Framework for Determining a Mutagenic Mode of Action (US EPA, Citation2007), target tissue mutation data provide the strongest evidence for a mutagenic MOA. To assess the potential for Cr(VI) to induce mutations in the small intestine following oral exposure to Cr(VI), we measured Kras codon 12 GAT MF in the duodenal epithelium of mice exposed to Cr(VI) for 90 days (O’Brien et al., Citation2012). Kras was selected because it is often mutated early in human intestinal tumors (Wicki et al., Citation2010), and codon 12 GGT to GAT mutation is one of the most commonly reported mutations in human duodenal tumors (Nishiyama et al., Citation2002; Sutter et al., Citation1996; Wicki et al., Citation2010; Younes et al., Citation1997), and accounts for 12.6% of colon tumors in the COSMIC database (Forbes et al., Citation2010). In addition, Kras mutation has been shown to contribute to small intestine tumorigenesis in mice (Castellano & Santos, Citation2011). Other mutations to consider include p53 and Apc. As discussed previously, toxicogenomic data from mice exposed to Cr(VI) for 90 days did not indicate changes in Apc or β-catenin. Mutations in p53 occur relatively late in tumorigenesis (Velho & Haigis, Citation2011), and thus p53 is not an ideal early somatic mutation target.
Importantly, although Kras is a single oncogene, several studies show that Kras codon 12 GAT mutation is amplified during carcinogenesis, even when the mutational specificity of the mutagen is other than the G to A mutation being measured. Significant and early dose-related increases in Kras codon 12 GAT mutation were observed in rodent tissues treated with chemicals expected to induce G to T mutation, specifically benzo[a]pyrene (BaP) and ethylene oxide. For example, an accumulation of Kras codon 12 GAT mutations was observed in the mouse lung following i.p. administration of BaP (Meng et al., Citation2010b). This study also found that Kras codon 12 GAT MF was positively correlated with BaP-DNA adduct formation. In mice, inhalation exposure to ethylene oxide for four weeks induced both codon 12 GAT and GTT mutations in their lungs, but Kras codon 12 GAT mutation was the most sensitive end point, more sensitive than the codon 12 GTT mutation which was expected to occur as a consequence of oxidative stress (Parsons et al., Citation2012). These results suggest that Kras codon 12 GAT MF represents a functional reporter (i.e. reporter of tumor initiation and/or progression) which is amplified with mutational loading that is not necessarily specific to a chemically induced DNA lesion of Kras codon 12 (Meng et al., Citation2010a; Parsons et al., Citation2010, Citation2012).
As standard sequencing techniques are not sufficiently sensitive to measure early mutations in preneoplastic tissue, allele-specific competitive blocker (ACB) PCR analysis was used to measure MF in scraped duodenal epithelium with an increased MF detection at very low levels (∼10−6) (O’Brien et al., Citation2012). No treatment-related effect on Kras codon 12 GAT MF was observed even at the high Cr(VI) doses that were carcinogenic in the 2-year bioassay and that increased crypt proliferation after 7 or 90 days of exposure (). Although Kras Codon 12 GAT is a single mutation in a single oncogene, DNA mutations in other oncogenes, if they occurred with Cr(VI) exposure, may be expected to amplify Kras mutations and be detected in the ACB-PCR assay. Moreover, the increase in crypt cells (), if preceded/induced by mutations, would likely increase the number of target Kras mutations thereby ensuring detection if present.
Figure 6. Mutagenicity analysis: (A) Kras codon 12 GGT to GAT mutation frequency (MF) in mouse duodenum assessed by ACB-PCR. The base 10 log of codon 12 GAF MF is plotted relative to the base 10 log of SDD concentration in mg/l. Error bars indicate the standard error of the geometric mean MF and (B) cluster analysis of expression values of 116 genes from four mutagenic (triangles) and four nonmutagenic (squares) carcinogens. The inner ellipse is the standard variance ellipse, and the outer ellipse is the 95% confidence boundary. The square with crosshair represents Cr(VI), indicating that the expression profile for Cr(VI) clusters more closely with nonmutagenic compounds (see text for further details). The four mutagenic carcinogens are 2-nitrofluorene, dimethylnitrosamine, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and aflatoxin B1; the four nonmutagenic carcinogens are methapyrilene, diethylstilbestrol, Wy-14643 and piperonylbutoxide. Panel B is adapted from Thompson et al. (Citation2012b).
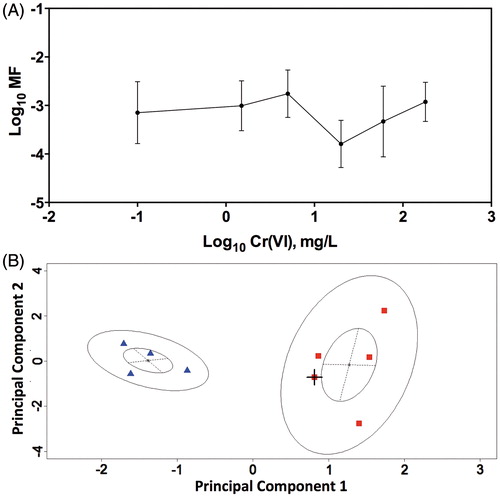
Changes in the DNA sequence are not readily detected by transcriptomics. However, some investigators have attempted to identify genomic fingerprints that may distinguish between mutagenic and nonmutagenic carcinogens. For example, Ellinger-Ziegelbauer et al. (Citation2005) compared gene expression data for four mutagenic and four nonmutagenic hepatic carcinogens after short-term exposure (≤2 weeks) in rats. Importantly, the mutagenic carcinogens were characterized as inducing DNA modification and causing mutations and physical distortion of DNA (Ellinger-Ziegelbauer et al., Citation2004, Citation2005), and have been characterized as mutagens by the US EPA (Citation2005b, Citation2007). Ellinger-Ziegelbauer et al. (Citation2005) identified 139 genes comprising seven toxicological categories that were useful for distinguishing the two classes of carcinogens. We compared the Cr(VI)-induced gene expression changes in the mouse duodenum with gene changes produced by the eight carcinogens – all after only 7 days of exposure. The reader is referred to Thompson et al. (Citation2012b) for further details of this analysis. Overall, the genomic response induced by 520 mg/l SDD at day 8 was more similar to that induced by nonmutagenic than by mutagenic carcinogens (; Thompson et al. Citation2012b). These findings lend additional support to the WOE indicating that the intestinal tumors in the mouse duodenum were the result of a nonmutagenic MOA.
In conclusion, the apparent absence of effects on Kras and Apc is consistent with the fact that early tumors, metastases and mortality were not observed in the NTP 2-year bioassay (NTP, Citation2008b), as well as the absence of preneoplastic (e.g. focal hyperplasia) or neoplastic lesions in any of the 90-day Cr(VI) drinking water studies (NTP, Citation2007; Thompson et al., Citation2011b, Citation2012c). It is also consistent with the absence of cytogenetic damage in the duodenal crypts. These findings support that Cr(VI) is not acting via a mutagenic MOA, specifically where mutation is an early key event, in the mouse small intestine.
Additional supporting information
In vitro MN formation
Previous studies have examined the ability of Cr(VI) to induce MN formation in vitro (see the section “Assessment of Cr(VI) genotoxicity in vitro”). As part of our Cr(VI) MOA research, Cr(VI) genotoxicity was assessed by high content screening methods in the CHO-K1 cell line (Thompson et al., Citation2012a), which is a cell model recommended by OECD (Citation2010). Treatment of CHO-K1 with Cr(VI) reduced viability and the number of bi-nucleated cells at ≥32 µM Cr(VI) – suggesting treatment-induced cell cycle arrest; at these concentrations Cr(VI) increased MN frequency (). In contrast, the positive control mitomycin-C (MMC) significantly (p < 0.001) increased MN frequency at concentrations that did not greatly reduce cell numbers or the percentage of bi-nucleated cells. Treatment of A549 (a human lung adenocarcinoma epithelial cell line) with Cr(VI) resulted in relatively small but statistically significant (p < 0.05) increases in cytotoxicity and decreases in the percentage of bi-nucleated cells at 3.2 µM. At this concentration, the frequency of MN in bi-nucleated cells slightly increased from 1.47 ± 0.50 to 2.12 ± 0.41% (). At higher Cr(VI) concentrations, cell death was extensive. The positive control MMC significantly increased MN frequency from 1.47 ± 0.50 to 6.89 ± 2.24%. These data indicate that Cr(VI) has very weak (or negligible) genotoxic potential at noncytotoxic concentrations.
Figure 7. MN formation in CHO-K1 (A) and A549 (B) cells. Cells were treated with the indicated concentrations of Cr(VI). MN formation in bi-nucleated cells is shown in the larger plots. The insets show cytotoxicity as indicated by decrease in the number of bi-nucleated cells. Data represent mean ± SEM, n = 9.
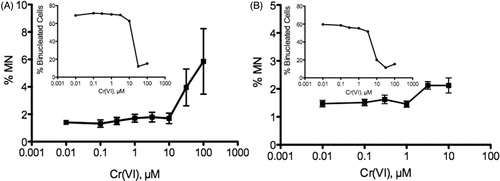
In vitro intestinal Caco-2 cell model
The genotoxicity of Cr(VI) was also assessed in Caco-2 cells, which are a well-accepted model for studying intestinal absorption, metabolism and cytotoxicity (Meunier et al., Citation1995; Natoli et al., Citation2011; Rawlinson et al., Citation2010; Sambruy et al., Citation2001; Shah et al., Citation2006; Smetanova et al., Citation2011). When initially plated and grown for ∼24 h, Caco-2 cells are undifferentiated and proliferating, and thus share some characteristics with crypt enterocytes. When grown to confluency (∼21 days), Caco-2 cells differentiate and exhibit structural (e.g. microvilli) and molecular (e.g. gene/protein expression) characteristics similar to villous enterocytes. This in vitro model was used to assess whether responses to Cr(VI) might differ between villous and crypt enterocytes of the small intestine.
Two forms of DNA damage were assessed in Caco-2 following 24-h treatment with Cr(VI). Oxidative DNA damage and DNA DSBs were assessed by nuclear staining intensity of 8-OHdG and phosphorylated histone variant H2AX (γ-H2AX) using high content imaging methods (Thompson et al., Citation2012a). H2AX phosphorylation is a sensitive indicator of DNA DSB that can result from chemical-induced DNA damage or damage introduced during DNA repair (Bonner et al., Citation2008; Kinner et al., Citation2008). Treatment with Cr(VI) for 24 h resulted in reduced cell numbers at ≥3 µM Cr(VI), as well as significant increases in nuclear staining of 8-OHdG and γ-H2AX in undifferentiated Caco-2 cells (). At noncytotoxic concentrations 0.1 and 0.3 µM, only 8-OHdG was significantly elevated (). In contrast to undifferentiated Caco-2 cells, differentiated Caco-2 cells were more resistant to Cr(VI). A reduction in cell numbers was only observed at 100 µM, and increased γ-H2AX nuclear staining was only significantly elevated at 30 and 100 µM (). Staining of 8-OHdG showed a similar pattern. These results suggest that at low µM Cr(VI) concentrations, oxidative DNA damage predominates, whereas more severe DNA damage (as indicated by H2AX phosphorylation) occurs at higher Cr(VI) concentrations.
Figure 8. Nuclear staining intensity of 8-OHdG (open bars) and γ-H2AX (filled bars) in Caco-2 after 24-h treatment with Cr(VI): (A) undifferentiated Caco-2: 8-OHdG was significantly elevated at ≥0.3 µM; γ-H2AX was significantly elevated at ≥3 µM (cytotoxic concentrations) and (B) differentiated Caco-2: 8-OHdG and γ-H2AX were significantly elevated at ≥30 µM (8-OHdG was also elevated at 0.3 µM). Nuclear staining represents mean ± SEM (n = 3); Cell # = mean ± SD (n = 3).
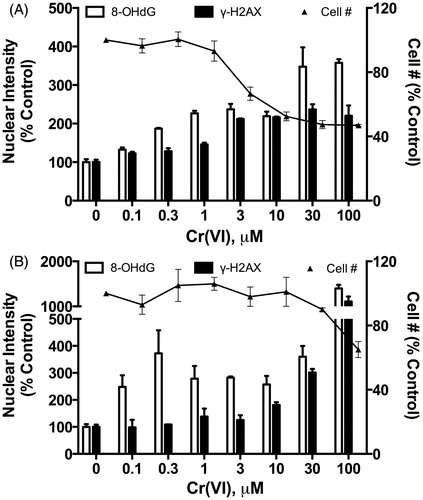
It should be noted that the SDD concentrations employed in the NTP study (14–516 mg/l) correspond to 100–3300 µM Cr(VI). The two clearly carcinogenic concentrations were effectively 1 and 3 mM. Although these concentrations were likely diluted and reduced by gastric contents, these concentrations entering into the intestinal lumen were likely well above cytotoxic concentrations observed in vitro in Caco-2 cells (), CHO-K1 cells (), A549 cells () and numerous other cell lines reported in the literature. Interestingly, the greater sensitivity of proliferating Caco-2 to Cr(VI) relative to differentiated Caco-2 predicts that Cr(VI) exposure in vivo might induce more adverse effects in crypt enterocytes than villous enterocytes. However, the in vivo Cr(VI) studies described in the section “Key event 2: Villous cytotoxicity” show that Cr(VI) causes toxicity to intestinal villi without apparent treatment-related effects on crypt enterocytes. These findings suggest that the in vitro model is either not a suitable model for the intestinal mucosa (with respect to Cr(VI) toxicity), or that crypt enterocytes were minimally (or not at all) exposed to Cr(VI) in vivo. This apparent protection against Cr(VI) exposure might be explained by mucus generated within the crypts (i.e. glands of Lieberkühn) that serves as a barrier to bacteria, viruses, particles and other toxins (DeSesso & Jacobson, Citation2001; Greaves, Citation2007; Talley, Citation2010). The finding is also consistent with basic intestinal function where mature differentiated villous enterocytes function to absorb dietary nutrients. There is also evidence that anion and solute transporter expression may be lower in crypts than villi (George et al., Citation2008).
Overall, the in vitro findings summarized in this section support that the intestinal tumors that arose following chronic Cr(VI) ingestion were due to prolonged increased cell turnover in an oxidized environment rather than direct DNA reactivity.
Concordance of dose–response
Following 90 days of exposure, chromium levels in duodenal tissue significantly increased at ≥14 mg/l SDD, and the GSH/GSSG ratio significantly decreased at these concentrations. Cytoplasmic vacuolization and other signs of villous toxicity significantly increased at ≥60 mg/l SDD, and crypt cell proliferation significantly increased at ≥170 mg/l SDD (Thompson et al., Citation2011b). demonstrates the dose concordance among relevant end points in the duodenum at both day 8 and day 91. The plots clearly support a sequence of events beginning with decreased GSH/GSSG ratio, followed by cytoplasmic vacuolization and crypt cell proliferation, and ultimately resulting in adenoma formation (which typically precedes carcinoma formation). A similar pattern occurs in the jejunum at day 91 (). depicts the sequence of key events in both our 90-day MOA studies and the NTP 2-year bioassay. In the 90-day study, tissue accumulation, changes in the GSH/GSSG ratio and villous cytotoxicity (cytoplasmic vacuolization) all precede crypt hyperplasia. In the NTP study, the term “diffuse hyperplasia” represented both villous cytotoxicity and crypt hyperplasia, and is therefore depicted adjacent to these two key events from the 90-day study. The incidence of diffuse hyperplasia can be seen to precede the incidence of tumor formation. It should be noted that the x-axis is shown in applied dose (mg/l SDD). We have shown the dose concordance within each study because we do not believe that it is possible to obtain dose concordance across studies for several reasons. This is due to several factors including possible colony differences from different vendors and over time (Farkas et al., Citation2009), differences in water intake between the studies and the large differences in study duration that potentially change the disposition of Cr(VI) over time. For example, these high concentrations of Cr(VI) can potentially alter gut microflora (Shrivastava et al., Citation2005). Nevertheless, taken together, the two Cr(VI) bioassays establish a plausible sequence of events whereby Cr(VI) absorption into the villous leads to cytotoxicity in the villi, sustained crypt proliferation due to chronic wounding, and ultimately tumorigenesis.
Figure 9. Dose concordance: (A) dose–response of effects in the mouse duodenum at day 8, (B) dose–response of effects in the mouse duodenum at day 91 and (C) dose–response of effects in the mouse jejunum at day 91.
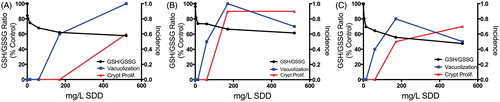
Figure 10. Comparison of key events in the 90-day MOA study and the NTP 2-year bioassay. The term “diffuse hyperplasia” includes both villous toxicity and crypt hyperplasia. Together, these studies establish that villous cytotoxicity is an antecedent to tumor formation in the mouse small intestine. This illustration is patterned after a generalized scheme in Meek & Klaunig (Citation2010).
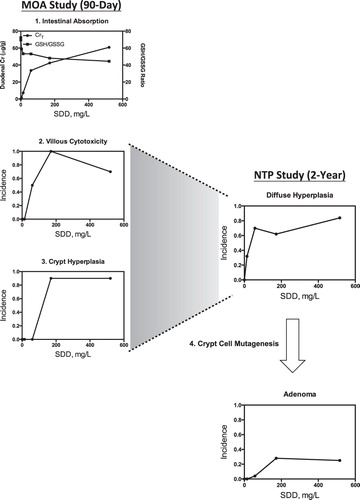
Temporal association
Key events that occur at the same time as tumors are not likely to contribute to tumor development (Boobis et al., Citation2006). In this regard, oxidative stress, toxicity to intestinal villi and crypt hyperplasia can be seen as early as 7 days of exposure (). After 90 days of exposure, damage to villi and crypt hyperplasia can be seen across multiple doses without tumors or preneoplastic lesions such as focal hyperplasia. Similar intestinal pathology (i.e. “diffuse hyperplasia”) was reported in the NTP 90-day bioassay (NTP, Citation2007). After 2 years of exposure, diffuse hyperplasia was observed in all treatment groups (i.e. ≥14 mg/l SDD) and tumors at ≥172 mg/l SDD relative to concurrent controls (≥57 mg/l relative to historical controls). These findings suggest that in the drinking water studies conducted to date, mice experienced increased cell proliferation and redox changes that began within the first week of exposure. The fact that neither preneoplastic lesions nor tumors were observed in the 90-day studies and that tumors did not occur until 450 days or later (NTP, Citation2007; Thompson et al., Citation2011b) suggests that it takes the majority of the 2-year mouse lifespan for these events to contribute to tumor formation. A highly proliferative tissue experiencing oxidative stress is expected to provide the ideal environment for an early mutation induced by a mutagenic compound – yet no increase in Kras codon 12 GAT MF or changes in Apc expression or signaling was observed with Cr(VI) administration.
Table 7. Temporal concordance.
Plausibility
In human sporadic colon cancer, tumor initiation occurs after the third decade of life, progresses for 10–20 years and results in carcinoma at a mean age of ∼68 years (Grady and Carethers, Citation2008). However, genetic disorders, that result in either accelerated initiation or progression can lead to earlier tumor development (Grady & Carethers, Citation2008). In this colon cancer paradigm, increasing the number of cell divisions within the intestinal crypt over a lifetime due to chronic intestinal wounding could accelerate the normal progression of spontaneous tumors. In this regard, the tumors observed in the NTP 2-year bioassay were similar to those that occur spontaneously, but only rarely in untreated animals (NTP, Citation2008b).
Cytotoxicity and subsequent regenerative hyperplasia is a well-known MOA (Ames et al., Citation1993; Boobis et al., Citation2009; Meek et al., Citation2003; US EPA, Citation2005a), and it has been suggested that cytotoxic and proliferative effects observable by 13 weeks of exposure can be predictive of effects in 2-year bioassays (Cohen, Citation2010; Gaylor, Citation2005; Slikker et al., Citation2004). Indeed, the NTP Cr(VI) studies followed this pattern; cytotoxicity and proliferation were observed in the intestine of mice after 13 weeks of exposure (NTP, Citation2007), and tumors were observed in the intestines after lifetime exposure (NTP, Citation2008b). In contrast, proliferation was not observed in the intestine of rats after 13 weeks of exposure (NTP, Citation2007), nor were intestinal tumors after lifetime exposure (NTP, Citation2008b). The small intestine is one of the most proliferative tissues in the body and it is likely that the additional proliferative pressure due to chronic high dose Cr(VI) exposure increased tumor initiation and promotion. Considering that Cr(VI) did not increase mortality and that the tumors did not appear to metastasize (NTP, Citation2008b), the WOE supports that the increase in tumors was the result of increased lifetime crypt proliferation as opposed to the activity of a direct acting mutagen. The lack of preneoplastic focal hyperplasia further suggests that pockets of clonal expansion that might arise from a mutagen did not occur.
Although we did not conduct any counterfactual experiments, i.e. experiments aimed at blocking key steps in the MOA to determine whether tumors could be prevented, several lines of evidence support that preventing villous cytotoxicity prevents Cr(VI)-induced intestinal carcinogenesis. First, rats exposed to equally high Cr(VI) concentrations in drinking water did not develop diffuse hyperplasia or tumors (NTP, Citation2008b). Second, the absence of lesions in the colon, which lacks a villus counterpart, also suggests a requirement of villous cytotoxicity. Subcutaneous injection of dimethylhydrazine (DMH) induces MN, crypt proliferation and tumors in both the duodenum and colon of mice (Goldberg et al., Citation1983; Lipkin & Deschner, Citation1976; Ronen and Heddle, Citation1984; Snow & Altmann, Citation1983; Sunter et al., Citation1978). These DMH-induced tumors likely arise from exposure in blood and/or bile (Pozharisski et al., Citation1975). If the Cr(VI)-induced intestinal hyperplasia and tumors in the mouse small intestine were mediated through the blood, it seems likely then that similar effects would have been observed in colonic crypts. The absence of colonic lesions indicates that either Cr(VI) was reduced to Cr(III) prior to reaching the colonic lumen or that the structure of the colon (namely absence of absorptive villi) prevented a cytotoxicity-induced proliferative pressure on the colonic crypts. It is well known that Cr(VI) uptake requires anion transporters, and it is reasonable to assume that uptake primarily occurs in the villus. Although the identity of individual Cr(VI) transporters remains uncertain, solute anion carriers are reported to be more highly expressed in the human villus than crypt (George et al., Citation2008). The observation that intestinal tumors in mice correlated with the location where nutrient absorption is highest in the intestine (duodenum and proximal jejunum) supports that villous toxicity is necessary step in the MOA.
It is also notable that the proliferative effects in the duodenum following chronic DMH treatment also lead to an increase in villus length and cell number (Snow & Altmann, Citation1983). This is in stark contrast to the villous vacuolization, blunting and atrophy following Cr(VI) exposure. DMH exposure also results in MN formation and fairly rapid onset (∼22 weeks) of intestinal tumors (Snow & Altmann, Citation1983; Sunter et al., Citation1978). These differences between DMH and Cr(VI) suggest very different MOAs for the two compounds. Indeed, as was discussed in the section “In vitro intestinal Caco-2 cell model”, our in vitro Caco-2 model predicts that Cr(VI) should be toxic to proliferating cells. As this was not observed in vivo, it appears that Cr(VI) is not directly affecting crypt cells.
Similarities with other chemicals that have caused small intestinal tumors in rodents in 2-year bioassays
Three chemicals investigated by NTP have been noted to cause intestinal tumors in mice: indium phosphide, o-nitrotoluene and captan (Chandra et al., Citation2010; Stout et al., Citation2009a). However, the increased incidence of intestinal tumors from indium phosphide was from an inhalation study, where the NTP study authors characterized the evidence for these tumors as marginal and “may have been related to exposure to indium phosphide” (NTP, Citation2001). Notably, lung tumors induced by indium phosphide are thought to involve oxidative stress (Gottschling et al., Citation2001; Upham & Wagner, Citation2001). O-nitrotoluene (2-nitrotoluene) caused increased incidences of neoplasms in the large intestine, not small intestine (NTP, Citation2002). Mechanistic studies suggest that o-nitrotoluene carcinogenicity may also involve oxidative mechanisms (Watanabe et al., Citation2010).
Captan and the structurally similar chemical folpet induce similar morphological changes in the intestine as Cr(VI) including cytotoxicity to villi, blunting of villi, regenerative crypt cell proliferation and adenomas and carcinomas in the mouse duodenum and, to a lesser extent, jejunum (Cohen et al., Citation2010; NCI, Citation1977). Furthermore, captan and folpet do not induce intestinal tumors in rats. Like Cr(VI), captan and folpet exhibit mutagenic activity in vitro, but evidence for in vivo mutagenicity is equivocal or negative (Arce et al., Citation2010; Bernard & Gordon, Citation2000). Interestingly, over three decades ago the mutagenicity of Cr(VI) and captan in bacteria was shown to be mitigated by S9 (De Flora, Citation1978), suggesting that they might share similar characteristics. Mechanistically, captan and folpet induce toxicity by reacting with thiols in GSH and proteins, which generates the highly reactive thiophosgene that also interacts with thiols and other cellular constituents (Cohen et al., Citation2010). Both chemicals have been shown to significantly deplete duodenal GSH within the first few hours of exposure and then significantly increase GSH by 24 h relative to untreated animals (Arce et al., Citation2010; Cohen et al., Citation2010). Although GSH levels were not measured in the intestine within 24 h of exposure to Cr(VI), Cr(VI) elicited dose-dependent increases in intestinal GSH at both day 8 and day 91 (Thompson et al., Citation2011b). Given that glutathione is one of the key intracellular agents responsible for reducing Cr(VI), it seems likely that Cr(VI) would also acutely deplete GSH levels before cells have sufficient time to adapt and synthesize more. All three chemicals also fail to induce MN in intestinal crypts (Arce et al., Citation2010; Chidiac & Goldberg, Citation1987; Harris et al., Citation2012).
In 2004, the US EPA classified captan as “not likely” to be a human carcinogen after an independent peer-review concluded that captan acted through a nonmutagenic MOA that “required prolonged irritation of the duodenal villi as the initial key event” (Gordon, Citation2007; US EPA, Citation2004). The similar effects induced by captan, folpet and Cr(VI) make it plausible that they share a common nonmutagenic MOA that likely involves point of contact cytotoxicity in the villi followed by sustained proliferative pressure on crypt cells to regenerate the mucosa. Over a lifetime of exposure to such cytotoxic chemicals, there is an increased risk of carcinogenesis.
Alternative MOAs
Alternative MOAs for Cr(VI)-induced intestinal cancer might include mitogenic and mutagenic MOAs. With regard to the former, there is no evidence to suggest that Cr(VI) stimulates crypt hyperplasia in the absence of cytotoxicity, as hyperplasia occurs subsequent to villous cytotoxicity in both dose and time. Moreover, a mitogenic effect would likely lead to elongated crypts without blunted villi. Prior to the conduct of the studies described herein, some investigators hypothesized that the intestinal tumors observed in mice arose by a mutagenic MOA – based primarily on data from nontarget tissues and in vitro systems (McCarroll et al., Citation2010). Specifically, it was hypothesized that hyperplasia occurred subsequent to DNA mutation (McCarroll et al., Citation2010). Consistent with this idea, mutations in Kras and Apc can result in increased crypt cell proliferation in the intestine (Barker et al., Citation2009; Feng et al., Citation2011; Luongo et al., Citation1994; Rizk & Barker, Citation2012; Shibata et al., Citation1997; Su et al., Citation1992). However, as described herein, we did not detect changes related to Apc expression or signaling, nor did we detect increases in Kras MF. Moreover, we believe that the high incidence (60%) of crypt proliferation after only seven days of exposure to Cr(VI) is unlikely to be the result of mutation – especially considering the lack of neoplasms at day 90, and the late tumor onset in the NTP studies. Furthermore, rats exposed to 60 mg/L SDD for 7 days developed crypt hyperplasia (Thompson et al., 2012c; see section “Data gaps”). That hyperplasia and tumors were not observed in the NTP 2-year study suggests that the Cr-induced hyperplasia is reversible and not the result of mutation.
It is difficult/impossible to completely disprove that Cr(VI) causes mutations directly. Unlike for the human, commercially available somatic mutation screening assays using competitive PCR methods are not readily available for mice; and other conventional sequencing approaches, such as Sanger sequencing, are not sufficiently sensitive to measure mutations in non-neoplastic tissue. Therefore, it is informative to assess the available data in the context of generally accepted characteristics for carcinogens that act through a mutagenic MOA. For example, the US EPA has proposed a general framework for assessing a mutagenic MOA in their draft guidance, Framework for Detecting a Mutagenic Mode of Action for Carcinogenicity (US EPA, Citation2007). Although still in draft form, the framework nevertheless provides insightful approaches for determining whether a chemical acts through a mutagenic MOA. First and foremost, the framework explicitly states the critical question, “Is mutation an early key event in the chemical’s induction of cancer?”. As part of determining a mutagenic MOA, one must (a) establish whether a chemical has mutagenic properties, (b) establish whether mutagenicity is relevant to the MOA in the tissue of interest by considering dose–response concordance, temporal concordance and plausibility and (c) consider alternative MOAs.
As shown in , data from the small intestine do not support dose–response concordance, temporal concordance or plausibility associated with a mutagenic MOA. With exposure to increasing concentrations of Cr(VI), there was no evidence of increased cytogenetic damage in crypt enterocytes, alteration in Apc expression or signaling, or mutations in Kras. In contrast, cytotoxicity, oxidative stress and crypt hyperplasia were all increased in a dose-dependent manner. With increased duration of exposure to Cr(VI), there was no evidence of increased cytogenetic damage in crypt enterocytes, alteration in Apc expression or signaling, or incidence of preneoplastic lesions. In contrast, oxidative stress, cytotoxicity and crypt hyperplasia were worse (or more prevalent) at day 91 relative to day 8. Thus, the hypothesis that mutation is an early initiating key event in the intestinal carcinogenesis is inconsistent with the MOA data collected from the target tissue, as well as the late tumor onset, lack of tumors outside the portal of entry and lack of increased mortality (NTP, Citation2008b). It should also be noted that most in vivo MN studies are conducted in proliferative tissues such as bone marrow, skin and intestine because proliferation facilitates the detection of genotoxicity (Morita et al., Citation2011). Because the small intestine is a highly proliferative tissue, it is likely that evidence of DNA damage and mutation would have been readily apparent if Cr(VI) tumorigenicity was mediated through a direct genotoxic or mutagenic MOA.
Table 8. US EPA mutagenic framework and application to Cr(VI) in the small intestine.
The US EPA mutagenic MOA framework provides a list of characteristics typical of mutagens – some of which are listed in . The data collected in target tissue and summarized in this review do not share these characteristics. For example, mutations found in genes that are associated with carcinogenesis and in the presence of low cytotoxicity increase the WOE for a mutagenic MOA. In this regard, we did not detect increases in Kras MF or changes in Apc/Wnt/β-catenin signaling, even at cytotoxic concentrations. The US EPA framework also indicates that mutagens often elicit tumor responses early in chronic studies (e.g. within 52 weeks), yet the tumors in the NTP Cr(VI) were observed late in the study (∼450 days) and did not increase mortality (NTP, Citation2008b). Boobis et al. (Citation2009) have also outlined general key events for chemicals with a mutagenic MOA (). Notably, clonal expansion of mutated cells often increases mutations in other key genes and leads to preneoplastic lesions. Thus, even if Cr(VI) did not specifically “target” Kras, a general increase in mutations would likely lead to increases in additional mutations in Kras codon 12, which were measurable, but unchanged by Cr(VI), using the highly sensitive ACB-PCR assay. Furthermore, the lack of preneoplastic lesions suggests that clonal expansion of cells with growth advantages (such as can be conferred by Kras mutations) were not present. Notably, Boobis et al. state that, “…carcinogens are considered in two clearly distinguished groups based upon their MOA—DNA-reactive (mutagenic) and nonDNA-reactive (e.g. receptor-mediated, mitogenic, cytotoxic, oxidative stress)”. The evidence herein supports involvement of both cytotoxicity and oxidative stress in the MOA for Cr(VI)-induced intestinal carcinogenesis. also provides a similar list of events/characteristics of DNA reactive compounds as described by Preston & Williams (Citation2005). These key events focus on effects in critical genes, in critical replicating target cells. The findings herein suggest that Cr(VI) is not directly affecting the target cells of the crypt.
Table 9. Select properties of mutagenicity as key events and application to the Cr(VI) in the small intestine.
Even without the new intestinal data summarized herein, much of the available literature indicates that Cr(VI) carcinogenicity is a high dose phenomenon and that nonphysiological exposure scenarios and high doses are required for carcinogenicity. Moreover, much of the available evidence for genotoxicity comes primarily from in vitro systems that lack pharmacokinetic defenses, and often involve already transformed cell lines. These views appear to be in direct contrast to those expressed by McCarroll et al. (Citation2010). However, that review was published before the conduct of the studies described in this article. In support of a mutagenic MOA, McCarroll et al. (Citation2010) point to the evidence for Cr(VI) carcinogenicity in multiple tissues and in multiple species. However, in each species, oral exposure resulted in only one tumor location (NTP, Citation2008b). Interestingly, NTP reported increases in chromium in the livers of both species, suggesting that Cr(VI) is present in portal circulation at the high doses administered in the NTP study, yet no increased incidence of liver tumors were observed. McCarroll et al. also state that mutagenesis in somatic cells is supported by “MN induction in fetuses”. This finding appears to be based on the work of De Flora et al. (Citation2006), who showed that a single i.p. injection of 50 mg/kg sodium chromate to pregnant mice induced MN in fetuses; however, in the same study De Flora et al. also showed that exposure to 10 mg/l sodium dichromate in drinking water throughout pregnancy did not induce MN in fetuses (see ). We have now shown that ingestion of Cr(VI) in drinking water does not induce MN in the target tissue (i.e. duodenal crypts) – even at carcinogenic concentrations.
Potentially susceptible subpopulations
In fasting conditions, gastric pH is normally low (typically between 1 and 2), and therefore the pH-dependent reduction of Cr(VI) to Cr(III) in the stomach lumen occurs rapidly (Kirman et al., Citation2012). This suggests that individuals with higher gastric pH might reduce Cr(VI) less efficiently, and as a result, have a higher tissue dose of Cr(VI) in the small intestine. Individuals with altered gastric secretion (e.g. achlorhydria or hypochlorhydria) adminstered Cr(VI) had increased Cr(VI) absorption, presumably due to impaired gastric reduction of Cr(VI) to Cr(III) (Donaldson & Barreras, Citation1966). Individuals taking proton pump inhibitors (PPIs) have an intragastric pH range of 4–10. This range is higher than that in healthy adults, who have fasting gastric pH of ∼1.5 and postprandial gastric pH of ∼5 (Amendola et al., Citation2007; Dressman et al., Citation1990; Gardner et al., Citation2004). Infants less than four weeks old have fasting gastric pH values of 3–4 and a postprandial gastric pH of 7 (Nagita et al., Citation1996). By age 2 or 3 years, gastric production and gastric pH reach adult values (Koren, Citation1997; Nagita et al., Citation1996). Although these life-stages and conditions may be associated with greater susceptibility to Cr(VI) exposure relative to healthy adults, PBPK models can incorporate differences in toxicokinetics imparted by life stage as well as pharmaceutical usage.
Individuals with intestinal diseases might also be more sensitive to Cr(VI) exposure. For example, people with celiac disease are at risk for adenocarcinomas (Green & Cellier, Citation2007), in part due to increased oxidative stress (Stojiljkovic et al., Citation2012). Nevertheless, a recent meta analysis of Cr(VI) found no evidence for increased risk of cancers of the alimentary canal from exposure to Cr(VI) (Gatto et al., Citation2010).
Data gaps
As with all datasets, there are some gaps in our knowledge of the detailed sequence of events. However, these gaps represent more of a deficiency in the detailed mechanism of action as opposed to the more general MOA needed for human health risk assessment (US EPA, Citation2005a). For example, the ability to distinguish between Cr(III) and Cr(VI) in biological samples could greatly enhance detailed understanding of Cr(VI) toxicity; to date, however, these forms cannot be readily distinguished. In addition to measures of the GSH/GSSG ratio, alternative measures of oxidative status, such as cysteine/cystine and NAD+/NADH ratios, and lipid oxidation could be informative. Further, measures of Cr-DNA adducts that can be reliably measured in vivo and differentiated between crypt and villi enterocytes could be helpful.
With regard to in vivo mutation analysis, access to paraffin embedded tumor samples from the NTP 2-year bioassay would allow for DNA extraction and determination of the specific mutations, if any, that might be more prevalent in Cr(VI)-induced intestinal tumors. Such knowledge would provide a means for more refined focus in the mutation analysis conducted in shorter term assays; to date, however, such information has not been published. Some studies have shown epigenetic changes in DNA methylation following chromium exposure (Klein et al., Citation2002; Sun et al., Citation2009; Takahashi et al., Citation2005); thus assessment of genomic methylation status might also provide additional useful information on chromium toxicity. It is also unclear whether the tumors that arise following Cr(VI) exposure are due simply to expansion of preexisting initiated cells as a consequence of the constant proliferative pressure, or whether cells become initiated due to new mutations that arise from constant proliferative pressure. However, the MOA is not meaningfully altered either way.
One of the goals of the 90-day MOA studies was to collect similar data in both mice and rats in order to better understand the species differences in tumor development. The previous NTP studies did not observe diffuse hyperplasia or tumors in the intestines of rats (NTP, Citation2007, Citation2008b). In contrast, Thompson et al. (Citation2012c) reported that rats exhibited apoptosis in the villi and crypt hyperplasia at ≥170 mg/l SDD. The reason for this difference likely relates to water intake, which was nearly 2-fold higher in Thompson et al. (Citation2012c), but consistent with published intake values (US EPA, Citation1988). These findings indicate that with increased Cr(VI) exposure, rats begin to exhibit non-neoplastic lesions similar to those observed in mice, and suggest that dosimetry is an important factor in the intestinal carcinogenesis of Cr(VI).
Human relevance
Human relevance addresses three fundamental questions: (1) Is the WOE sufficient to establish the MOA in animals?, (2) Are the key events plausible in humans? and (3) Are the key events plausible in humans after accounting for pharmacokinetics and pharmacodynamics? (Boobis et al., Citation2006; Meek et al., Citation2003). In our initial MOA assessment (conducted in 2010), it was concluded that the WOE to establish the MOA in animals was not sufficient and that additional data were needed, in particular from the target tissues (Thompson et al., Citation2011a). As such, an effort was undertaken to gather data necessary to fill the data gaps identified. Based on findings summarized herein, we now conclude that the WOE is sufficient to establish the MOA in mice, which likely involves the key events described in . These key events are listed in , along with available evidence in mice, rats and humans. The evidence in rats is primarily from the 90-day and 2-year studies conducted by NTP; however, as described in the section “Data gaps”, rats with higher Cr(VI) intake than in the NTP studies did develop intestinal lesions in Thompson et al. (Citation2012c). These findings suggest that the MOA in mice might be applicable to rats under greater exposure scenarios. This also suggests that the MOA is relevant to humans provided that the dose is sufficient. This leads to the third of the aforementioned questions, i.e. Are the key events plausible in humans after accounting for pharmacokinetics and pharmacodynamics?
Table 10. Concordance table for human relevance.
Although the gastrointestinal tract is, in general, similar between rodents and humans, there are several factors that influence pharamacokinetics. Cr(VI) reduction rate is pH dependent, with greater Cr(VI) reduction capacity in fed than fasted conditions (Kirman et al., 2012; Proctor et al., Citation2012). The pH of the various sections of the gastrointestinal tract differs across tissues and species, as do feeding patterns and gastric motility (Kirman et al., Citation2012, accepted; McConnell et al., Citation2008). Thus, the only way to predict the pharmacokinetics of Cr(VI) with any certainty is through PBPK models. Recently developed PBPK models for Cr(VI) will be useful for quantifying pharmacokinetic differences of Cr(VI) across species (Kirman et al., Citation2012, accepted; Proctor et al., Citation2012). PBPK models can also be used to investigate potentially susceptible populations, such as individuals taking PPIs.
Equally important as pharmacokinetics is exposure. The Cr(VI) concentrations that induced tumors in mice (≥20 mg/l Cr(VI)) are three orders of magnitude higher than typical drinking water exposures, which average 0.001–0.005 mg Cr(VI)/l (AWWA, Citation2004; CDPH, Citation2011). Thus, human pharmacokinetics (or dynamics) would have to differ substantially from mice in order for typical exposures to pose a cancer risk. It should also be noted that rodents can synthesize their own ascorbate, whereas humans must acquire ascorbate in their diet (Linster & Van Schaftingen, 2007). It has been argued that intracellular ascorbate-mediated Cr(VI) reduction may be more deleterious than other forms of Cr(VI) reduction (Reynolds et al., Citation2012). If true, then the inability of humans to synthesize ascorbate may be “beneficial”; furthermore, diets rich in ascorbate imply that chromium present in the diet would more likely be in the reduced form, i.e. Cr(III), and less likely to be absorbed in the intestine. Importantly, one should not disregard the fact that there is little convincing epidemiological evidence that Cr(VI) increases risk of intestinal cancer among individuals exposed in the workplace (Gatto et al., Citation2010); nor is there evidence of elevated overall cancer rates among individuals exposed environmentally via groundwater (Morgan, Citation2011).
There are several human conditions that result in small intestinal damage. Villus blunting and crypt proliferation are a nonspecific reaction to various types of intestinal injury including viral, bacterial and parasitic intestinal infections (Greaves, Citation2007; Serra & Jani, Citation2006). Celiac disease is characterized by villus atrophy, crypt hyperplasia and infiltrations into the lamina propria. This pathology is due, in part, to inflammatory responses to gluten in the diet, which leads to inflammation, oxidative stress, villus atrophy and crypt hyperplasia. Individuals with inflammatory diseases of the small intestine such as celiac disease are at risk for adenocarcinomas (Kumar et al., Citation2005). Nevertheless, small intestinal cancer is a relatively rare disease (DeVita et al., Citation2011; Talley, Citation2010). Although there is widespread exposure to low levels of Cr(VI) in drinking water, it seems unlikely that low levels of Cr(VI) pose an additional risk for intestinal cancer in patients with intestinal diseases.
Conclusion
MOA analysis is a key element in human health risk assessment (Boobis et al., Citation2006, Citation2009; Meek et al., Citation2003; Seed et al., Citation2005; US EPA, 2005a). When a general MOA has been well established, it “lowers the barrier” of acceptance when such an MOA is proposed for another chemical/tumor site of interest (Boobis et al., Citation2006). In this regard, Cr(VI) shares similar toxicological and carcinogenic characteristics as captan and folpet, which have previously been determined to act via a cytotoxic nonmutagenic MOA (Arce et al., Citation2010; Cohen et al., Citation2010; Gordon, Citation2007; US EPA, Citation2004). The relative rarity of intestinal tumors means that risk assessors have had comparatively less experience analyzing MOAs for carcinogenesis of the small intestine. In fact, 80–90% of cancers observed in rodent bioassays occur in eight tissues/systems, and the small intestine is not among these (Gold et al., Citation2001). Even though Cr(VI) and captan/folpet do not share structural similarity, their toxicological and carcinogenic phenotypes begin to establish a general MOA for intestinal neoplasms induced by environmental chemicals that involves chronic wounding and healing of the intestinal mucosa.
The newly developed target tissue-specific mechanistic data provide a substantially stronger and more scientifically robust basis for assessing the MOA for Cr(VI)-induced intestinal carcinogenesis than has been available previously. The WOE from these studies strongly supports that Cr(VI) acts by a cytotoxic MOA in the development of rodent small intestinal tumors. It is important to recognize that this MOA is specific to the small intestine, and although it may be informative to other tissues, it is not necessarily applicable to all tumors caused by Cr(VI). Chemicals may act by multiple MOAs, at different tumor sites and potentially at different doses (US EPA, 2005a); however, the MOA that is specific to the tumor response used for risk assessment is critical for low-dose extrapolation. Because mouse small intestinal cancers have been used as the basis of a number of quantitative risk assessments for ingested Cr(VI) (McCarroll et al., Citation2010; OEHHA, Citation2011; Stern, Citation2010; US EPA, Citation2010), these MOA findings specifically inform the most appropriate approach for low-dose extrapolations for these and other risk assessments on oral exposure to Cr(VI).
Declaration of interest
The authors employment affiliation is as shown on the cover page. Both ToxStrategies and Summit Toxicology are private consulting firms providing services to private and public organizations on toxicology and risk assessment issues. The authors [CT, MH, DP, CK, LH] have presented study findings in meetings with regulators including public meetings on behalf of the Cr(VI) Panel of the American Chemistry Council (ACC). DP has also been an expert in litigation involving Cr(VI), which was unrelated to this research or ACC.
This work was supported by the Cr(VI) Panel of the ACC. ACC had no role in study design, data collection and analysis, decision to publish or preparation of any manuscript. The funders were given the opportunity to review the draft study design as it went through an external peer review process and draft manuscripts at the time of external peer review. The purpose of such review was to allow input on the clarity of the science presented but not in interpretation of the research findings. The contents of this review reflect solely the view of the authors.
Acknowledgements
The authors would like to thank the Toxicology Excellence for Risk Assessment (TERA) Expert Panel for overseeing the Cr(VI) MOA Research Program. The panel report is available at http://www.tera.org/Peer/Chromium/Chromium.htm.
Notes
1Calculation: (0.59 mg/kg × 0.0225 kg ÷ 0.00005 l) = 265 mg/l.
2These increases were on the order of 1.5- to 2-fold.
References
- Alexander J, Aaseth, J. (1995). Uptake of chromate in human red blood cells and isolated rat liver cells: the role of the anion carrier. Analyst, 120, 931–3
- Amendola R, Santos MC, Massa JM, et al. (2007). Effects of a single low dose of ranitidine and effervescent antacids on intragastric acidity in healthy volunteers. Acta Gastroenterol Latinoam, 37, 231–7
- Ames BN, McCann J, Yamasaki E. (1975). Methods for detecting carcinogens and mutagens with the Salmonella/mammalian-microsome mutagenicity test. Mutat Res, 31, 347–64
- Ames BN, Shigenaga MK, Gold LS. (1993). DNA lesions, inducible DNA repair, and cell division: three key factors in mutagenesis and carcinogenesis. Environ Health Perspect, 101, 35–44
- Anderson RA. (2000). Chromium in the prevention and control of diabetes. Diabetes Metab, 26, 22–7
- Arce GT, Gordon EB, Cohen SM, Singh, P. (2010). Genetic toxicology of folpet and captan. Crit Rev Toxicol, 40, 546–74
- AWWA. (2004). Occurrence survey of boron and hexavalent chromium. Denver (CO): American Water Works Association Research Foundation
- Azad N, Iyer AK, Wang L, et al. (2010). Nitric oxide-mediated bcl-2 stabilization potentiates malignant transformation of human lung epithelial cells. Am J Respir Cell Mol Biol, 42, 578–85
- Azzolini C, Fiorani M, Guidarelli A, Cantoni O. (2011). Studies with low micromolar levels of ascorbic and dehydroascorbic acid fail to unravel a preferential route for vitamin C uptake and accumulation in U937 cells. Br J Nutr, 107, 691–6
- Bagchi D, Balmoori J, Bagchi M, et al. (2002). Comparative effects of TCDD, endrin, naphthalene and chromium (VI) on oxidative stress and tissue damage in the liver and brain tissues of mice. Toxicology. Comp Biochem Physiol C Pharmacol Toxicol Endocrinol, 175, 73–82
- Bagchi D, Hassoun EA, Bagchi M, et al. (1995a). Oxidative stress induced by chronic administration of sodium dichromate [Cr(VI)] to rats. Comp Biochem Physiol C Pharmacol Toxicol Endocrinol, 110, 281–7
- Bagchi D, Hassoun EA, Bagchi M, et al. (1995b). Chromium-induced excretion of urinary lipid metabolites, DNA damage, nitric oxide production, and generation of reactive oxygen species in Sprague-Dawley rats. Comp Biochem Physiol C Pharmacol Toxicol Endocrinol, 110, 177–87
- Bagchi D, Vuchetich PJ, Bagchi M, et al. (1997). Induction of oxidative stress by chronic administration of sodium dichromate [chromium VI] and cadmium chloride [cadmium II] to rats. Free Radic Biol Med, 22, 471–8
- Barker N, Ridgway RA, van Es JH, et al. (2009). Crypt stem cells as the cells-of-origin of intestinal cancer. Nature, 457, 608–11
- Beaver LM, Stemmy EJ, Schwartz AM, et al. (2009). Lung inflammation, injury, and proliferative response after repetitive particulate hexavalent chromium exposure. Environ Health Perspect, 117, 1896–902
- Bennicelli C, Camoirano, A, Petruzzelli S, et al. (1983). High sensitivity of Salmonella TA102 in detecting hexavalent chromium mutagenicity and its reversal by liver and lung preparations. Mutat Res, 122, 1–5
- Berlanga-Acosta J, Playford RJ, Mandir N, Goodlad RA. (2001). Gastrointestinal cell proliferation and crypt fission are separate but complementary means of increasing tissue mass following infusion of epidermal growth factor in rats. Gut, 48, 803–7
- Bernard B, Gordon E. (2000). An evaluation of the common mechanism approach to the food quality protection act: captan and four related fungicides, a practical example. Int J Toxicol, 19, 43–61
- Blakey DH, Duncan AM, Wargovich MJ, et al. (1985). Detection of nuclear anomalies in the colonic epithelium of the mouse. Cancer Res, 45, 242–9
- Bonner WM, Redon CE, Dickey JS, et al. (2008). GammaH2AX and cancer. Nat Rev Cancer, 8, 957–67
- Boobis AR, Cohen SM, Dellarco V, et al. (2006). IPCS framework for analyzing the relevance of a cancer mode of action for humans. Crit Rev Toxicol, 36, 781–92
- Boobis AR, Doe JE, Heinrich-Hirsch B, et al. (2008). IPCS framework for analyzing the relevance of a cancer mode of action for humans. Crit Rev Toxicol, 38, 87–96
- Boobis AR, Daston GP, Preston RJ, Olin SS. (2009). Application of key events analysis to chemical carcinogens and noncarcinogens. Crit Rev Food Sci Nutr, 49, 690–707
- Buttner B, Beyersmann D. (1985). Modification of the erythrocyte anion carrier by chromate. Xenobiotica, 15, 735–41
- Campbell CE, Gravel RA, Worton RG. (1981). Isolation and characterization of Chinese hamster cell mutants resistant to the cytotoxic effects of chromate. Somatic Cell Genet, 7, 535–46
- Castellano E, Santos E. (2011). Functional specificity of ras isoforms: so similar but so different. Genes Cancer, 2, 216–31
- CDPH. (2011). Chromium-6 in drinking water: an overview of sampling results. California: California Department of Public Health
- Celik A, Mazmanci B, Camlica Y, et al. (2005). Induction of micronuclei by lambda-cyhalothrin in Wistar rat bone marrow and gut epithelial cells. Mutagenesis, 20, 125–9
- Chandra SA, Nolan MW, Malarkey DE. (2010). Chemical carcinogenesis of the gastrointestinal tract in rodents: an overview with emphasis on NTP carcinogenesis bioassays. Toxicol Pathol, 38, 188–97
- Chen Q, Espey MG, Krishna MC, et al. (2005). Pharmacologic ascorbic acid concentrations selectively kill cancer cells: action as a pro-drug to deliver hydrogen peroxide to tissues. Proc Natl Acad Sci USA, 102, 13604–9
- Cheng L, Sonntag DM, de Boer J, Dixon, K. (2000). Chromium(VI)-induced mutagenesis in the lungs of big blue transgenic mice. J Environ Pathol Toxicol Oncol, 19, 239–49
- Chidiac P, Goldberg MT. (1987). Lack of induction of nuclear aberrations by captan in mouse duodenum. Environ Mutagen, 9, 297–306
- Chiu A, Shi XL, Lee WK, et al. (2010). Review of chromium (VI) apoptosis, cell-cycle-arrest, and carcinogenesis. J Environ Sci Health. Part C, Environ Carcinog Ecotoxicol Rev, 28, 188–230
- Circu ML, Aw TY. (2011). Redox biology of the intestine. Free Radic Res, 45, 1245–66
- Cohen SM. (2010). An enhanced thirteen-week bioassay as an alternative for screening for carcinogenesis factors. Asian Pac J Cancer Prev, 11, 15–7
- Cohen SM, Gordon EB, Singh P, et al. (2010). Carcinogenic mode of action of folpet in mice and evaluation of its relevance to humans. Crit Rev Toxicol, 40, 531–45
- Collins BJ, Stout MD, Levine KE, et al. (2010). Exposure to hexavalent chromium resulted in significantly higher tissue chromium burden compared with trivalent chromium following similar oral doses to male F344/N rats and female B6C3F1 mice. Toxicol Sci, 118, 368–79
- Coogan TP, Motz J, Snyder CA, et al. (1991). Differential DNA-protein crosslinking in lymphocytes and liver following chronic drinking water exposure of rats to potassium chromate. Toxicol Appl Pharmacol, 109, 60–72
- Crott JW, Fenech M. (1999). Effect of vitamin C supplementation on chromosome damage, apoptosis and necrosis ex vivo. Carcinogenesis, 20, 1035–41
- Cupo DY, Wetterhahn KE. (1985). Binding of chromium to chromatin and DNA from liver and kidney of rats treated with sodium dichromate and chromium(III) chloride in vivo. Cancer Res, 45, 1146–51
- Dahm LJ, Jones DP. (2000). Rat jejunum controls luminal thiol-disulfide redox. J Nutr, 130, 2739–45
- Dana Devi K, Rozati R, Saleha Banu B, et al. (2001). In vivo genotoxic effect of potassium dichromate in mice leukocytes using comet assay. Food Chem Toxicol, 39, 859–65
- De Flora S. (1978). Metabolic deactivation of mutagens in the Salmonella-microsome test. Nature, 271, 455–6
- De Flora S. (2000). Threshold mechanisms and site specificity in chromium(VI) carcinogenesis. Carcinogenesis, 21, 533–41
- De Flora S, Badolati GS, Serra D, et al. (1987). Circadian reduction of chromium in the gastric environment. Mutat Res, 192, 169–74
- De Flora S, Bagnasco M, Serra D, Zanacchi, P. (1990). Genotoxicity of chromium compounds. A review. Mutat Res, 238, 99–172
- De Flora S, Bianchi V, Levis AG. (1984). Distinctive mechanisms for interaction of hexavalent and trivalent chromium with DNA? Toxicol Environ Chem, 8, 287–94
- De Flora S, Camoirano A, Bagnasco M, et al. (1997). Estimates of the chromium(VI) reducing capacity in human body compartments as a mechanism for attenuating its potential toxicity and carcinogenicity. Carcinogenesis, 18, 531–7
- De Flora S, D’Agostini F, Balansky R, et al. (2008). Lack of genotoxic effects in hematopoietic and gastrointestinal cells of mice receiving chromium(VI) with the drinking water. Mutat Res, 659, 60–7
- De Flora S, Iltcheva M, Balansky RM. (2006). Oral chromium(VI) does not affect the frequency of micronuclei in hematopoietic cells of adult mice and of transplacentally exposed fetuses. Mutat Res, 610, 38–47
- De Flora S, Morelli A, Basso C, et al. (1985). Prominent role of DT-diaphorase as a cellular mechanism reducing chromium(VI) and reverting its mutagenicity. Cancer Res, 45, 3188–96
- DeSesso JM, Jacobson CF. (2001). Anatomical and physiological parameters affecting gastrointestinal absorption in humans and rats. Food Chem Toxicol, 39, 209–28
- DeVita VT, Lawrence TS, Rosenberg SA. (2011). DeVita, Hellman, and Rosenberg's cancer: principles and practice of oncology. 9th ed. Philadelphia (PA): Lippincott Williams & Wilkins
- Di Bona, KR, Love S, Rhodes NR, et al. (2011). Chromium is not an essential trace element for mammals: effects of a “low-chromium” diet. J Biol Inorg Chem, 16, 381–90
- Donaldson RM Jr, Barreras RF. (1966). Intestinal absorption of trace quantities of chromium. J Lab Clin Med, 68, 484–93
- Dressman JB, Berardi RR, Dermentzoglou LC, et al. (1990). Upper gastrointestinal (GI) pH in young, healthy men and women. Pharm Res, 7, 756–61
- Duncan AM, Heddle JA, Blakey DH. (1985). Mechanism of induction of nuclear anomalies by gamma-radiation in the colonic epithelium of the mouse. Cancer Res, 45, 250–2
- Ellinger-Ziegelbauer H, Stuart B, Wahle B, et al. (2004). Characteristic expression profiles induced by genotoxic carcinogens in rat liver. Toxicol Sci, 77, 19–34
- Ellinger-Ziegelbauer H, Stuart B, Wahle B, et al. (2005). Comparison of the expression profiles induced by genotoxic and nongenotoxic carcinogens in rat liver. Mutat Res, 575, 61–84
- Farkas B, Boldizsar F, Tarjanyi O, et al. (2009). BALB/c mice genetically susceptible to proteoglycan-induced arthritis and spondylitis show colony-dependent differences in disease penetrance. Arthritis Res Ther, 11, R21
- Febel H, Szegedi B, Huszar S. (2001). Absorption of inorganic, trivalent and hexavalent chromium following oral and intrajejunal doses in rats. Acta Vet Hung, 49, 203–9
- Fenech M, Kirsch-Volders M, Natarajan AT, et al. (2011). Molecular mechanisms of micronucleus, nucleoplasmic bridge and nuclear bud formation in mammalian and human cells. Mutagenesis, 26, 125–32
- Feng Y, Bommer GT, Zhao J, et al. (2011). Mutant KRAS promotes hyperplasia and alters differentiation in the colon epithelium but does not expand the presumptive stem cell pool. Gastroenterology, 141, 1003–1013, e1–10
- Forbes SA, Tang G, Bindal N, et al. (2010). COSMIC (the Catalogue of Somatic Mutations in Cancer): a resource to investigate acquired mutations in human cancer. Nucleic Acids Res, 38, D652–7
- Franco R, Cidlowski JA. (2009). Apoptosis and glutathione: beyond an antioxidant. Cell Death Differ, 16, 1303–14
- Gardner JD, Sloan S, Robinson M, Miner PB Jr. (2004). Frequency analyses of gastric pH in control and gastro-oesophageal reflux disease subjects treated with a proton-pump inhibitor. Aliment Pharmacol Ther, 20, 1381–6
- Gatto NM, Kelsh MA, Mai DH, et al. (2010). Occupational exposure to hexavalent chromium and cancers of the gastrointestinal tract: a meta-analysis. Cancer Epidemiol, 34, 388–99
- Gaylor DW. (2005). Are tumor incidence rates from chronic bioassays telling us what we need to know about carcinogens? Regul Toxicol Pharmacol, 41, 128–33
- George MD, Wehkamp J, Kays RJ, et al. (2008). In vivo gene expression profiling of human intestinal epithelial cells: analysis by laser microdissection of formalin fixed tissues. BMC Genomics, 9, 209
- Gibb HJ, Lees PS, Pinsky PF, Rooney, BC. (2000). Clinical findings of irritation among chromium chemical production workers. Am J Ind Med, 38, 127–31
- Glaser U, Hochrainer D, Kloppel H, Oldiges H. (1986). Carcinogenicity of sodium dichromate and chromium (VI/III) oxide aerosols inhaled by male Wistar rats. Toxicology, 42, 219–32
- Go YM, Jones DP. (2010). Redox control systems in the nucleus: mechanisms and functions. Antioxid Redox Signal, 13, 489–509
- Gold, LS, Manley NB, Slone TH, Ward JM. (2001). Compendium of chemical carcinogens by target organ: results of chronic bioassays in rats, mice, hamsters, dogs, and monkeys. Toxicol Pathol, 29, 639–52
- Goldberg MT, Blakey DH, Bruce WR. (1983). Comparison of the effects of 1,2–dimethylhydrazine and cyclophosphamide on micronucleus incidence in bone marrow and colon. Mutat Res, 109, 91–8
- Gordon E. (2007). Captan: transition from ‘B2’ to 'not likely’. How pesticide registrants affected the EPA Cancer Classification Update. J Appl Toxicol, 27, 519–26
- Gottschling BC, Maronpot RR, Hailey JR, et al. (2001). The role of oxidative stress in indium phosphide-induced lung carcinogenesis in rats. Toxicol Sci, 64, 28–40
- Grady WM, Carethers JM. (2008). Genomic and epigenetic instability in colorectal cancer pathogenesis. Gastroenterology, 135, 1079–99
- Greaves P. (2007). Histopathology of preclinical toxicity studies. 3rd ed. London: Elsevier-Academic Press
- Green PH, Cellier C. (2007). Celiac disease. N Engl J Med, 357, 1731–43
- Hagen TM, Wierzbicka GT, Bowman BB, et al. (1990). Fate of dietary glutathione: disposition in the gastrointestinal tract. Am J Physiol, 259, G530–5
- Haney JT, Erraguntla N, Sielken RL, Valdez-Flores C. (2012). Development of a cancer-based chronic inhalation reference value for hexavalent chromium based on a nonlinear-threshold carcinogenic assessment. Regul Toxicol Pharmacol, 64, 466–80
- Harris MA, Thompson CM, Wolf JC, et al. (2012). Assessment of genotoxic potential of Cr(VI) in the intestine via in vivo intestinal micronucleus assay and in vitro high content analysis in differentiated and undifferentiated Caco-2. Society of Toxicology. San Francisco, CA
- Henics T, Wheatley DN. (1999). Cytoplasmic vacuolation, adaptation and cell death: a view on new perspectives and features. Biol Cell, 91, 485–98
- Hirose T, Kondo K, Takahashi Y, et al. (2002). Frequent microsatellite instability in lung cancer from chromate-exposed workers. Mol Carcinog, 33, 172–80
- Holmes AL, Wise SS, Wise JP Sr. (2008). Carcinogenicity of hexavalent chromium. Indian J Med Res, 128, 353–72
- Howard WR, Hoffman SA, Kochhar TS. (1992). Enhanced production of micronuclei by hexavalent chromium in cultured CHO cells. Bull Environ Contam Toxicol, 49, 535–40
- IARC. (1990). Chromium, nickel and welding, IARC Monogr. Eval Carcinog Risks Hum, 49, 1–648
- Itoh S, Shimada H. (1998). Bone marrow and liver mutagenesis in lacZ transgenic mice treated with hexavalent chromium. Mutat Res, 412, 63–7
- Itzkovitz S, Lyubimova A, Blat IC, et al. (2012). Single-molecule transcript counting of stem-cell markers in the mouse intestine. Nat Cell Biol, 14, 106–14
- Jennette KW. (1979). Chromate metabolism in liver microsomes. Biol Trace Elem Res, 1, 55–62
- John LJ, Fromm M, Schulzke JD. (2011). Epithelial barriers in intestinal inflammation. Antioxid Redox Signal, 15, 1255–70
- Joseph P, He Q, Umbright C. (2008). Heme-oxygenase 1 gene expression is a marker for hexavalent chromium-induced stress and toxicity in human dermal fibroblasts. Toxicol Sci, 103, 325–34
- Jung KA, Kwak MK. (2010). The Nrf2 system as a potential target for the development of indirect antioxidants. Molecules, 15, 7266–91
- Kargacin B, Squibb KS, Cosentino S, et al. (1992). Comparison of the uptake and distribution of chromate in rats and mice. Biol Trace Elem Res, 36, 307–318
- Katz SA, Salem H. (1993). The toxicology of chromium with respect to its chemical speciation: a review. J Appl Toxicol, 13, 217–24
- Kerger BD, Paustenbach DJ, Corbett GE, Finley BL. (1996). Absorption and elimination of trivalent and hexavalent chromium in humans following ingestion of a bolus dose in drinking water. Toxicol Appl Pharmacol, 141, 145–58
- Kim J, Cha YN, Surh YJ. (2010). A protective role of nuclear factor-erythroid 2-related factor-2 (Nrf2) in inflammatory disorders. Mutat Res, 690, 12–23
- Kinner A, Wu W, Staudt C, Iliakis G. (2008). Gamma-H2AX in recognition and signaling of DNA double-strand breaks in the context of chromatin. Nucleic Acids Res, 36, 5678–94
- Kirman CR, Aylward LL, Suh M, et al. (accepted). Physiologically based pharmacokinetic model for humans orally exposed to chromium. Chem Biol Interact
- Kirman CR, Hays SM, Aylward LL, et al. (2012). Physiologically based pharmacokinetic model for rats and mice orally exposed to chromium. Chem Biol Interact, 200, 45–64
- Kirsch-Volders M, Elhajouji A, Cundari E, Van Hummelen P. (1997). The in vitro micronucleus test: a multi-endpoint assay to detect simultaneously mitotic delay, apoptosis, chromosome breakage, chromosome loss and non-disjunction. Mutat Res, 392, 19–30
- Kirsch-Volders M, Plas G, Elhajouji A, et al. (2011). The in vitro MN assay in 2011: origin and fate, biological significance, protocols, high throughput methodologies and toxicological relevance. Arch Toxicol, 85, 873–99
- Klaunig JE, Kamendulis LM. (2008). Chemical carcinogens (C. D. Klaassen). 7th ed. New York (NY): McGraw-Hill
- Klaunig JE, Kamendulis LM, Hocevar BA. (2010). Oxidative stress and oxidative damage in carcinogenesis. Toxicol Pathol, 38, 96–109
- Klein CB, Su L, Bowser D, Leszczynska J. (2002). Chromate-induced epimutations in mammalian cells. Environ Health Perspect, 110, 739–43
- Knudsen I. (1980). The mammalian spot test and its use for the testing of potential carcinogenicity of welding fume particles and hexavalent chromium. Acta Pharmacol Toxicol (Copenh), 47, 66–70
- Kondo K, Hino N, Sasa M, et al. (1997). Mutations of the p53 gene in human lung cancer from chromate-exposed workers. Biochem Biophys Res Commun, 239, 95–100
- Kopec AK, Kim S, Forgacs AL, et al. (2012a). Genome-wide gene expression effects in B6C3F1 mouse intestinal epithelia following 7 and 90 days of exposure to hexavalent chromium in drinking water. Toxicol Appl Pharmacol, 259, 13–26
- Kopec AK, Thompson CM, Kim S, et al. (2012b). Comparative toxicogenomic analysis of oral Cr(VI) exposure effects in rat and mouse small intestinal epithelia. Toxicol Appl Pharmacol, 262, 124–38
- Koren G. (1997). Therapeutic drug monitoring principles in the neonate. National Academy of Clinical Biochemistry. Clin Chem, 43, 222–7
- Krishnaja AP, Sharma NK. (2003). Ascorbic acid potentiates mitomycin C-induced micronuclei and sister chromatid exchanges in human peripheral blood lymphocytes in vitro. Teratog Carcinog Mutagen, Suppl 1, 99–112
- Kruidenier L, Verspaget HW. (2002). Review article: oxidative stress as a pathogenic factor in inflammatory bowel disease–radicals or ridiculous? Aliment Pharmacol Ther, 16, 1997–2015
- Kumar V, Abbas A, Fausto N. (2005). Robins and cotran pathological basis of disease. 7th ed. Philadelphia (PA): Elsevier Saunders
- Le Hegarat L, Jacquin AG, Bazin E, Fessard V. (2006). Genotoxicity of the marine toxin okadaic acid, in human Caco-2 cells and in mice gut cells. Environ Toxicol, 21, 55–64
- Levin DE, Hollstein M, Christman MF, et al. (1982). A new Salmonella tester strain (TA102) with A X T base pairs at the site of mutation detects oxidative mutagens. Proc Natl Acad Sci USA, 79, 7445–9
- Levina A, Lay PA. (2008). Chemical properties and toxicity of chromium(III) nutritional supplements. Chem Res Toxicol, 21, 563–71
- Lipkin M, Deschner E. (1976). Early proliferative changes in intestinal cells. Cancer Res, 36, 2665–8
- Lister CL, Van Schaftingen E. (2007). Vitamin C. Biosynthesis, recycling and degradation in mammals. FEBS J, 274, 1–22
- Liu KJ, Shi X. (2001). In vivo reduction of chromium (VI) and its related free radical generation. Mol Cell Biochem, 222, 41–7
- Long LH, Halliwell B. (2012). The effects of oxaloacetate on hydrogen peroxed generation from ascorbate and epigallocatechin gallate in cell culture media: potential for altering cell metabolism. Biochem Biophys Res Commun, 417, 446–450
- Lu YY, Yang JL. (1995). Long-term exposure to chromium(VI) oxide leads to defects in sulfate transport system in Chinese hamster ovary cells. J Cell Biochem, 57, 655–65
- Luippold RS, Mundt KA, Austin RP, et al. (2003). Lung cancer mortality among chromate production workers. Occup Environ Med, 60, 451–7
- Luippold RS, Mundt KA, Dell LD, Birk T. (2005). Low-level hexavalent chromium exposure and rate of mortality among US chromate production employees. J Occup Environ Med, 47, 381–5
- Luongo C, Moser AR, Gledhill S, Dove WF. (1994). Loss of Apc+ in intestinal adenomas from Min mice. Cancer Res, 54, 5947–52
- Machle W, Gregorius F. (1948). Cancer of the respiratory system in the United States chromate producing industry. Public Health Rep, 63, 1114–27
- Markovich D. (2001). Physiological roles and regulation of mammalian sulfate transporters. Physiol Rev, 81, 1499–533
- Marnett LJ. (2000). Oxyradicals and DNA damage. Carcinogenesis, 21, 361–70
- Martensson J, Jain A, Meister A. (1990). Glutathione is required for intestinal function. Proc Natl Acad Sci USA, 87, 1715–9
- Martensson J, Meister A. (1992). Glutathione deficiency increases hepatic ascorbic acid synthesis in adult mice. Proc Natl Acad Sci USA, 89, 11566–8
- McCann J, Choi E, Yamasaki E, Ames BN. (1975). Detection of carcinogens as mutagens in the Salmonella/microsome test: assay of 300 chemicals. Proc Natl Acad Sci USA, 72, 5135–9
- McCarroll N, Keshava N, Chen J, et al. (2010). An evaluation of the mode of action framework for mutagenic carcinogens case study II: chromium (VI). Environ Mol Mutagen, 51, 89–111
- McConnell EL, Basit AW, Murdan S. (2008). Measurements of rat and mouse gastrointestinal pH, fluid and lymphoid tissue, and implications for in-vivo experiments. J Pharm Pharmacol, 60, 63–70
- Meek ME, Bucher JR, Cohen SM, et al. (2003). A framework for human relevance analysis of information on carcinogenic modes of action. Crit Rev Toxicol, 33, 591–653
- Meek ME, Klaunig JE. (2010). Proposed mode of action of benzene-induced leukemia: interpreting available data and identifying critical data gaps for risk assessment. Chem Biol Interact, 184, 279–85
- Meister A, Anderson ME. (1983). Glutathione. Annu Rev Biochem, 52, 711–760
- Meng F, Bermudez E, McKinzie PB, et al. (2010a). Measurement of tumor-associated mutations in the nasal mucosa of rats exposed to varying doses of formaldehyde. Regul Toxicol. Pharmacol 57, 274--83
- Meng F, Knapp GW, Green T, et al. (2010b). K-Ras mutant fraction in A/J mouse lung increases as a function of benzo[a]pyrene dose. Environ Mol Mutagen, 51, 146–55
- Meunier V, Bourrie M, Berger Y, Fabre G. (1995). The human intestinal epithelial cell line Caco-2; pharmacological and pharmacokinetic applications. Cell Biol Toxicol, 11, 187–94
- Miller JB. (1953). Effect of chromates on nose, throat, and ear. AMA Arch Otolaryngol, 58, 172–8
- Mimnaugh EG, Xu W, Vos M, et al. (2006). Endoplasmic reticulum vacuolization and valosin-containing protein relocalization result from simultaneous hsp90 inhibition by geldanamycin and proteasome inhibition by velcade. Mol Cancer Res, 4, 667–81
- Mirsalis JC, Hamilton CM, O’Loughlin KG, et al. (1996). Chromium (VI) at plausible drinking water concentrations is not genotoxic in the in vivo bone marrow micronucleus or liver unscheduled DNA synthesis assays. Environ Mol Mutagen, 28, 60–3
- Morgan JW. (2011). Preliminary assessment of cancer occurrence in the Hinkley Census Tract, 1996–2008. Loma Linda (CA): California Cancer Registry, Loma Linda University Medical Center
- Moriarty-Craige SE, Jones DP. (2004). Extracellular thiols and thiol/disulfide redox in metabolism. Annu Rev Nutr, 24, 481–509
- Morita T, MacGregor JT, Hayashi M. (2011). Micronucleus assays in rodent tissues other than bone marrow. Mutagenesis, 26, 223–30
- Nagita A, Amemoto K, Yoden A, et al. (1996). Diurnal variation in intragastric pH in children with and without peptic ulcers. Pediatr Res, 40, 528–32
- Natoli M, Leoni BD, D’Agnano I, et al. (2011). Cell growing density affects the structural and functional properties of Caco-2 differentiated monolayer. J Cell Physiol, 226, 1531–43
- NCI. (1977). Bioassay of Captan for Possible Carcinogenicity, CAS No. 133-06-2. National Cancer Institute CARCINOGENESIS Technical Report Series 15
- Neal MD, Richardson WM, Sodhi CP, et al. (2011). Intestinal stem cells and their roles during mucosal injury and repair. J Surg Res, 167, 1–8
- Nickens KP, Patierno SR, Ceryak S. (2010). Chromium genotoxicity: a double-edged sword. Chem Biol Interact, 188, 276–88
- Nishiyama K, Yao T, Yonemasu H, et al. (2002). Overexpression of p53 protein and point mutation of K-ras genes in primary carcinoma of the small intestine. Oncol Rep, 9, 293–300
- NTP (2001). NTP technical report on the toxicology and carcinogenesis studies of indium phosphide (CAS No. 22398-80-7) in F344/N rats and B6C3F1 mice (inhalation studies), NTP TR 499. NIH Publication No. 01-4433
- NTP (2002). NTP technical report on the toxicology and carcinogenesis studies of o-nitrotoluene (CAS No. 88-72-2) in F344/N rats and B6C3F1 mice (feed studies), NTP TR 504. NIH Publication No. 02-4438
- NTP (2007). National Toxicology Program technical report on the toxicity studies of sodium dichromate dihydrate (CAS No. 7789-12-0) administered in drinking water to male and female F344/N rats and B6C3F1 mice and male BALB/c and am3-C57BL/6 mice. NTP Toxicity Report Series Number 72, NIH Publication No. 07-5964
- NTP (2008a). National Toxicology Program technical report on the toxicology and carcinogenesis studies of chromium picolinate monohydrate (CAS NO. 27882-76-4) in F344/N rats and B6C3F1 mice (feed studies). NIH Publication No. 08-5897
- NTP (2008b). National Toxicology Program technical report on the toxicology and carcinogenesis studies of sodium dichromate dihydrate (CAS No. 7789-12-0) in F344/N rats and B6C3F1 mice (drinking water studies), NTP TR 546. NIH Publication No. 08-5887
- O’Brien TJ, Ceryak S, Patierno SR. (2003). Complexities of chromium carcinogenesis: role of cellular response, repair and recovery mechanisms. Mutat Res, 533, 3–36
- O’Brien TJ, Ding H, Suh M, et al. (2012). K-Ras codon 12 GGT to GAT mutation is not elevated in the duodenum of mice subchronically exposed to hexavalent chromium in drinking water. Presented at the Society of Toxicology, 2012 March 11–15. San Francisco, CA
- O’Brien TJ, Witcher P, Brooks B, Patierno SR. (2009). DNA polymerase zeta is essential for hexavalent chromium-induced mutagenesis. Mutat Res, 663, 77–83
- OECD (1997). OECD Guideline for the testing of chemicals: bacterial reverse mutation test (#471). Organisation for Economic Co-operation and Development. OECD Publishing
- OECD (2010). OECD Guideline for the testing of chemicals: in vitro mammalian cell micronucleus test (#487). 3rd ed. Organisation for Economic Co-operation and Development. OECD Publishing
- OEHHA (2011). Final technical support document on public health goal for hexavalent chromium in drinking water. Oakland (CA): Pesticide and Environmental Toxicology Branch, Office of Environmental Health Hazard Assessment, California Environmental Protection Agency
- Osburn WO, Karim B, Dolan PM, et al. (2007). Increased colonic inflammatory injury and formation of aberrant crypt foci in Nrf2-deficient mice upon dextran sulfate treatment. Int J Cancer, 121, 1883–91
- Park RM, Bena JF, Stayner LT, et al. (2004). Hexavalent chromium and lung cancer in the chromate industry: a quantitative risk assessment. Risk Anal, 24, 1099–108
- Parry EM, Parry JM, Corso C, et al. (2002). Detection and characterization of mechanisms of action of aneugenic chemicals. Mutagenesis, 17, 509–21
- Parsons BL, Manjanath MB, Myers MB, et al. (2012). Induction of cII and K-Ras mutation in lung DNA of Big Blue mice exposed to ethylene oxide by inhalation. Bellevue (WA): Environmental Mutagens Society
- Parsons BL, Myers MB, Meng F, et al. (2010). Oncomutations as biomarkers of cancer risk. Environ Mol Mutagen, 51, 836–50
- Pastides H, Austin R, Lemeshow S, et al. (1994). A retrospective-cohort study of occupational exposure to hexavalent chromium. Am J Ind Med, 25, 663–75
- Percy AJ, Moore, N, Chipman, JK. (1989). Formation of nuclear anomalies in rat intestine by benzidine and its biliary metabolites. Toxicology, 57, 217–23
- Peterson-Roth, E, Reynolds M, Quievryn G, Zhitkovich A. (2005). Mismatch repair proteins are activators of toxic responses to chromium-DNA damage. Mol Cell Biol, 25, 3596–607
- Petrilli FL, De Flora S. (1978a). Metabolic deactivation of hexavalent chromium mutagenicity. Mutat Res, 54, 139–47
- Petrilli FL, De Flora S. (1978b). Oxidation of inactive trivalent chromium to the mutagenic hexavalent form. Mutat Res, 58, 167–73
- Petrilli FL, De Flora S. (1982). Interpretations on chromium mutagenicity and carcinogenicity. Prog Clin Biol Res, 109, 453–64
- Potten CS, Booth C, Pritchard DM. (1997). The intestinal epithelial stem cell: the mucosal governor. Int J Exp Pathol, 78, 219–43
- Potten CS, Loeffler M. (1990). Stem cells: attributes, cycles, spirals, pitfalls and uncertainties. Lessons for and from the crypt. Development, 110, 1001–20
- Pozharisski KM, Kapustin YM, Likhachev AJ, Shaposhnikov JD. (1975). The mechanism of carcinogenic action of 1,2-dimethylhydrazine (SDMH) in rats. Int J Cancer, 15, 673–83
- Preston RJ, Williams GM. (2005). DNA-reactive carcinogens: mode of action and human cancer hazard. Crit Rev Toxicol, 35, 673–83
- Proctor DM, Suh M, Aylward LL, et al. (2012). Hexavalent chromium reduction kinetics in rodent stomach contents. Chemosphere, 89, 487–493
- Proctor DM, Thompson CM, Suh M, Harris MA. (2011). A response to “A quantitative assessment of the carcinogenicity of hexavalent chromium by the oral route and its relevance to human exposure”. Environ Res, 111, 468–70; discussion 471–2
- Rahman I, MacNee W. (2000). Oxidative stress and regulation of glutathione in lung inflammation. Eur Respir J, 16, 534–54
- Rawlinson LA, O’Brien PJ, Brayden DJ. (2010). High content analysis of cytotoxic effects of pDMAEMA on human intestinal epithelial and monocyte cultures. J Control Release, 146, 84–92
- Reynolds M, Armknecht S, Johnston T, Zhitkovich A. (2012). Undetectable role of oxidative DNA damage in cell cycle, cytotoxic and clastogenic effects of Cr(VI) in human lung cells with restored ascorbate levels. Mutagenesis, 27, 437–43
- Reynolds M, Stoddard L, Bespalov I, Zhitkovich A. (2007). Ascorbate acts as a highly potent inducer of chromate mutagenesis and clastogenesis: linkage to DNA breaks in G2 phase by mismatch repair. Nucleic Acids Res, 35, 465–76
- Rizk, P, Barker N. (2012). Gut stem cells in tissue renewal and disease: methods, markers, and myths. Wiley interdisciplinary reviews. Syst Biol Med, 4, 475–96
- Roberts RA, Laskin DL, Smith CV, et al. (2009). Nitrative and oxidative stress in toxicology and disease. Toxicol Sci, 112, 4–16
- Rodrigues CF, Urbano AM, Matoso E, et al. (2009). Human bronchial epithelial cells malignantly transformed by hexavalent chromium exhibit an aneuploid phenotype but no microsatellite instability. Mutat Res, 670, 42–52
- Ronen A, Heddle JA. (1984). Site-specific induction of nuclear anomalies (apoptotic bodies and micronuclei) by carcinogens in mice. Cancer Res, 44, 1536–40
- Salnikow K, Zhitkovich A. (2008). Genetic and epigenetic mechanisms in metal carcinogenesis and cocarcinogenesis: nickel, arsenic, and chromium. Chem Res Toxicol, 21, 28–44
- Sambruy Y, Ferruzza S, Ranaldi G, De Angelis I. (2001). Intestinal cell culture models: applications in toxicology and pharmacology. Cell Biol Toxicol, 17, 301–17
- Sarkar D, Sharma A, Talukder G. (1993). Differential protection of chlorophyllin against clastogenic effects of chromium and chlordane in mouse bone marrow in vivo. Mutat Res, 301, 33–8
- Schafer FQ, Buettner GR. (2001). Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic Biol Med, 30, 1191–1212
- Sedelnikova OA, Redon CE, Dickey JS, et al. (2010). Role of oxidatively induced DNA lesions in human pathogenesis. Mutat Res, 704, 152–9
- Sedman RM, Beaumont J, McDonald TA, et al. (2006). Review of the evidence regarding the carcinogenicity of hexavalent chromium in drinking water. J Environ Sci Health C Environ Carcinog Ecotoxicol Rev, 24, 155–82
- Seed J, Carney EW, Corley RA, et al. (2005). Overview: using mode of action and life stage information to evaluate the human relevance of animal toxicity data. Crit Rev Toxicol, 35, 664–72
- Seoane AI, Dulout FN. (2001). Genotoxic ability of cadmium, chromium and nickel salts studied by kinetochore staining in the cytokinesis-blocked micronucleus assay. Mutat Res, 490, 99–106
- Seoane AI, Guerci AM, Dulout FN. (2002). Malsegregation as a possible mechanism of aneuploidy induction by metal salts in MRC-5 human cells. Environ Mol Mutagen, 40, 200–6
- Serra S, Jani PA. (2006). An approach to duodenal biopsies. J Clin Pathol, 59, 1133–50
- Shah P, Jogani V, Bagchi T, Misra, A. (2006). Role of Caco-2 cell monolayers in prediction of intestinal drug absorption. Biotechnol Prog, 22, 186–98
- Shibata H, Toyama K, Shioya H, et al. (1997). Rapid colorectal adenoma formation initiated by conditional targeting of the Apc gene. Science, 278, 120–3
- Shindo Y, Toyoda Y, Kawamura K, et al. (1989). Micronucleus test with potassium chromate(VI) administered intraperitoneally and orally to mice. Mutat Res, 223, 403–6
- Shrivastava R, Upreti RK, Chaturvedi UC. (2003). Various cells of the immune system and intestine differ in their capacity to reduce hexavalent chromium. FEMS Immunol Med Microbiol, 38, 65–70
- Shrivastava R, Kannan A, Upreti RK, Chaturvedi UC. (2005). Effects of chromium on the resident gut bacteria of rat. Toxicol Mech Methods, 15, 211–8
- Shumilla JA, Broderick RJ, Wang Y, Barchowsky A. (1999). Chromium(VI) inhibits the transcriptional activity of nuclear factor-kappaB by decreasing the interaction of p65 with cAMP-responsive element-binding protein-binding protein. J Biol Chem, 274, 36207–12
- Slikker W Jr, Andersen ME, Bogdanffy MS, et al. (2004). Dose-dependent transitions in mechanisms of toxicity: case studies. Toxicol Appl Pharmacol, 201, 226–94
- Smetanova L, Stetinova V, Svoboda Z, Kvetina J. (2011). Caco-2 cells, biopharmaceutics classification system (BCS) and biowaiver. Acta Medica, 54, 3–8
- Snow AD, Altmann GG. (1983). Morphometric study of the rat duodenal epithelium during the initial six months of 1,2-dimethylhydrazine carcinogenesis. Cancer Res, 43, 4838–49
- Sonich-Mullin C, Fielder R, Wiltse J, et al. (2001). IPCS conceptual framework for evaluating a mode of action for chemical carcinogenesis. Regul Toxicol Pharmacol, 34, 146–52
- Steinhoff D, Gad SC, Hatfield GK, Mohr U. (1986). Carcinogenicity study with sodium dichromate in rats. Exp Pathol, 30, 129–41
- Stern AH. (2010). A quantitative assessment of the carcinogenicity of hexavalent chromium by the oral route and its relevance to human exposure. Environ Res, 110, 798–807
- Stojiljkovic V, Pejic S, Kasapovic J, et al. (2012). Glutathione redox cycle in small intestinal mucosa and peripheral blood of pediatric celiac disease patients. Anais da Academia Brasileira de Ciencias, 84, 175–84
- Stout MD, Herbert RA, Kissling GE, et al. (2009a). Hexavalent chromium is carcinogenic to F344/N rats and B6C3F1 mice after chronic oral exposure. Environ Health Perspect, 117, 716–22
- Stout MD, Nyska A, Collins BJ, et al. (2009b). Chronic toxicity and carcinogenicity studies of chromium picolinate monohydrate administered in feed to F344/N rats and B6C3F1 mice for 2 years. Food Chem Toxicol, 47, 729–33
- Su LK, Kinzler KW, Vogelstein B, et al. (1992). Multiple intestinal neoplasia caused by a mutation in the murine homolog of the APC gene. Science, 256, 668–70
- Sun H, Zhou X, Chen H, et al. (2009). Modulation of histone methylation and MLH1 gene silencing by hexavalent chromium. Toxicol Appl Pharmacol, 237, 258–66
- Sun H, Clancy HA, Kluz T, et al. (2011). Comparison of gene expression profiles in chromate transformed BEAS-2B cells. PLoS One, 6, e17982
- Sunter JP, Appleton DR, Wright NA, et al. (1978). Kinetics of changes in the crypts of the jejunal mucosa of dimethylhydrazine-treated rats. Br J Cancer, 37, 662–72
- Sutter T, Arber N, Moss SF, et al. (1996). Frequent K-ras mutations in small bowel adenocarcinomas. Dig Dis Sci, 41, 115–8
- Swenberg JA, Fryar-Tita E, Jeong YC, et al. (2008). Biomarkers in toxicology and risk assessment: informing critical dose-response relationships. Chem Res Toxicol, 21, 253–65
- Takahashi Y, Kondo K, Hirose T, et al. (2005). Microsatellite instability and protein expression of the DNA mismatch repair gene, hMLH1, of lung cancer in chromate-exposed workers. Mol Carcinog, 42, 150–8
- Takahashi-Yanaga F, Sasaguri T. (2007). The Wnt/beta-catenin signaling pathway as a target in drug discovery. J Pharmacol Sci, 104, 293–302
- Talley NJ. (2010). Practical gastroenterology and hepatology: small and large intestine and pancreas. Hoboken (NJ): Wiley-Blackwell
- Thomas RS, Allen BC, Nong A, et al. (2007). A method to integrate benchmark dose estimates with genomic data to assess the functional effects of chemical exposure. Toxicol Sci, 98, 240–8
- Thomas RS, Clewell HJ, 3rd, Allen BC, et al. (2011). Application of transcriptional benchmark dose values in quantitative cancer and noncancer risk assessment. Toxicol Sci, 120, 194–205
- Thompson CM, Haws LC, Harris MA, et al. (2011a). Application of the U.S. EPA mode of action Framework for purposes of guiding future research: a case study involving the oral carcinogenicity of hexavalent chromium. Toxicol Sci, 119, 20–40
- Thompson CM, Fedorov Y, Brown DD, et al. (2012a). Assessment of Cr(VI)-induced cytotoxicity and genotoxicity using high content analysis. PLoS One, 7, e42720
- Thompson CM, Gregory Hixon J, Proctor DM, et al. (2012b). Assessment of genotoxic potential of Cr(VI) in the mouse duodenum: an in silico comparison with mutagenic and nonmutagenic carcinogens across tissues. Regul Toxicol Pharmacol, 64, 68–76
- Thompson CM, Proctor DM, Haws LC, et al. (2011b). Investigation of the mode of action underlying the tumorigenic response induced in B6C3F1 mice exposed orally to hexavalent chromium. Toxicol Sci, 123, 58–70
- Thompson CM, Proctor DM, Suh M, et al. (2012c). Comparison of the effects of hexavalent chromium in the alimentary canal of F344 rats and B6C3F1 mice following exposure in drinking water: implications for carcinogenic modes of action. Toxicol Sci, 125, 79–90
- Tsapakos MJ, Hampton TH, Jennette KW. (1981). The carcinogen chromate induces DNA cross-links in rat liver and kidney. J Biol Chem, 256, 3623–6
- Upham BL, Wagner JG. (2001). Toxicant-induced oxidative stress in cancer. Toxicol Sci, 64, 1–3
- US EPA. (1988). Recommendations for and Documentation of Biological Values for Use in Risk Assessment. Cincinnati, OH: Office of Health and Environmental Assessment, Office of Research and Development, U.S. Environmental Protection Agency
- US EPA. (1991). National primary drinking water regulations-synthetic organic chemicals and inorganic chemicals; monitoring for unregulated contaminants; national primary drinking water regulations implementation; national secondary drinking water regulations. Final rule. Fed Regist, 56, 3526–3597
- US EPA. (1998). Toxicological review of hexavalent chromium in support of summary information on the integrated risk information system (IRIS). Washington (DC): US Environmental Protection Agency
- US EPA. (2004). Captan; cancer reclassification; amendment of reregistration eligibility decision; notice of availability. Fed Regist, 69, 68357–60
- US EPA. (2005a). Guidelines for carcinogen risk assessment, EPA/630/P-03/001F. Washington, DC: Risk Assessment Forum, U.S. Environmental Protection Agency
- US EPA. (2005b). Supplemental guidance for assessing susceptibility from early-life exposure to carcinogens, EPA/630/R-03/003F. Washington, DC: Risk Assessment Forum, US Environmental Protection Agency
- US EPA. (2007). Framework for determining a mutagenic mode of action for carcinogenicity: using EPA’s 2005 Cancer Guidelines and Supplemental Guidance for Assessing Susceptibility from Early-Life Exposure to Carcinogens. Washington, DC: US Environmental Protection Agency
- US EPA. (2009). An approach to using toxicogenomic data in U.S. EPA Human Health Risk Assessments: A Dibutyl Phthalate case study, EPA/600/R-09/028F. Washington, DC: US Environmental Protection Agency
- US EPA. (2010). Toxicological review of hexavalent chromium in support of summary information on the integrated risk information system (IRIS). External Review DRAFT. Washington, DC: US Environmental Protection Agency
- Vanhauwaert A, Vanparys P, Kirsch-Volders M. (2001). The in vivo gut micronucleus test detects clastogens and aneugens given by gavage. Mutagenesis, 16, 39–50
- Velho S, Haigis KM. (2011). Regulation of homeostasis and oncogenesis in the intestinal epithelium by Ras. Exp Cell Res, 317, 2732–9
- Vincent JB. (2000). The biochemistry of chromium. J Nutr, 130, 715–8
- Vissers MC, Bozonet SM, Pearson JF, Braithwaite LJ. (2011). Dietary ascorbate intake affects steady state tissue concentrations in vitamin C-deficient mice: tissue deficiency after suboptimal intake and superior bioavailability from a food source (kiwifruit). Am J Clin Nutr, 93, 292–301
- Wang X, Son YO, Chang Q, et al. (2011). NADPH oxidase activation is required in reactive oxygen species generation and cell transformation induced by hexavalent chromium. Toxicol Sci, 123, 399–410
- Watanabe C, Egami T, Midorikawa K, et al. (2010). DNA damage and estrogenic activity induced by the environmental pollutant 2-nitrotoluene and its metabolite. Environ Health Prev Med, 15, 319–26
- Whittle BJ, Varga C. (2010). New light on the anti-colitic actions of therapeutic aminosalicylates: the role of heme oxygenase. Pharmacol Rep, 62, 548–56
- Wicki A, Herrmann R, Christofori G. (2010). Kras in metastatic colorectal cancer. Swiss Med Wkly, 140, w13112
- Wild D. (1978). Cytogenetic effects in the mouse of 17 chemical mutagens and carcinogens evaluated by the micronucleus test. Mutat Res, 56, 319–27
- Williams GM. (2008). Application of mode-of-action considerations in human cancer risk assessment. Toxicol Lett, 180, 75–80
- Wilson VS, Keshava N, Hester S, et al. (2011). Utilizing toxicogenomic data to understand chemical mechanism of action in risk assessment. Toxicol Appl Pharmacol, (in press)
- Yan KS, Chia LA, Li X, et al. (2012). The intestinal stem cell markers Bmi1 and Lgr5 identify two functionally distinct populations. Proc Natl Acad Sci USA, 109, 466–71
- Yao H, Guo L, Jiang BH, et al. (2008). Oxidative stress and chromium(VI) carcinogenesis. J Environ Pathol Toxicol Oncol, 27, 77–88
- Yilmaz OH, Katajisto P, Lamming DW, et al. (2012). mTORC1 in the Paneth cell niche couples intestinal stem-cell function to calorie intake. Nature, 486, 490–5
- Younes N, Fulton N, Tanaka R, et al. (1997). The presence of K-12 ras mutations in duodenal adenocarcinomas and the absence of ras mutations in other small bowel adenocarcinomas and carcinoid tumors. Cancer, 79, 1804–8
- Zhitkovich A. (2005). Importance of chromium-DNA adducts in mutagenicity and toxicity of chromium(VI). Chem Res Toxicol, 18, 3–11
- Zhitkovich A. (2011). Chromium in drinking water: sources, metabolism, and cancer risks. Chem Res Toxicol, 24, 1617–29
- Zhu X, Fan WG, Li DP, et al. (2011). Heme oxygenase-1 system and gastrointestinal inflammation: a short review. World J Gastroenterol, 17, 4283–8