
Abstract
Inhalation of naphthalene causes olfactory epithelial nasal tumors in rats (but not in mice) and benign lung adenomas in mice (but not in rats). The limited available human data have not identified an association between naphthalene exposure and increased respiratory cancer risk. Assessing naphthalene's carcinogenicity in humans, therefore, depends entirely on experimental evidence from rodents. We evaluated the respiratory carcinogenicity of naphthalene in rodents, and its potential relevance to humans, using our Hypothesis-Based Weight-of-Evidence (HBWoE) approach. We systematically and comparatively reviewed data relevant to key elements in the hypothesized modes of action (MoA) to determine which is best supported by the available data, allowing all of the data from each realm of investigation to inform interpretation of one another. Our analysis supports a mechanism that involves initial metabolism of naphthalene to the epoxide, followed by GSH depletion, cytotoxicity, chronic inflammation, regenerative hyperplasia, and tumor formation, with possible weak genotoxicity from downstream metabolites occurring only at high cytotoxic doses, strongly supporting a non-mutagenic threshold MoA in the rat nose. We also conducted a dose–response analysis, based on the likely MoA, which suggests that the rat nasal MoA is not relevant in human respiratory tissues at typical environmental exposures. Our analysis illustrates how a thorough WoE evaluation can be used to support a MoA, even when a mechanism of action cannot be fully elucidated. A non-mutagenic threshold MoA for naphthalene-induced rat nasal tumors should be considered as a basis to determine human relevance and to guide regulatory and risk-management decisions.
Introduction
Naphthalene is a natural constituent of coal tar and crude oil, from which it is produced and sold commercially. It is a white solid that readily evaporates and is present in several consumer products, such as mothballs and moth crystals, and in household deodorant blocks. It is also used in making dyes, resins, tanning agents, and insecticides. It is released into the air in smoke from burning wood or tobacco.
The International Agency for Research on Cancer (IARC) classifies naphthalene as “Possibly carcinogenic to humans (Group 2B)” (CitationIARC 2002), stating that “There is sufficient evidence in experimental animals for the carcinogenicity of naphthalene,” but “There is inadequate evidence in humans for the carcinogenicity of naphthalene” (CitationIARC 2002). The National Toxicology Program (NTP) more recently classified naphthalene as “Reasonably anticipated to be a human carcinogen” based on inadequate epidemiology studies but “sufficient evidence from studies in experimental animals” (CitationNTP 2011). In their reviews, neither NTP nor IARC included a discussion of a possible mode of action (MoA) for naphthalene carcinogenesis and the relevance to humans.
Inhalation of naphthalene causes olfactory and respiratory epithelial nasal tumors in rats (but not in mice) (CitationNTP, 1992, Citation2000); there was a significant increase in neuroblastomas of the olfactory tissue in females and a significant trend with dose in both sexes, and males exhibited a significant increase in adenomas of the nasal respiratory epithelium at all exposure levels. Inhalation of naphthalene also caused a significant increase in benign lung adenomas in female mice (but not in rats, or in male mice) (CitationNTP 1992, Citation2000). There are no other animal inhalation carcinogenesis studies for naphthalene. Although there are no systematic epidemiology studies relating naphthalene exposure and cancer, studies of respiratory tract cancers in humans and their potential causative agents have not identified naphthalene exposure as associated with tumor risk (CitationGriego et al. 2008; CitationLewis 2012). In fact, there is strong evidence indicating a lack of a tumorigenic effect in humans (particularly nasal tumors), despite the lack of systematic investigation, for occupationally-exposed people and for people in the general population. The question of naphthalene's carcinogenicity in humans, therefore, depends entirely on the experimental evidence from rats and mice, along with mechanistic information that bears on whether the rat nasal tumors or the mouse lung tumors are likely to be indicative of a potential for human respiratory cancer risk. Nasal tumors are rare in humans; therefore, one would expect to see an increase in these tumors if present. However, lung tumors are not rare, and therefore, the lack of evidence for naphthalene's lung carcinogenicity in humans is a less compelling refutation.
There are several important observations with respect to naphthalene-induced tumors in rats and mice from the existing NTP bioassays. First, the tumors are confined to specific epithelial tissues of the respiratory tract that are directly exposed to naphthalene vapors, suggesting a very specific and local mechanism of action. In mice, adenomas are localized to bronchioles, and in rats, distinct but similar tumors are located in nasal olfactory and respiratory epithelia (CitationNTP 1992, Citation2000).
Second, in both rats and mice, there is widespread cytotoxicity and inflammation at all doses evaluated in the tissues where tumors occur, likely due to exceedance of the maximum tolerated dose (MTD) (CitationNorth et al. 2008). Tissues beyond the nasal and lung epithelia do not show such cytotoxicity and hyperplasia, and they do not have tumors. Target tissue toxicity with cell-killing and regenerative hyperplasia is thought to be the immediate and primary carcinogenic process for many carcinogens, followed by cell proliferation, clonal expansion of somatic mutations, and tumor promotion. In the NTP inhalation bioassay, the tumors occur only where there is marked tissue toxicity (nasal and lung epithelia), strongly suggesting a causal role (CitationNorth et al. 2008).
Third, in the nasal (rats and mice) and lung (mice) tissues where toxicity occurs, there is concentrated and localized metabolic activity toward naphthalene (CitationBogen et al. 2008). In these tissues, naphthalene is metabolized to its reactive 1,2-epoxide by cytochrome P450 (CYP). The epoxide can be further conjugated with glutathione (GSH) and detoxified, but at high naphthalene exposures, GSH can become depleted, resulting in toxicity, possibly from downstream toxic metabolites such as 1,2-naphthoquinone. In fact, inhibiting naphthalene metabolism eliminates respiratory cytotoxicity. Further, naphthalene is metabolized in other tissues as well, such as the liver; in these tissues, however, there is no evident tissue injury following naphthalene exposure in the NTP bioassays, likely because GSH levels and resynthesis are sufficient to prevent significant GSH depletion. Exposure to naphthalene by intraperitoneal (IP) injection—either a single injection, or repeatedly at high enough concentrations where mouse lung tissue does not become tolerant (CitationBuckpitt et al. 2002)—results in metabolic activation and cytotoxicity in the same respiratory tract epithelia as in inhalation studies, indicating that the localization of effects in rats and mice is attributable to localized high metabolic activity rather than to the direct inhalation exposure of the tissues (CitationVan Winkle et al. 1995, Citation1997; CitationBuckpitt et al. 2002; CitationPlopper et al. 1992a,Citationb). It is also notable that CYP2F is largely localized in the tissues where tumors occur and has considerable metabolic activity toward naphthalene, raising the possibility that the localization of tumor response may be dependent on this particular CYP isozyme (CitationBogen et al. 2008).
There are also observations that clearly reflect differences across species and tissues. A key difference is that naphthalene metabolism is very active in mouse nasal tissue (CitationBogen et al. 2008), resulting in nasal toxicity but, unlike the rat nose, there are no nasal tumors in mice. If tissue toxicity from local naphthalene metabolism is thought to be critical and sufficient for generation of rat nasal tumors, then one must account for why the tissue toxicity in the mouse nose does not also lead to tumors. It is notable that a CYP isozyme other than CYP2F (i.e., CYP2A5) has been shown to be primarily responsible for naphthalene metabolism in the mouse nose (CitationLi et al. 2011; CitationHu et al. 2014). Perhaps this difference accounts for mouse nasal toxicity but not tumors; further investigation would help to elucidate this possibility.
Overall, the high degree of localization of naphthalene metabolic enzymes, in combination with GSH depletion and cytotoxicity in tissues where tumors occur, along with the lack of tumors in tissues where these events do not occur, suggest that these events are involved in naphthalene's carcinogenic MoA in rodents. Moreover, the balance and types of the activities of the specific CYPs involved in naphthalene metabolism in target tissues, in combination with enzymes responsible for detoxification or further metabolism to toxic naphthalene metabolites, is ultimately what determines the potential for naphthalene to cause tissue injury, and this balance will vary across tissues and species. The extent to which the tumor responses come from a common underlying mechanism across species provides support for the overall MoA.
The distinction between mechanism of action and MoA is important. A MoA can be thought of as a biological change at the cellular level, with intermediate complexity between molecular events and physiological outcomes, whereas a mechanism of action describes specific biological changes at the molecular level. Different chemicals may have similar modes of action but very different underlying mechanisms of action for carcinogenesis. There are a number of carcinogenesis models that have been proposed in the history of carcinogenesis that are not necessarily exclusive for a given chemical (CitationVineis et al. 2010). For the purposes of our analysis, we have divided the general models into two categories (or two modes of action for carcinogenesis) based on the relative doses likely required for each. One category (which we call a “mutagenic MoA”) involves direct reaction of the chemical or metabolites of the chemical with DNA, which can lead to mutations that are not reversible, and, therefore, possibly leads to tumor formation. Importantly, this MoA can potentially occur at low exposure concentrations. The other category (which we call a “non-mutagenic MoA”) involves mechanisms of carcinogenesis that require a higher level of exposure to the chemical before detoxification mechanisms are saturated, and there is enough exposure to the cells and tissues that other types of toxicity, which are often reversible (i.e., not involving direct reaction of the parent compound or one of its metabolites with DNA and subsequent mutations), can occur, which can lead to secondary mutations and tumor formation. The “non-mutagenic MoA” category includes mechanisms involving inflammation, cytotoxicity, regenerative hyperplasia, genomic instability, epigenetics, mitogenesis, and apoptosis, all leading to cell proliferation and clonal expansion, possible selection of spontaneous mutations, and tissue disorganization (see a recent discussion of models of carcinogenesis in CitationVineis et al. 2010). Understanding the MoA is most important for regulatory decision making since critical doses for one or the other MoA may be very different, driving very different health- protective exposure concentrations and potential risks. Therefore, although ideally one would want to understand both the mechanism and mode of action for a chemical of concern, when data are not sufficient to fully understand the mechanism, it still may be possible to sufficiently understand the MoA and apply that understanding to regulatory and risk-management decisions. Our analysis includes consideration of mechanistic data and attempts to determine a potential mechanism of action for naphthalene carcinogenesis. In the end, however, following integration of all of the relevant data, we conclude that the mechanism of action for nasal carcinogenesis in rats is still not entirely clear, and instead focus on the MoA for our dose–response analysis.
Although naphthalene is known to cause adverse non-cancer effects beyond respiratory tissue (i.e., cataracts—predominantly in rodents, and hemolytic anemia—predominantly in humans) (CitationATSDR 2005), our analysis focuses on cancer endpoints and the non-cancer events that bear on the carcinogenic mechanism of action and MoA. Although there is not always concordance of tumor location across species, our analysis suggests that naphthalene-induced tumors in other organs in humans is not likely. Therefore, our evaluation focuses on the respiratory carcinogenicity of naphthalene in rodents, and its potential relevance to human respiratory cancers, using our Hypothesis-Based Weight-of-Evidence (HBWoE) approach. Our HBWoE approach is outlined in several recent weight-of-evidence evaluations conducted by our group, one of which is an evaluation of naphthalene (CitationRhomberg et al. 2010, Citation2011; CitationPrueitt et al. 2011; CitationBailey et al. 2012). In the present paper, we provide an update to the earlier evaluation of CitationRhomberg et al. (2010), incorporating new data that have been published since 2010. We then use the results of our HBWoE evaluation to estimate human equivalent concentrations (HECs) for naphthalene carcinogenicity based on a recently developed rat/human physiologically-based pharmacokinetic (PBPK) model (CitationCampbell et al. 2014) that predicts metabolized doses of naphthalene in respiratory tissue.
Methods
Literature reviews and data collection
We included all studies that were reviewed and discussed in our earlier paper (CitationRhomberg et al. 2010), along with more recent studies identified through PubMed literature searches. For our current analysis, we conducted similar literature searches in PubMed for more recent epidemiology, animal, toxicokinetic, genotoxicity, and other mechanistic studies using the search terms “naphthalene,” “genotoxicity,” “metabolism,” and “toxicokinetic.” These searches were conducted weekly throughout the duration of our analysis, to ensure that we were including the most current information. In order to remain aware of new results relevant to naphthalene as soon as they became available, we worked closely with the Naphthalene Research Committee and with researchers sponsored by this Committee who were involved in projects investigating naphthalene toxicity and metabolism.
Hypothesis-based weight-of-evidence (HBWoE) methodology
The ultimate goal of the HBWoE approach is to weigh all of the data from each realm of investigation (i.e., epidemiology, animal data, toxicokinetics, genotoxicity, and other mechanistic data), allowing the data sets to inform interpretation of one another. Our approach is to look systematically and comparatively at the various key elements in the metabolism and the hypothesized MoAs for inhaled naphthalene, to try to identify the responsible and necessary elements for carcinogenesis. For each key event, and for each species and tissue, we apply three key lines of questioning:
What is necessary in the proposed naphthalene MoA? What is sufficient, and are other elements also necessary?
For those events or processes proposed as critical to the observed carcinogenic effects of naphthalene, what other observable manifestations should they have (in other tissues or species)? Are these other manifestations indeed found?
If either the operation or the necessity of these proposed critical events were disproven, how else would one account for the array of outcomes?
The steps in our HBWoE approach are illustrated in . The first two steps are presented in the first five main sections of this paper where we examined the data for particular endpoints across studies within each realm of investigation (epidemiology, animal bioassays, toxicokinetics, genotoxicity, other mechanistic data), considering study quality, and evaluating consistency, specificity, and reproducibility of the reported outcomes.
Figure 1. The seven key aspects of the Hypothesis-Based Weight-of-Evidence (HBWoE) approach.

For Step 3, we articulate the hypotheses that have been put forth within the scientific community regarding the MoA for naphthalene-induced respiratory carcinogenesis.
The data for each realm of investigation are summarized later in the HBWoE section in the context of the proposed MoAs (Step 4) and in the context of all of the data combined (Step 5), so that all realms of investigation are allowed to inform interpretation of one another.
Next, in Step 6, we compare two alternative contentions for the nature of the MoA and ask, for each one, if it were the true characterization of the underlying processes, how (and how well) it would serve to explain the patterns of outcomes among the available studies. We formulate these sets of explanations of the observed results across all lines of evidence into two competing accounts. One account lays out the reasoning and explanations contingent on accepting one overarching MoA hypothesis. The other account lays out the alternative explanations and reasoning, based on the same array of studies, that would be needed if the other overarching hypothesis were true.
Briefly, one overarching hypothesis proposes that naphthalene causes tumors in rodents via a “mutagenic MoA”—that is, by direct interaction of the agent or its metabolites with DNA to cause somatic mutations that are the basis of cell transformation. Such a process would be presumed to operate qualitatively in humans and at lower exposure levels, at least to some degree. One can consider that direct mutagenesis is the sole relevant effect; alternatively, one could consider variants of the hypothesis that suggest direct mutagenesis acts alone at low exposures, but at higher exposures, its effect may be exacerbated by co-occurring cytotoxicity or other cellular dysfunction, or diminished by mutations occurring in dying cells. Each variant would be presumed to produce low-dose risk, but their dose–response patterns would be expected to differ.
The alternative overarching hypothesis is a “non-mutagenic MoA” that identifies cytotoxicity, or other marked cellular dysfunction, occurring once a threshold tissue exposure is exceeded, as the necessary factor in inducing added risk of cellular transformation. Again, one can consider this dysfunction to be the sole relevant factor, or one can consider a variant in which the effects are exacerbated by mutagenic effects at high doses, either because mutations are secondary to high-dose cellular toxicity or because the mutagenic metabolites are generated only at doses that are also sufficient to cause cytotoxicity, inflammation, or hyperplasia. Such a MoA would make the relevance of high-exposure rodent tumors to the assessment of lower-exposure human risk less clear. It also opens possibilities that species-, sex-, and dose-specific factors that influence the susceptibility to such high-dose tissue toxicity are also important to the interpretation of relevance of animal bioassay results to typical human exposures.
In the evaluation of these alternatives, we consider how each experimental result could have arisen under the operation of each alternative overarching MoA hypothesis. This includes tentative explanations for uncertainties and inconsistencies among the data sets, as well as any ad hoc assumptions that may be required in order to reconcile an observation with the overarching MoA. The various competing accounts are weighed by comparing the reasoning for each account, including ad hoc assumptions and overall plausibility of explanations needed for each, and how likely it is that additional studies will support a given explanation. The most compelling account is the one requiring the fewest number of ad hoc assumptions and implausible explanations of the data.
Dose–response analysis and human equivalent concentrations
We applied United States Environmental Protection Agency's (US EPA's) Benchmark Dose Software (BMDS) Version 2.3.1 (CitationUS EPA 2012a, Citation2012b) to conduct the dose–response modeling and to estimate points of departure (PODs) from the incidence of cytotoxic lesions of concern in rat nasal tissue. In all analyses of benchmark dose (BMD), a benchmark response (BMR) of 10% increase in extra risk was used as the basis for the BMD, with the corresponding 95% lower confidence limit on the benchmark dose (BMDL10) also calculated (CitationUS EPA 2012a). The average of all BMDL10 models that adequately fit the data was selected as the POD. As discussed later in this paper, for the data used in our dose–response analysis, the average and best-fit BMDL10 models were very similar.
We applied a recently developed computational fluid dynamic (CFD)–PBPK model (CitationCampbell et al. 2014) that predicts metabolized doses of naphthalene in rat and human respiratory tissue, to estimate HECs for naphthalene carcinogenicity.
Human studies
There is very little information about cancer risk in humans associated with naphthalene exposure. The human data are limited to a few case reports and one nested case-control study of oral/oropharyngeal cancer. To our knowledge, there are no cohort or case-control studies of naphthalene and lung or nasal cancer risk. Lung cancer has been addressed in several occupational studies of industries where naphthalene exposure may occur, but the interpretation of these studies is limited due to low or unquantified naphthalene exposure and concurrent exposure to other chemical agents.
Case reports
Since nasal cancer is rare in humans, case reports may be useful for identifying possible associations. To date, there have been no case reports of nasal cancer in association with naphthalene exposure. Reviews by CitationGriego et al. (2008)Citation and Lewis (2012) have identified two reports of naphthalene exposure and cancer cases involving either laryngeal cancer or colorectal cancer (in the latter report, the patients had ingested naphthalene). The former report described four cases of laryngeal cancer among 15 workers at a German naphthalene purification plant. All of the cases were smokers, however, and were exposed to other potentially cancer-causing chemicals. Smoking is known to be highly associated with laryngeal cancer (CitationBosetti et al. 2006; CitationTalamini et al. 2002).
Case-control studies
CitationOlsson et al. (2010) conducted a case-control study of 433 lung cancer cases and 1,253 controls, nested within a cohort of 38,296 European and Israeli asphalt workers, as an update of an earlier study by CitationBoffetta et al. (2003a). Although naphthalene exposure was not studied specifically, naphthalene may account for up to 90% of occupational inhalation exposure to polycyclic aromatic hydrocarbons (PAHs) for asphalt workers (CitationNIOSH 2000). The authors found no significant association between lung cancer and any exposure to asphalt fumes (OR = 1.12; 95% CI = 0.84–1.49) when adjusted for tobacco smoking and exposure to coal tar. In addition, there were no significant trends for lung cancer risk and exposure duration, average exposure, or cumulative exposure.
CitationMerletti et al. (1991) conducted a population-based case-control study of 86 oral or oropharyngeal cancer cases and 373 controls in Italy. This group evaluated the risk associated with 40 occupations, 41 industries, and 16 specific chemicals, including naphthalene. Naphthalene was not associated with the risk of oral or oropharyngeal cancer in this study. The results must be interpreted with caution, however, due to the small number of cases and the large number of comparisons.
Occupational cohort studies
CitationLewis (2012) reviewed several studies of lung cancer risk in connection with industries and occupations in which naphthalene exposure is common, including the petroleum (CitationWong and Raabe, 2000; CitationConsonni et al. 1999; CitationLewis et al. 2003; CitationRushton, 1993), asphalt (CitationBoffetta et al. 2003a,Citationb; CitationOlsson et al. 2010; CitationFayerweather 2007), and creosote industries (CitationWong and Harris, 2005), and jet-fuel handlers (CitationD’Mello and Yamane, 2007; CitationYamane, 2006). No association with lung or nasal cancer was reported in any of the studies. CitationLewis (2012) noted that all of the studies had limitations such as lack of exposure data, low levels of naphthalene exposure, exposure to other chemical agents, and in some studies, small numbers of cancer cases. CitationLewis (2012) concluded that the insufficiencies in the data did not allow for ruling out a potential association. He did note, however, that none of the studies provided any evidence of an association between naphthalene and lung or nasal cancer.
Overall analysis of data quality and consistency of results from human studies
All of the studies described here (with the exception of two case studies) were negative for an association between naphthalene and cancer. There are many data gaps, however, in the epidemiology literature for naphthalene. While case reports can be informative, they cannot be regarded as proof of an association. Only one case report has been identified that involves naphthalene inhalation exposure and any cancer (laryngeal). The four cases described in this report all had confounding factors, however, including smoking and exposure to other potentially cancer-causing chemicals. Two case-control studies revealed no association between lung or oral/oropharyngeal cancers and exposure to asphalt fumes or naphthalene. While both of these studies provide evidence of no association, they each have limitations such as low numbers of cases and a lack of exposure quantifications. Most of these studies were occupational, with exposures likely ranging from 10–3,000 μg/m3 (0.002 to 0.6 ppm) (CitationGriego et al. 2008). Background and residential naphthalene exposure levels are much lower, ranging from 0.001–10 μg/m3 (CitationGriego et al. 2008). The occupational cohort studies, which provide no evidence of naphthalene exposure and lung cancer, also have limitations that preclude drawing any definitive conclusions. None of these studies addressed naphthalene exposure specifically, and all involved exposure to additional chemical agents.
Animal studies
Mice
In mice, following inhalation, the respiratory tract is the target for naphthalene toxicity. NTP conducted a 2-year bioassay (CitationNTP 1992; CitationAbdo et al. 1992; also summarized by CitationNorth et al. 2008) in which B6C3F1 mice were exposed via inhalation to 0, 10, or 30 parts per million (ppm) naphthalene for 6 h/day, 5 days/week, for 104 weeks. Only females exhibited a significant increase in combined incidence of alveolar/bronchiolar adenomas and carcinomas at the highest dose (7/70, 18/69, 34/135 [male] and 5/69, 2/65, 29/135 [female]) (See summary tables of neoplastic and non-neoplastic effects in rats and mice in CitationRhomberg et al. 2010). Chronic inflammation of alveolar/bronchiolar lung tissue was observed in both sexes at both naphthalene exposures. There were no significant increases in lung tissue hyperplasia at any exposure. There was no evidence of nasal tumors in either sex at any exposure; however, all treatment groups exhibited significant increases in nasal inflammation, metaplasia of the olfactory epithelium, and hyperplasia of the respiratory epithelium. Based on the outcome of this assay, NTP concluded that there was no evidence of carcinogenic activity in male mice, and some evidence of carcinogenic activity in female mice.
CitationAdkins et al. (1986) exposed strain A/J mice to 0, 10, or 30 ppm naphthalene by inhalation, 6 h/day, 5 days/week, for 6 months. The authors noted no effects on the total number of lung tumors per mouse, but there was a significant increase in the number of lung tumors per tumor-bearing mouse in both the 10 and 30 ppm exposure groups.
One study evaluated subchronic (13-week) exposure to naphthalene in mice via oral gavage (12.5, 25, 50, 100, and 200 mg/kg) (CitationBattelle 1980a) and observed little to no toxicity in lungs. Since nasal tissue was not evaluated, it is not clear whether toxicity occurred. The major effects were roughened hair coat and decreased body weight gain. As discussed in a review by CitationBuckpitt et al. (2002), single naphthalene doses as low as 50 mg/kg did lead to lung toxicity (CitationPlopper et al. 1992b), with nasal toxicity also occurring at single doses of 400 mg/kg. The lack of toxicity in the mouse lung following a 13-week exposure at 200 mg/kg in the Battelle study is consistent with studies discussed by CitationBuckpitt et al. (2002), where mouse lung tolerance was observed following intraperitoneal doses of 200 mg/kg or less for 7 days. Buckpitt referred to another study by Shop et al. (1984, cited in CitationBuckpitt et al. 2002), in which the authors observed no lung toxicity at 267 mg/kg for 14 days and 133 mg/kg for 90 days in mice; the authors did not look at doses higher than 133 mg/kg for 90 days.
CitationVan Winkle et al. (1995, Citation1997) monitored Club cell injury and repair in mice following acute (single dose) IP injections of naphthalene (200 mg/kg). This group observed regenerative repair (hyperplasia) of lung tissue during the first few days following injections.
Rats
In rats, aside from the occurrence of cataracts in some studies, the respiratory tract was observed to be the target for naphthalene inhalation toxicity (CitationATSDR 2005). NTP conducted a 2-year bioassay study in F344/N rats (CitationNTP 2000; CitationAbdo et al. 2001; also summarized by CitationNorth et al. 2008). Rats were exposed by inhalation to 0, 10, 30, or 60 ppm naphthalene for 6 h/day, 5 days/week, for 105 weeks. In nasal tissue, there was a significant increase in neuroblastomas of the olfactory tissue in females in the 60 ppm exposure group, and there was a significant trend with dose in both sexes (0/49, 0/49, 4/48, 3/48 [male] and 0/49, 2/49, 3/49, 12/49 [female]). Males exhibited a significant increase in adenomas of the nasal respiratory epithelium at all exposure levels (0/49, 6/49, 8/48, 15/48 [male] and 0/49, 0/49, 4/49, 2/49 [female]). For both sexes, at all exposure levels, there were significant increases in inflammation and hyperplasia in both olfactory and respiratory epithelial tissues. In lung tissue, there was some evidence of hyperplasia in females but not in males, and some inflammation in males but not females. NTP noted that it was not clear whether these changes were exposure-related, since minimal inflammatory foci are often found in chamber control rats. No increases in lung tumors were observed. Based on the outcome of this assay, NTP concluded that there was clear evidence of carcinogenic activity in male and female rats.
CitationSchmahl (1955) exposed 28 BD I and BD III rats to 10–20 mg/day (equivalent to approximately 25–50 mg/kg-d) of naphthalene via ingestion (added to food) and observed no increased incidence of toxicity or tumor formation following up to 700 days of exposure. In the same publication, the author reported no increase in tumors and no toxicity following weekly subcutaneous or IP injections of 20 mg/rat, for up to 40 weeks. Another study evaluated subchronic (13-week) exposure to naphthalene in rats via oral gavage (25, 50, 100, 200, and 400 mg/kg) (CitationBattelle 1980b) and observed little to no toxicity in lung tissue. Since nasal tissue did not appear to be evaluated, it is not clear whether nasal toxicity occurred in this study. The major effects were roughened hair coat, decreased body weight gain, renal tubular necrosis in one male rat at 400 mg/kg, and depletion of thymic lymphocytes in two female rats at 400 mg/kg. Although the exposure period was longer in the Schmahl study, results from both studies were consistent with very little to no lung toxicity in short-term studies where rats were given single IP injections of naphthalene ranging from 200 to 1,600 mg/kg (CitationBuckpitt et al. 2002; CitationPlopper et al. 1992b). These results are consistent with the lack of toxicity in rat lung following inhalation of naphthalene. As discussed by CitationBuckpitt et al. (2002), rat nasal toxicity was observed from single oral exposures of 200–1,600 mg/kg in a study by CitationPlopper et al. (1992b).
Shorter-term inhalation studies in rats have been conducted by CitationDodd et al. (2010, Citation2012). These authors conducted 1-day and 5-day studies to assess nasal toxicity of naphthalene in Fischer 344 (F344) and Sprague-Dawley (SD) rats (CitationDodd et al. 2010). In the 1-day study, rats were exposed via inhalation to 0, 0.1, 0.3, 1, 10, or 30 ppm naphthalene for 6 h. Necrosis of the olfactory tissue occurred in a concentration-dependent manner, starting at the lowest naphthalene concentration in SD rats (0.1 ppm), but only at exposures of 1 ppm or higher in F344 rats. Necrosis of the respiratory epithelium occurred in all rats at the two highest concentrations only (10 and 30 ppm). In the 5-day study, rats were exposed via inhalation to 0, 0.1, 1, or 10 ppm naphthalene for 6 h/day. In this study, concentration-dependent necrosis of olfactory tissue occurred in all exposure groups for SD rats but only at exposures of 1 ppm or higher in F344 rats. It is not clear why there is a difference in effects for the two rat strains; the authors concluded that “overall the strain difference was minimal.” Since the NTP carcinogenicity assay was conducted on F344 rats, 90-day exposure studies were conducted in this strain.
CitationDodd et al. (2012) conducted a 90-day study to observe the toxicity of various concentrations of naphthalene on nasal tissue in F344 rats. Rats were exposed via inhalation to 0, 0.1, 1, 10, or 30 ppm naphthalene for 6 h/day, 5 days/week. After 90 days, no toxicity was observed in the 0.1 ppm exposure group, and minimal hyperplasia of the respiratory epithelium was observed in the 1 ppm group. At 10 and 30 ppm, there was mild hyperplasia and metaplasia of the respiratory epithelium; degeneration, necrosis, and basal cell hyperplasia of the olfactory tissue; and hypertrophy/hyperplasia of goblet cells in the nasopharyngeal duct. Some recovery of the olfactory tissue was observed after a 4-week recovery period, but some degeneration and basal cell hyperplasia were still evident. Recovery of the respiratory epithelium at 4 weeks was nearly complete at all doses. The authors concluded that the threshold for cytotoxicity seen in this study may indicate a threshold for tumorigenicity as well.
CitationCichocki et al. (2014) observed an increase in cytotoxicity (via membrane permeability to ethidium homodimer-1) in rat nasal epithelium and olfactory mucosa following exposures to 15 and 30 ppm naphthalene for 6 h.
Primates
There are no in vivo naphthalene bioassays in primates. A preliminary study by CitationVan Winkle et al. (2014) examined cytotoxic effects in nasal epithelial tissue explants of male and female rhesus monkey following exposures to 10, 50, 100, and 500 μM naphthalene in a tissue culture medium for 3 h. The highest dose of 500 μM is likely equivalent to an inhaled concentration greater than 10 ppm, based on predicted nasal epithelial tissue concentrations in mice for a range of naphthalene inhalation exposure concentrations (CitationMorris 2013). CitationVan Winkle et al. (1996) established the reflection of in vivo conditions in an explant model for mouse lung, which likely would reflect in vivo conditions in rat and primate respiratory tissue as well. Tissue incubation and preparation conditions (other than a slightly different exposure time and dose) were the same as those described in CitationDeStefano-Shields et al. (2010), in which naphthalene metabolism was shown to occur via observation of naphthalene metabolite protein adducts. CitationVan Winkle et al. (2014) also included a positive control for GSH activity (acrolein). The authors found that naphthalene caused only minimal cytotoxicity in primate nasal epithelium.
Although explant studies can be quite informative with regard to a qualitative understanding of local metabolism and effects in a given tissue, there are several issues that should be considered with respect to how well explant studies reflect the in vivo environment. One drawback is that only local metabolism can be measured. As discussed in the toxicokinetics section, the majority of inhaled naphthalene in humans is metabolized in the liver (approximately 90%); therefore, the explant model does not reflect in vivo clearance of naphthalene through the liver. Further, although the explant assays are designed to closely reflect the in vivo environment (CitationVan Winkle et al. 1996), there is some uncertainty as to how well the explant studies compare to in vivo physiology in the primate nose (tissue oxygenation, blood supply, GSH cycling, CYP450 activity, etc.), and the results, therefore, should be interpreted carefully.
Overall analysis of data quality and consistency of results from animal studies
While the NTP's chronic exposure studies of mouse (CitationNTP 1992) and rat (CitationNTP 2000) provided evidence of naphthalene carcinogenicity, this occurred concurrently with cytotoxicity in the target tissues (mouse lung and rat nose). In fact, cytotoxicity was observed at all doses in both assays. Cytotoxicity was also observed in mouse nasal tissue, but without tumor formation. Since naphthalene was tested for carcinogenicity only at dose levels that were above the maximum tolerated dose, the results must be interpreted with caution. Shorter-term rat studies by CitationDodd et al. (2012), at inhalation exposures that included the NTP dose range, confirmed that cytotoxicity occurs in rat nasal tissue at doses lower than those used in the NTP study. The Dodd studies had the advantage of testing a wider range of doses than those that were associated with cytotoxicity in the NTP rat study, and they identified a cytotoxicity threshold for nasal toxicity in rats that was well below the lowest exposures of the NTP studies. These results should be taken into consideration when extrapolating from effects of naphthalene in rodents to potential effects in humans.
Toxicokinetics
Absorption and distribution
Upon inhalation, naphthalene is presumed to undergo absorption by passive diffusion across the alveolar membranes (CitationATSDR 2005). Two pharmacokinetic models have been developed to describe the distribution of naphthalene following inhalation in rats and mice (CitationWillems et al. 2001) and in humans (CitationCampbell et al. 2014).
Metabolism
The toxicity of naphthalene is dependent on its metabolism to reactive species. Because the patterns of toxicity are observed regardless of the route of administration (as discussed above), it is presumed that differences in the toxic response in different species and in various tissues within a species are due to differences in metabolism and bioactivation in the target tissues.
Naphthalene is metabolized by a variety of CYP isozymes. These include CYP2F2, CYP1A1, CYP1A2, CYP2A5, and CYP2E1 in mice; CYP2F4, CYP2E1, CYP1A2, and CYP2B in rats; CYP1A1, CYP2B1, and CYP2E1 in rhesus monkeys; and CYP2F1, CYP2A13, CYP1A1, CYP2A6, CYP2E1, CYP2B6, and CYP2S1 in humans (CitationBuckpitt et al. 2002; CitationBogen et al. 2008; CitationBoland et al. 2004; CitationFukami et al. 2008; CitationCruzan et al. 2009; CitationLi et al. 2011; CitationLewis et al. 2009; CitationGenter et al. 2006; CitationGreen et al. 2001; CitationKarlgren et al. 2005; CitationSimmonds et al. 2004; CitationThornton-Manning and Dahl 1997).
Naphthalene metabolism is complex, and the ultimate toxic metabolite(s) is/are not known. The first step in naphthalene metabolism is CYP-mediated formation of 1,2-naphthalene epoxide. This epoxide may react directly with cellular nucleophiles to form covalent adducts, or it may be transformed to other reactive metabolites (CitationBogen 2008). Alternatively, the epoxide may undergo detoxification via GSH conjugation and subsequent elimination in the urine as mercapturic acids (CitationBogen 2008; CitationBuckpitt et al. 2002). The latter pathway is likely predominant at low levels of naphthalene exposure. At higher levels of exposure, GSH may be depleted, allowing for the formation of reactive metabolites. One such pathway is the spontaneous rearrangement of naphthalene 1,2-epoxide to 1-naphthol (a major naphthalene metabolite) and subsequent metabolism to 1,4-naphthoquinone and other reactive metabolites. Another is the epoxide hydrolase-catalyzed formation of 1,2-dihydroxy-1,2-dihydronaphthalene (or dihydrodiol) and subsequent formation of 1,2-naphthoquinone and other reactive metabolites (see ). The relative amounts of intermediate metabolites, generation of reactive species, and the relative importance of the alternate pathways will vary depending on the species and type of tissue in which the metabolism is taking place.
Figure 2. Proposed scheme for naphthalene metabolism and reactive metabolites (adapted from CitationATSDR 2005). CYP450 = Cytochrome P450 Enzyme(s); GSH = Reduced Glutathione; SG = Glutathione.
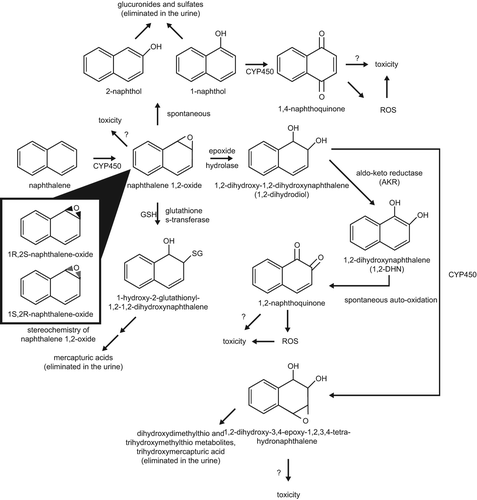
The relative amounts of different enantiomers of reactive metabolites produced in different tissues and species also appear to play a role in naphthalene toxicity. For example, the rate of formation of the 1R,2S-epoxide enantiomer correlates well with the toxicity of naphthalene among species, tissues, and cell types. The 1S,2R-epoxide is not as clearly associated with toxicity (CitationBuckpitt et al. 1992). In the mouse lung, 1R,2S-epoxide is the predominant enantiomer (CitationEUR 2003; CitationBuckpitt et al. 1992; CitationCruzan et al. 2009), correlating with toxicity. In nasal mucosa, the 1R,2S-epoxide is predominant in mice, rats, and hamsters (CitationBuckpitt et al. 1992), correlating with toxicity in mice and rats. In rat, hamster, monkey, and human lung tissue, the 1S,2R-epoxide is predominant (CitationCruzan et al. 2009; CitationBuckpitt et al. 1992). The 1R,2S-epoxide has been shown in mouse hepatocytes to be metabolized to the dihydrodiol at a much faster rate than the 1S,2R-epoxide (CitationBuckpitt et al. 2002). This may contribute to the greater cytotoxicity of the 1R,2S-epoxide compared to the 1S,2R-epoxide.
Mice
Club cells (nonciliated bronchiolar epithelial cells) are the primary target for naphthalene toxicity in mouse lung tissue, and this is true regardless of the route of exposure (CitationBuckpitt et al. 2002; CitationPlopper et al. 1992a,Citationb). Within the mouse lung, Club cells have the highest capacity to metabolize naphthalene compared to other cell types (CitationBogen et al. 2008).
CYP2F2 isolated from mouse liver catalyzes naphthalene epoxidation (CitationNagata et al. 1990), and the enzyme is believed to be predominantly responsible for the metabolism of naphthalene to naphthalene 1,2-epoxide in the mouse lung (CitationBuckpitt et al. 2002; CitationBogen et al. 2008). The concentration of CYP2F2 in mouse airway subcompartments is 6–40 times higher than the concentration of CYP2F4, the homologous enzyme, in rat airway subcompartments (CitationBaldwin et al. 2004). CYP2F2 has high activity in the mouse lung, with the highest concentration of the enzyme found in the distal airways. It is highly localized in mouse Club cells (CitationBuckpitt et al. 1992) and is stereoselective for the formation of the 1R,2S-naphthalene epoxide enantiomer (CitationBuckpitt et al. 2002). The rate of naphthalene metabolism is much higher in the mouse lung than in the rat lung, and, consequently, the rate of GSH depletion upon exposure to naphthalene is also higher (CitationBuckpitt et al. 2002). Depletion of GSH in the mouse lung leads to greater toxicity to Club cells, indicating that GSH conjugation is a major detoxification pathway in mouse Club cells (CitationWarren et al. 1982; CitationWest et al. 2000; CitationPhimister et al. 2004). This is evidenced by the fact that covalent binding of reactive naphthalene metabolites to mouse lung proteins is only observed after GSH is depleted (CitationBuckpitt and Warren 1983; CitationWarren et al. 1982; CitationPhimister et al. 2004).
Naphthalene is extensively metabolized in mouse nasal tissue, and the metabolism contributes to its uptake (CitationMorris 2013). In this study, upper respiratory tract uptake was concentration-dependent, with more efficient uptake at lower exposure concentrations (90% at 0.5 ppm) and less efficient uptake at higher exposure concentrations (50% at 30 ppm), with elimination of the effect upon treatment with 5-phenyl-1-pentyne CYP inhibitor, indicating saturation of naphthalene uptake and metabolism at high exposure concentrations. Mouse nasal tissue GSH is depleted upon exposure to naphthalene, indicating the involvement of GSH in the metabolic pathway for this tissue (CitationPhimister et al. 2004). There is some evidence that CYP enzymes other than CYP2F2 may be involved in naphthalene metabolism in mouse nasal tissue. Although the mouse nasal olfactory epithelium is high in CYP2F2, it also is abundant in other CYP enzymes, such as CYP2A5 (CitationCruzan et al. 2009). CitationLi et al. (2011) developed a Cyp2f2-null mouse strain. Upon exposure to naphthalene, the null mice were protected against lung toxicity but not against nasal toxicity (olfactory mucosa, or OM). The authors concluded that bioactivation of naphthalene by CYP2F2 is not necessary for OM toxicity and suggested that CYP2A5, which is abundant in mouse OM tissue, may be involved in naphthalene metabolism and toxicity in the mouse OM (CitationLi et al. 2011). Recently, the same researchers developed a Cyp2a5-null mouse strain and conducted a similar experiment to determine whether mouse CYP2A5 plays a role in the toxicity of naphthalene in the mouse OM (CitationHu et al. 2014). The authors found that upon naphthalene exposure, the null mice were more resistant than the wild type to OM toxicity but not to lung toxicity, indicating that CYP2A5 plays an essential role in naphthalene-induced OM toxicity in the mouse.
CitationGenter et al. (2006) have ruled out the involvement of CYP1A1 and CYP1A2 in mouse olfactory naphthalene metabolism and toxicity. This group developed Cyp1a1-null and Cyp1a2-null mice, and tested them for naphthalene toxicity. Neither strain of knockout mice were protected against toxicity. When these mice were treated with 5-phenyl-1- pentyne, a CYP2F enzyme inhibitor, they did not exhibit nasal toxicity from naphthalene exposure. The authors suggested that CYP2F is involved in naphthalene toxicity in mouse nasal tissue. This conclusion is inconsistent with the results from the more recent study by CitationHu et al. (2014); however, CYPs other than CYP2F are sensitive to 5-phenyl-1-pentyne inhibition (including CYP2E1 and CYP2A5) (CitationRoberts et al. 1998; CitationGreen et al. 2001).
CitationBuckpitt et al. (2013) measured the kinetics of naphthalene metabolism in microsomes isolated from mouse nasal and airway subcompartments and compared the metabolism rates to those in microsomes isolated from comparable tissues in rats and rhesus monkeys. Similar high rates of metabolism were observed in mouse nasal olfactory (Km = 50.2 μM, Vmax = 36.5 min− 1) and airway tissues (Km = 81.9 μM, Vmax = 48.3 min− 1), and these were comparable to the rates in the rat olfactory epithelium and much higher than rates in monkey nasal and monkey and rat airway tissue (discussed below). The rates of naphthalene metabolism in mouse tissues correlated well with their susceptibility to naphthalene toxicity.
CitationKedderis et al. (2014) conducted an in vitro study using lung, nasal epithelial, and liver cells from F344 rats, B6C3F1 mice, and humans to evaluate dose–response relationships for naphthalene-induced GSH depletion and cytotoxicity, and generation of naphthalene metabolites (dihydrodiol, 1,2-naphthoquinone, 1,4-naphthoquinone, naphthalene diol epoxide). Although there were some intriguing differences in generation of these metabolites across species and tissues (e.g., generation of 1,2-naphthoquinone in rat but not mouse nasal epithelial cells), the results of this study are difficult to interpret given the extremely high incubation concentrations applied (i.e., above saturation for CYP-mediated epoxidation at 500–2000 μM) and results that are inconsistent with observations in vivo. For example, rat nasal epithelial and mouse lung cells (where toxicity and tumor formation have been observed in vivo) and mouse nasal epithelial cells (where toxicity has been observed in vivo), although showing GSH depletion and metabolism to toxic metabolites of naphthalene, showed no statistically significant decrease in cell survival, even with concentrations as high as 2000 μM (likely equivalent to an inhaled concentration > 40 ppm in mice [CitationMorris 2013]). Rat lung cells (where toxicity and tumors have not been observed in vivo) also exhibited GSH depletion and metabolism to toxic metabolites of naphthalene with little decrease in cell survival. The results from human lung and nasal cells are also inconsistent; these cells exhibited GSH depletion and decreased cell survival but with no detectable metabolites of naphthalene. Although data suggest very low metabolism of naphthalene in these tissues in humans (discussed below), the GSH depletion suggests some metabolic capacity, yet no observable metabolites. The authors suggested that the results in human cells may have been due to a smaller GSH pool than in rodents.
Rats
In rats, as discussed above, the main target of naphthalene toxicity is the nasal tissue. Rat lung tissue is not a target, and rat Club cells are not affected by naphthalene administered by IP injection (CitationBuckpitt et al. 2002; CitationPlopper et al. 1992a,Citationb) or by inhalation exposure (CitationWest et al. 2001). The olfactory tissue has higher concentrations of CYP proteins than any other tissue in the rat (CitationBaldwin et al. 2004), and the rate of naphthalene metabolism in the olfactory epithelium is 40 times higher than that in the septal non-olfactory epithelium (CitationMorris and Buckpitt 2009). The rat enzyme CYP2F4 is homologous to the mouse CYP2F2 and is present in high concentrations in rat nasal tissue (CitationBaldwin et al. 2004). The concentration of CYP2F4 is much lower in rat lung than in nasal tissue, corresponding to the lower toxicity in the rat lung (CitationBaldwin et al. 2004). Another CYP enzyme, CYP2E1, is also concentrated in rat nasal tissue (CitationCruzan et al. 2009). The relative contributions of these two enzymes to naphthalene metabolism in the rat nose is not yet known. In rats, as in mice, the uptake and metabolism of naphthalene in nasal tissue is greatly reduced by the inhibition of CYP metabolism (CitationMorris and Buckpitt 2009).
Injury to the rat nasal olfactory tissue occurs regardless of the route of naphthalene administration. The pattern of injury, however, differs by route. When naphthalene is administered as an IP injection, the injury to the olfactory cells is evenly distributed throughout the nasal mucosa. When naphthalene is administered via inhalation, the amount of injury correlates with the amount of airflow that reaches the different nasal regions (CitationLee et al. 2005).
CitationLee et al. (2005) also monitored the metabolism of naphthalene in incubations with microsomes from different nasal regions. They found that naphthalene was metabolized at high rates by microsomes from the olfactory mucosa of the septum and of the ethmoturbinates, but at much lower rates by microsomes from the non-olfactory region of the septum. These rates correlated with the amount of CYP enzymes present in the tissues. The majority of metabolites in all incubations were GSH conjugates of naphthalene-1,2-epoxide. The primary metabolite in all three regions, accounting for approximately 70–78% of naphthalene metabolites, was 1R-hydroxy-2R-gluththionyl-1,2-dihydronapthalene, which is derived from the 1R,2S-naphthalene epoxide.
CitationBuckpitt et al. (2013) also measured rates of naphthalene metabolism in microsomes from rat tissue subcompartments. The authors found a high rate of metabolism in rat nasal respiratory (Km = 11.6 μM, Vmax = 8.8 min− 1) and olfactory (Km = 70 μM, Vmax = 42.5 min− 1) tissue, but a much lower rate in rat lung airway tissue (Km = 3.1 μM, Vmax = 0.45 min− 1). The differences in naphthalene metabolism rates correlate well with the toxicity of naphthalene in these tissues.
CitationCichocki et al. (2014) observed a significant reduction in GSH levels in both male and female rat nasal olfactory and respiratory epithelial tissue following naphthalene inhalation exposure concentrations of 1, 3, 10, and 30 ppm for 4 and 6 h, with greater loss in the respiratory epithelium than olfactory. These results indicate that GSH conjugation is a major detoxification pathway in rat nose.
Humans and other primates
The human enzyme CYP2F1 shares 82% homology with mouse CYP2F2 and is also found in the lung. Unlike mouse CYP2F2, CYP2F1 has a slight stereoselectivity for the formation of the 1S,2R-naphthalene epoxide (CitationBuckpitt et al. 2002). The rate of human CYP2F1 metabolism of naphthalene is also less than 0.1% that of mouse CYP2F2 (CitationBuckpitt et al. 2002), and human Club cells have barely detectable amounts of CYP2F1 (CitationCruzan et al. 2009). Recombinant human lung CYP2F1 has been shown to metabolize naphthalene to naphthalene epoxide in human lymphoblastoid cells at very low rates (CitationLanza et al. 1999; CitationBogen et al. 2008). CYP2F1 messenger ribonucleic acid (mRNA) has been identified in human respiratory tissue, but results in much lower expression than CYP2F4 in rats (CitationBogen et al. 2008; CitationRaunio et al. 1999; CitationDing and Kaminsky, 2003).
Another human CYP enzyme, CYP2A13, has also been shown to catalyze the metabolism of naphthalene. This enzyme is predominantly expressed in the respiratory tract, with the highest concentrations in the nasal mucosa, followed by the lung and trachea (CitationLewis et al. 2009; CitationFukami et al. 2008; CitationSu et al. 2000). In an in vitro cell-free assay, CYP2A13 catalyzed the conversion of naphthalene preferentially to 1-naphthol rather than 2-naphthol, and the conversion of 1-naphthol to 1,2- and 1,4-naphthoquinone (CitationFukami et al. 2008). CYP1A1, CYP1A2, CYP2A6, CYP2D6, CYP2E1, CYP2S1, and CYP3A4 are also expressed in human respiratory tissue and may contribute to naphthalene metabolism, although the relative quantities of these enzymes are not known (CitationChang et al. 2006; CitationDing and Kaminsky, 2003; CitationFukami et al. 2008; CitationKarlgren et al. 2005). CitationCho et al. (2006) showed that in liver microsomes, CYP2E1 activated naphthalene to 1-naphthol and 2-naphthol. This group also showed that CYP1A2 was the most active isoform for producing the dihydrodiol and 1-naphthol metabolites in liver microsomes, CYP1A2 and 2D6*1 were the most active isoform for producing 1,4-naphthoquinone, CYP3A4 was most effective for 2-naphthol production, and CYP2A6 and CYP3A4 were most active in metabolizing the dihydrodiol. The relative activities of these processes in lung tissue are unknown.
CitationKlotz et al. (2011) investigated the urinary naphthalene metabolites of 55 occupationally exposed workers. These authors detected 1,2-dihydroxynaphthalene (1,2-DHN) as the main urinary metabolite in 54 of the 55 subjects, at approximately 10-fold the amounts of 1- and 2-naphthol. In control subjects, the relative amounts of all three metabolites were comparable to each other. This may provide evidence for saturation of the metabolic pathways that produce 1-naphthol and 2-naphthol. 1,2-DHN is a precursor to 1,2-naphthoquinone (see ).
Other investigators have detected 1- and 2-naphthol in the urine of urban children (CitationOrjuela et al. 2012) and workers exposed to bitumen (asphalt) fumes (CitationMarczynski et al. 2011). These groups did not report on levels of urinary 1,2-DHN. Naphthalene excretion will be further discussed later in this paper.
Rhesus macaques have been used as models for human metabolism. In rhesus macaque lung microsomes, the rate of naphthalene metabolism is very slow compared to the rates in rodents, and is nearly identical to the rate observed in human lung microsomes (CitationBuckpitt et al. 1992). In both species, the 1S,2R-naphthalene epoxide enantiomer is preferentially formed (CitationBuckpitt et al. 1992; CitationBuckpitt and Bahnson, 1986) and has been shown in mouse hepatocytes to be metabolized to the dihydrodiol at slower rates than the 1R,2S enantiomer that is formed in other rodents (CitationBuckpitt et al. 2002). Microsomes isolated from rhesus macaque lungs metabolize naphthalene at rates 100-fold lower than mouse lung microsomes and 10-fold lower than rat lung microsomes (CitationBuckpitt et al. 1992). Rhesus macaque nasal tissue has far less CYP2F than rodent nasal tissue, with one-tenth the amount of that in rats and one-twentieth the amount in mice (CitationBaldwin et al. 2004). CYP2F was undetected in rhesus macaque pulmonary tissue in immunolocalization studies (CitationBaldwin et al. 2004).
The rates of naphthalene metabolism in rhesus macaque nasal and airway tissue microsomes were measured by CitationBuckpitt et al. (2013). The rates in rhesus macaque tissues were low; alveolar subcompartments (Km = 1.14 μM, Vmax = 0.019 min− 1) were well below (Vmax 2500-fold lower) those from mouse lung airway, and nasal compartments (Vmax ranged from 0.1 to 1.49 min− 1) were well below those from rat and mouse nasal tissue (Vmax 10- to 400-fold lower). These results suggest that primate nasal and lung tissue do not metabolize naphthalene as extensively as mouse and rat nasal and mouse lung tissue.
A preliminary study by CitationVan Winkle et al. (2014) examined GSH depletion in nasal epithelial tissue explants of male and female rhesus monkey following exposures to 10, 50, 100, and 500 μM naphthalene for 3 h. The authors found that naphthalene only began to deplete GSH at the highest concentration (500 μM), which is likely equivalent to an inhaled naphthalene concentration of greater than 10 ppm (based on predictions in the mouse nose) (CitationMorris 2013). As discussed later in this paper, naphthalene exposure concentrations in the general population are approximately 0.95 μg/m3 (0.00017 ppm) (CitationATSDR 2005).
A recent study by CitationDing et al. (2014) evaluated expression and activity of CYP2F1 toward naphthalene in a CYP2A13/2F1 humanized mouse (on a Cyp2abfgs-null background). The authors found that CYP2A13 and/or CYP2F1 were active toward naphthalene in the humanized mouse, with CYP2F1 contributing to metabolism primarily in the lung and CYP2A13 contributing to metabolism primarily in the nasal mucosa.
Covalent binding of metabolites to proteins
The ultimate cause of naphthalene toxicity is not completely understood, but evidence suggests that it could be related to covalent binding of reactive metabolites to cellular constituents, especially proteins (CitationBogen 2008). Protein adducts of naphthalene metabolites have been observed in cell-free systems in vitro (CitationPham et al. 2012a,Citationb), in isolated mouse Club cells in culture (CitationCho et al. 1994), tissue preparations of mouse and monkey lung (CitationBoland et al. 2004; CitationCho et al. 1994; CitationLin et al. 2006), rat and monkey nasal tissue (CitationDeStefano-Shields et al. 2010), and the mouse trachea (CitationCho et al. 1994). The doses in these studies were fairly high (250 to 500 μM), likely comparable to inhalation concentrations of greater than 10 ppm in mice (CitationVan Winkle et al. 2014; CitationMorris 2013).
Covalent binding of metabolites to DNA is discussed in the genotoxicity section.
Mice
The toxicity of naphthalene to mouse Club cells correlates well with the amount of covalent binding of reactive metabolites to proteins in these cells compared to non-target cells (CitationCho et al. 1994). CitationCho et al. (1994) observed highly selective binding of reactive naphthalene metabolites to proteins of specific molecular weights in Club cells in vitro compared to other lung cell types. CitationCho et al. (1994) also measured naphthalene metabolite binding in dissected mouse lung airway subcompartments. They found that the greatest amount of binding occurred in the subcompartment that included the distal bronchioles. CitationLin et al. (2005) studied the binding of naphthalene metabolites to mouse lung airway proteins in an in situ model. These investigators found that the adducted proteins included several involved in protein folding and translocation, mitochondrial proteins associated with production of adenosine triphosphate (ATP), and antioxidant enzymes. Any or all of these adducts may be involved in the selective toxicity to Club cells. CitationZheng et al. (1997) reported that 1,2-naphthoquinone covalently bound to proteins in mouse Club cells after exposure in vitro, and CitationWaidyanatha and Rappaport (2008) observed albumin and hemoglobin adducts of naphthalene-1,2-epoxide, 1,2-naphthoquinone, and 1,4-naphthoquinone in mouse blood after IP injection of these chemicals.
Rats
CitationDeStefano-Shields et al. (2010) measured the rates of naphthalene metabolite protein adduct formation in rat nasal tissue explants. They found that the rates were similar to those found in the mouse distal airway epithelium (CitationCho et al. 1994), suggesting that protein binding correlates well with toxicity. Adducted proteins in the rat nose included structural and catalytic proteins and some involved in the unfolded protein response. CitationCho et al. (1994) observed that protein adduct formation in rat tracheal tissue occurred at very low rates compared to adduct formation in mouse trachea and Club cells (less than 1% of the amount in Club cells and less than 10% of the amount in isolated mouse trachea). These differences also correlate with the relative toxicities of naphthalene in these tissues.
CitationWaidyanatha et al. (2002) measured the rates of covalent binding of naphthalene-1,2-epoxide, 1,2-naphthoquinone, and 1,4-naphthoquinone to albumin and hemoglobin of rats after IP injection of these chemicals. The authors found that all three naphthalene metabolites formed adducts with both proteins in a dose-dependent manner.
Humans and other primates
CitationDeStefano-Shields et al. (2010) also incubated naphthalene (250 μM) with nasal epithelium explants isolated from rhesus macaques. The rate of formation of covalently bound metabolites in rhesus macaque nasal epithelium was similar to that in the rat and lower than the rate in mouse distal airway epithelium by about half. This is in contrast to the relative amounts of CYP2F enzymes and the rates of naphthalene metabolism, which are much lower in rhesus macaques than in rodent airway tissues (CitationBuckpitt et al. 1992; CitationBaldwin et al. 2004). Similarly, in isolated lung tissue incubated with 500 μM naphthalene, CitationBoland et al. (2004) showed that rates of reactive naphthalene metabolite covalent protein binding were only 2- to 3-fold lower in rhesus macaque lung tissue than in mouse lung tissue (by comparison to CitationCho et al. 1994), although the rates of formation of naphthalene metabolites in the rhesus macaque airway epithelium were about 70-fold lower than those in the mouse. A naphthalene incubation concentration of 500 μM in the explant studies is roughly equivalent to naphthalene concentrations in mouse nasal tissue following inhalation concentrations of greater than 10 ppm (CitationMorris 2013). Therefore, exposure concentrations in these explant studies are high and could contribute to high levels of in vitro protein binding that may not occur in vivo at lower exposure concentrations (see more discussion in the HBWoE section).
The role of adduct formation with specific proteins in the toxicity of naphthalene is not clear. CitationLin et al. (2006) investigated the specific adducts formed in rhesus macaque vs mouse airway tissue. The only adducted proteins these authors found in common in both rhesus macaque and mouse tissue were actin, HSP70, and α-1-anti-trypsin precursor. Rhesus macaque adducts included more cytoskeletal, chaperone, and metabolic enzyme proteins, while mouse adducts included more proteins involved in folding and translation, ATP synthase, and redox protection. These findings suggest that differences in protein targets among species may contribute to the differences in species susceptibility to naphthalene toxicity.
CitationLin et al. (2009) also observed adducts of 1,2- and 1,4- naphthoquinone with serum albumin in the blood of human subjects. Levels of 1,2-naphthoquinone adducts were 5–6 times higher than those of 1,4-naphthoquinone. In contrast, the authors calculated cumulative tissue doses of 1,4-naphthoquinone to be about 3-fold higher than those of 1,2-naphthoquinone.
As discussed earlier, however, despite relatively high levels of protein binding in the primate nasal epithelium observed by CitationDeStefano-Shields et al. (2010), a preliminary study by CitationVan Winkle et al. (2014) found minimal toxicity in primate nasal tissue under similar experimental conditions.
Cell-free binding
CitationPham et al. (2012b) observed covalent binding of naphthalene metabolites naphthalene epoxide, naphthalene diol epoxide, 1,2-naphthoquinone, and 1,4- naphthoquinone to model peptides in a cell-free system. The binding occurred on cysteine, lysine, and histidine residues, and on the N-terminus of the peptides. Both quinone metabolites formed covalent bonds at higher rates than the epoxides, suggesting a greater reactivity for the quinones. The same metabolites incubated with the model proteins actin and protein disulfide isomerase (PDI) in a cell-free system also exhibited covalent binding to cysteine, lysine, and histidine residues on these proteins (CitationPham et al. 2012a). In this study, naphthalene epoxide bound at fewer sites than did naphthalene diol epoxide or the quinones. When the authors incubated naphthalene with actin or PDI in the presence of microsomes from a target tissue (rat nasal) or a non-target tissue (mouse liver), they again observed covalent binding to the model proteins but were unable to ascertain specific binding sites on the proteins or specific adducted metabolites of naphthalene (CitationPham et al. 2012a).
Excretion
Naphthalene, in the form of various metabolites, is mainly excreted in the urine. One of the major urinary metabolites is the glucuronide conjugate of 1-naphthol. Naphthalene conjugated to GSH is converted to premercapturic and mercapturic acids, and also excreted in urine. This is a major pathway of excretion in rodents, but its importance in primates is unclear (CitationATSDR 2005).
There is little information on the excretion of naphthalene in humans after inhalation exposure. In naphthalene-exposed workers, urinary levels of 1-naphthol (a major urinary metabolite of naphthalene) reached a peak at 1 h after the end of the workers’ shifts (CitationBieniek 1994, Citation1997). The mean excretion rate of 1-naphthol in these workers was 0.57 mg/h, and the half-time for urinary excretion was about 4 h (CitationBieniek 1994, Citation1997). CitationKlotz et al. (2011) compared urinary metabolites of naphthalene-exposed workers to those of control subjects and found differing ratios of metabolites 1,2-dihydroxynaphthalene (1,2-DHN), 1-naphthol, and 2-naphthol in the 2 groups. This study is discussed in more detail above. CitationWu et al. (2005) also found evidence of metabolite 1,2-DHN, as well as other DHNs, and 1- and 2-naphthol in the urine of workers and controls. One- and 2-naphthol have also been detected in the urine of urban children (CitationOrjuela et al. 2012) and of workers exposed to bitumen fumes (CitationMarczynski et al. 2011). To date, there are no readily available studies of excretion following inhalation exposure in rodents.
Toxicokinetic models
A hybrid computational fluid dynamic (CFD)–PBPK model for naphthalene in rats and humans was recently developed (CitationCampbell et al. 2014) to describe naphthalene tissue concentrations and rates of metabolism in the upper respiratory tract (nasal respiratory and olfactory epithelium, and lung) and liver. As discussed by the authors, the model predictions provided good concordance with measurements in vivo (i.e., naphthalene blood concentrations following intravenous and inhalation exposures in the rat NTP studies, in addition to upper respiratory tract extraction data in naïve rats and rats treated with a CYP2F inhibitor [5-phenyl pentene]). The model also predicted rat naphthalene olfactory and respiratory tissue concentrations consistent with those reported in a mouse hybrid CFD–PBPK model (CitationMorris 2013). For our purposes, two limitations of the model are (1) that only the first metabolic step (the generation of the epoxide) is modeled, with no description of the fate of this or subsequent metabolites; and (2) there is no similar version of the model describing metabolism and disposition in the mouse nose, lung, and liver.
As illustrated in , the amount of naphthalene metabolized in the rat nose is about 5-fold higher than that from the same inhaled dose in humans, based on the PBPK model results. Based on naphthalene tissue concentrations following 0.1 ppm naphthalene exposure in rats (6 h/day, 5 days/wk)—the concentration considered to be a no-observed-adverse-effect level (NOAEL) for rat nasal lesions in the CitationDodd et al. (2012) 90-day rat study—CitationCampbell et al. (2014) estimated regional gas dosimetry ratios (RGDRs) of 0.18 for dorsal olfactory tissue, 0.93 for ventral respiratory tissue, and 0.73 for the lung. RGDRs for the NOAEL, however, are much higher if based on the amount of naphthalene metabolized (i.e., 5.56 for dorsal olfactory tissue, 21 for ventral respiratory tissue, and 20.5 for the lung), reflecting the much lower metabolic activity toward naphthalene in the primate upper respiratory tract compared to rats.
Figure 3. Amount of naphthalene metabolized in rat and human dorsal olfactory, ventral respiratory, lung, and liver tissue per inhaled dose in accordance with the hybrid CFD–PBPK model for naphthalene (CitationCampbell et al. 2014).
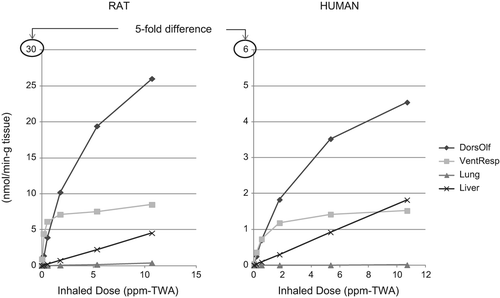
The CFD–PBPK model reports metabolized dose in rate “per gram tissue.” We estimated the total amount of naphthalene metabolized in each of the tissues shown in (dorsal olfactory, ventral respiratory, liver, and lung) based on information provided in CitationCampbell et al. (2014) with respect to percent bodyweight for each organ and an assumption of 300 g and 70 kg total body weight for rats and humans, respectively. Our estimates suggest that the majority of naphthalene is metabolized in the liver in both rats and humans (i.e., 5%, 14%, 0.3%, and 69% in rat dorsal olfactory, ventral respiratory, lung, and liver tissue, respectively; and 0.02%, 0.4%, 0.3%, and 89% in human dorsal olfactory, ventral respiratory, lung, and liver tissue, respectively). The totals add up to approximately 90%; the remaining naphthalene is likely exhaled or metabolized in other areas of the respiratory tract (e.g., trachea). Lack of toxicity in the liver is most likely due to high GSH levels in this tissue.
Overall analysis of data quality and consistency of results from toxicokinetic studies
There is a multitude of carefully conducted studies, produced by several different groups of investigators, showing clear consistencies in the data for naphthalene metabolism and toxicity. One of the most striking consistencies seen across studies is the role of CYP2F in mediating naphthalene toxicity and carcinogenicity. In rodent species and tissues with susceptibility to naphthalene toxicity and tumors (rat nose and mouse lung) at high doses, high concentrations of CYP2F are observed (CitationBuckpitt et al. 1992; CitationMorris and Buckpitt 2009; CitationBaldwin et al. 2004). A recent study by CitationLi et al. (2011) indicated that although CYP2F is likely involved in mouse lung toxicity, CYP2F is not involved in mouse nasal toxicity, where tumors are not observed.
The relationship is less well defined in rhesus monkeys and humans, but both primate species have lower amounts of CYP2F in airway tissues (CitationBuckpitt et al. 1992, Citation2002; CitationBaldwin et al. 2004; CitationCruzan et al. 2009) and may also exhibit less toxicity. It is also evident that the formation of the 1R,2S-epoxide correlates well with toxicity across species and tissues, and that the 1S,2R-epoxide, the predominant isomer in primates, is less relevant to toxicity (CitationBuckpitt et al. 1992, Citation2002; CitationCruzan et al. 2009). Studies have also consistently shown that the depletion of GSH in target tissues is a necessary step before toxicity can occur (CitationBuckpitt and Warren, 1983; CitationWarren et al. 1982; CitationWest et al. 2000; CitationPhimister et al. 2004).
The recent CFD–PBPK model (CitationCampbell et al. 2014) addresses species differences in the metabolism of naphthalene in the upper respiratory tract of rats and humans. The model suggests that metabolism of naphthalene is very low in the rat lung, consistent with little toxicity in that tissue, and higher in the rat nose where toxicity and tumors are observed. The model indicates that naphthalene metabolism in human nose and lung tissue is also very low, suggesting little toxicity in these tissues.
The relationship between covalent binding of naphthalene metabolites to proteins and tissue toxicity is uncertain, given the discrepancy between the rates of naphthalene metabolism in primates and rodents and the amount of protein binding observed in their respective tissues. While rodents exhibit 70-fold higher rates of naphthalene metabolism compared to primates (humans and rhesus monkey), the observed differences in protein adduct levels in rodents compared to rhesus monkeys are only 1- to 3-fold higher (CitationBoland et al. 2004; CitationDeStefano-Shields et al. 2010). Differences in protein targets among species, however, as shown by CitationLin et al. (2006), may contribute to the differences in species susceptibility to naphthalene toxicity. Further, it is also possible that these results are an artifact of the explant model and do not reflect the in vivo environment (see further discussion in the HBWoE section).
Genotoxicity
A considerable number of published studies reveal little evidence indicating naphthalene to be mutagenic. In 2005, the Agency for Toxic Substances and Disease Registry (ATSDR) reviewed the genotoxicity studies that were available at that time. The majority of these studies (close to 80%) reported negative results. In a review of 15 assays of reverse mutation of bacterial genes, CitationATSDR (2005) reported 14 negative and 1 weakly positive result. The weak positive result was from an assay for 1,2-naphthoquinone (CitationFlowers-Geary 1996), a reactive metabolite of naphthalene. In other types of bacterial gene mutation assays, 6 studies reported negative results both with and without rat S9 activation, and one study reported a positive result with S9 activation only. Out of 16 assays using eukaryotic cells in vitro, four reported positive results for genotoxic effects (sister chromatid exchange or chromosomal aberrations) and the rest reported negative results, including those for gene mutation and cell transformation. Among six in vitro assays that were conducted on human cells, only one showed a positive result for sister chromatid exchange associated with 1,2- and 1,4-naphthoquinone in human mononuclear leukocytes (CitationWilson et al. 1996). Among in vivo studies with eukaryotic organisms, five assays yielded negative results and five yielded positive results. Most of the studies tested genotoxicity rather than mutagenicity. Genotoxicity refers to DNA damage (e.g., DNA adducts or abasic sites). However, this damage can often be repaired before DNA replication occurs and therefore will not always lead to mutations. Observations of genotoxicity, therefore, cannot be taken to equal observations of mutagenicity. Further, most of the studies that were positive were found at high concentrations of naphthalene. These studies indicate that positive genotoxic results may be the result of: 1) saturation of detoxification mechanisms and generation of downstream genotoxic metabolites (likely to occur only at high concentrations that deplete detoxification mechanisms); and/or 2) secondary genotoxic effects due to cytotoxicity.
Reviews of naphthalene genotoxicity data by CitationSchreiner (2003), CitationBrusick et al. (2008),Citation and Brusick (2008) discussed the lack of evidence for naphthalene genotoxicity. CitationBrusick (2008) recently reviewed the studies described above and identified several factors that may influence the interpretation of the genotoxicity assay results. Notably, CitationBrusick (2008) concluded that most of the positive assays were either technically unsuited for testing the class of compounds to which naphthalene belongs, thereby generating unreliable data, or were subject to secondary genotoxic effects due to the cytotoxicity of naphthalene or its metabolites.
A number of naphthalene genotoxicity studies have become available since the ATSDR review was published. Most of these, discussed below, also point to a cytotoxic effect for naphthalene.
Recent rodent assays
CitationMeng et al. (2011) looked at the ability of naphthalene to induce mutations in the p53 tumor suppressor gene in rat nasal tissue in vivo after 90 days of exposure at low (noncytotoxic) and high (cytotoxic) concentrations (0.1, 1.0, 10, and 30 ppm naphthalene). There were no increases in p53 mutations at any dose, but there was a significant decreasing trend in p53 mutations with increasing naphthalene dose in male respiratory epithelia. The authors concluded that these results were most consistent with a loss of spontaneous p53 mutations due to chronic cytotoxicity.
CitationKaragiannis et al. (2012) injected naphthalene at 200 mg/kg (single dose) into mice (intraperitoneally), and recorded lung function, airway epithelial damage, and DNA strand breaks in lung tissue, using γH2AX as a molecular marker for DNA damage, at time points up to 72 h after injection. Significant airway epithelial damage was observed, with a maximum at 12 h and a decrease in damage at later time points. Significant increase in DNA strand breaks occurred, but not until 24 h, with a maximum at 48 h. The results of this study suggest that genotoxicity occurs subsequent to cytotoxicity in the mouse lung following naphthalene exposure, reaching a maximum only after the maximum cytotoxic damage was observed.
Recent mammalian in vitro assays
CitationRecio et al. (2012) conducted an in vitro assay to determine dose–response associations for naphthalene-induced micronuclei and cytotoxicity in human lymphoblasts. This group also looked at the effects of GSH on the dose–response relationships. Significant increases in micronuclei were only observed at naphthalene concentrations that also induced cytotoxicity, as measured by cell survival. The no-observed-effect level (NOEL) for induction of micronuclei was determined to be between 2.5 and 10 μM. In the presence of 5 mM GSH, however, no genotoxicity or cytotoxicity was observed at naphthalene concentrations up to 500 μM. The authors interpreted these results as evidence for a NOEL for naphthalene genotoxicity, indicating a cytotoxic MoA. They also concluded that naphthalene can be effectively detoxified by biological levels of GSH.
CitationKapuci et al. (2012) reported that naphthalene, 1-naphthol, and 2-naphthol all induced DNA fragmentation, as measured by the terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) assay, at concentrations that were not cytotoxic in human lymphocytes in culture. These authors found that the genotoxic effect occurred in the presence of 10–100 μM naphthalene or 2-naphthol, and in the presence of 50–100 μM 1-naphthol. These results are inconsistent with the CitationRecio et al. (2012) and CitationKaragiannis et al. (2012) studies described above, which showed genotoxicity only at cytotoxic concentrations of naphthalene. The human lymphoblast micronucleus assay employed by CitationRecio et al. (2012) has high predictivity for genotoxicity in vivo (CitationPfuhler et al. 2011). The TUNEL assay used by CitationKapuci et al. (2012) is generally used to measure DNA fragmentation that occurs during apoptosis, not to specifically evaluate genotoxic events (CitationMorley et al. 2006), and has a high degree of false positives as an apoptosis assay (CitationRibble et al. 2005). Therefore, it is likely that the genotoxicity at low doses in the CitationKapuci et al. (2012) study is due to problems with the assay and should not necessarily be attributed to naphthalene. This would explain the inconsistencies between results from CitationKapuci et al. (2012) and the CitationRecio et al. (2012) and CitationKaragiannis et al. (2012) studies.
Recent human sudies
Three recent studies have reported on the genotoxic effects of naphthalene in humans. CitationOrjuela et al. (2012) investigated the relationship between chromosomal aberrations in the lymphocytes of 5-year-old urban minority children and their urinary levels of 1- and 2-naphthol. The authors reported a statistically significant increase in chromosomal aberrations and translocations with increasing levels of naphthalene metabolites. They also measured three other categories of urinary PAH metabolites (pyrene, fluoranthene, and phenanthrene) and found no associations between any of these and lymphocyte chromosomal aberrations and translocations. The measurements of naphthalene metabolites and chromosomal aberrations did not vary according to whether or not the child was in the presence of a smoker during the 48 h before urine collection. It should be noted, however, that total PAH metabolites were not measured, nor were any other urinary components measured that might impact lymphocyte genotoxicity. Therefore, it is not possible to attribute the increase in chromosomal aberrations to naphthalene exposure.
CitationKrieg et al. (2012) studied genotoxicity in leukocytes of jet fuel-exposed workers, who are routinely exposed to naphthalene as a component of jet fuel. The authors found no relationship between naphthalene exposure as measured in breath samples and in air during work shifts, and leukocyte genotoxicity, as measured by the comet assay.
CitationMarczynski et al. (2011) looked at the relationship between occupational exposure to bitumen and DNA damage (oxidative and strand breaks via comet assay) in lymphocytes. Although exposed workers had statistically significant increases in DNA damage compared to unexposed workers, the amount of damage was within the range for unexposed and healthy populations, and there were no exposure–response relationships with ambient air measurements of bitumen fumes/aerosols or with urinary metabolites of naphthalene. Because of the lack of exposure–response relationships, the authors did not attribute the DNA damage in lymphocytes of workers to bitumen exposure.
DNA adducts
There is very little information about DNA adduct formation with naphthalene metabolites. CitationSaeed et al. (2007, Citation2009) showed that 1,2-naphthoquinone can react with DNA in vitro and in vivo in mouse skin to form depurinating N3- adenine and N7-guanine adducts. The significance of this is unclear, since a recent review by CitationBoysen et al. (2009) suggests that the unstable N7-guanine adducts, or their apurinic sites, are not the cause of mutagenesis in cells and tissues, and that stable adducts may be more relevant. Studies by CitationKim et al. (1995) and CitationKim et al. (2000) suggest that naphthalene can induce stable N6 adducts of deoxyadenosine from the tetrahydro diol epoxide (“diol epoxide”) of naphthalene. These adducts are well downstream of the initial naphthalene epoxide and would not likely form until GSH has been depleted and cytotoxicity has already occurred. Further, the presence of DNA adducts does not equal mutagenesis since there is an opportunity for the damage to be repaired prior to DNA replication.
Overall analysis of data quality and consistency of results from genotoxicity studies
The majority of genotoxicity studies for naphthalene have yielded negative results. The positive results were mainly observed in assays for genotoxicity rather than mutagenicity. Most cell culture and in vivo studies that were positive for genotoxicity were also positive for cytotoxicity at the same doses, casting doubt on the interpretation of these results as being due to direct reaction of naphthalene metabolites with DNA at less than cytotoxic doses. In addition, many of the genotoxicity assays were determined by CitationBrusick (2008) to be technically unsuitable and thus of little use in determining the genotoxicity of naphthalene. A more recent study by CitationKapuci et al. (2012) also applied a technically unsuitable method: the authors used DNA fragmentation that is generally used to measure apoptosis, not genotoxicity. Thus, the evidence for genotoxicity and/or mutagenicity of naphthalene is not strong. Unfortunately, most of the investigations into the genotoxicity of naphthalene were not conducted on target tissues (mouse lung and rat nasal). The two studies that were conducted on rodent target tissue found either no increase in genotoxicity (CitationMeng et al. 2011), or genotoxicity only in connection with cytotoxicity (CitationKaragiannis et al. 2012).
Of the four recent genotoxicity studies of naphthalene in human cells or tissue, three found either no evidence of increased DNA damage due to naphthalene exposure (CitationKrieg et al. 2012; CitationMarczynski et al. 2011), or DNA damage only in the presence of cytotoxicity (CitationRecio et al. 2012). The human studies by CitationKrieg et al. (2012) and CitationMarczynski et al. (2011) were well conducted and included analyses of exposure– response relationships. The study by CitationRecio et al. (2012) was an examination of naphthalene effects in vitro using a range of dose levels. Although the CitationRecio et al. (2012) study is consistent with a cytotoxic MoA (with a possible genotoxic component only at cytotoxic doses), all three studies were conducted using lymphocytes/lymphoblasts, and did not provide specific information about naphthalene effects on human respiratory tissue. The study by CitationOrjuela et al. (2012), also on lymphocytes, found an association between chromosomal aberrations and urinary levels of 1- and 2-naphthol. Again, results in lymphocytes do not provide information on potential human respiratory target tissues. Further, the authors did not measure total urinary PAH metabolites or other agents that might be genotoxic to lymphocytes.
Potential mechanisms of action
Several potential mechanisms have been proposed for naphthalene carcinogenesis to support a more general MoA. In addition to cytotoxicity, which has always been observed in target tissues for tumor formation, evidence has been reported for protein adducts and oxidative damage by reactive oxygen species, with limited evidence for direct DNA damage by naphthalene-specific metabolites. While all of these potential mechanisms may be present in target tissue, it is not clear how, or if, they are involved in the initiation of tumor formation.
Potential mechanisms of cytotoxicity and genotoxicity are described above and will not be reiterated here. Here, we summarize the potential involvement of protein adducts and reactive oxygen species in naphthalene carcinogenesis based on the data previously discussed, and also present recent toxicogenomic evidence of potential mechanisms of action. The potential mechanisms in the context of the MoA are evaluated more fully in our HBWoE evaluation in the following section.
Protein adducts
Evidence from several studies suggests that naphthalene toxicity could be related to covalent binding of reactive metabolites to cellular constituents, especially proteins (CitationBogen 2008). Protein adducts of naphthalene metabolites have been observed in various cultured cells (CitationCho et al. 1994), target respiratory tissues in rodents and non-human primates (CitationBoland et al. 2004; CitationCho et al. 1994; CitationLin et al. 2006; CitationDeStefano-Shields et al. 2010), and cell-free systems (CitationPham et al. 2012a,Citationb).
The amount of adducted proteins has been observed to correlate with the degree of naphthalene toxicity in both target and non-target tissues in rodents (CitationCho et al. 1994; CitationDeStefano-Shields et al. 2010). As discussed above, there is a discrepancy between the rates of naphthalene metabolism in primates and rodents and the amount of protein binding observed in their respective tissues. While rodents exhibit much higher rates of naphthalene metabolism in respiratory tissues compared to primates (humans and rhesus monkey), the levels of protein adduct in monkey and rodent lung and nasal respiratory tissues are similar (Boland et al.; 2004; CitationDeStefano-Shields et al. 2010).
The targeted proteins in the rat nose appear to be involved in protein folding and repair, including heat-shock proteins (HSP60 and HSP70), potentially leading to thiol oxidation and protein unfolding (CitationDeStefano-Shields et al. 2010). Although a specific mechanism is unclear, such disruption of functions by key regulatory proteins might result in cytotoxicity and or tumor promotion. Proteins found to covalently bind naphthalene metabolites specifically in the mouse lung are those involved in protein folding and translocation, production of ATP, and redox regulation proteins (CitationLin et al. 2005). Proteins found to covalently bind naphthalene metabolites specifically in rhesus monkey airway tissue are cytoskeletal, chaperone, and metabolic enzyme proteins (CitationLin et al. 2006). The only proteins modified in both species by naphthalene metabolites are actin, HSP70 (heat-shock protein), and α-1 anti-trypsin precursor (CitationLin et al. 2006). It is not clear how, or if, binding of naphthalene metabolites (including which specific naphthalene metabolites) to these proteins is involved in toxicity.
Reactive oxygen species
The generation of reactive oxygen species (ROS) has also been suggested as a possible factor in tumorigenesis of target tissues following administration of naphthalene (CitationUS EPA 2004; CitationBagchi et al. 1998a,Citationb, Citation2000, Citation2002). Generation of ROS may occur by redox cycling of naphthoquinone, or as a result of cytotoxicity and inflammation at the target site. ROS generation may lead to lipid peroxidation, depletion of reducing equivalents, and DNA oxidation or strand breaks (CitationATSDR 2005).
GSH depletion has been observed with high levels of administered naphthalene, such as those that have been associated with tumor formation in the mouse lung, (CitationPhimister et al. 2004). A significant reduction in GSH levels has also been observed in the rat olfactory and nasal respiratory epithelium at naphthalene inhalation exposure concentrations as low as 1 ppm (CitationCichocki et al. 2014). GSH protects cells against reactive species, such as reactive xenobiotic metabolites and oxygen radicals that are generated from those metabolites, or from naturally occurring intracellular processes (CitationBergamini et al. 2004; CitationPhimister et al. 2004). Thus, GSH depletion could lead to cellular injury via naphthalene-metabolite-induced protein adducts and/or oxidative stress, both of which may be precursors to cytotoxicity-induced tumor formation.
CitationCichocki et al. (2014) also examined potential differences in reduced (GSH) and oxidized glutathione (GSSG) levels in nasal respiratory and olfactory mucosa of male and female rats exposed to naphthalene vapor (1, 3, 10, or 30 ppm) for 4 or 6 h. The authors found no accumulation of GSSG in either tissue at any time point or dose, indicating no overwhelming response to naphthalene-induced oxidative stress. These results suggest that redox cycling of the 1,2-naphthoquinone, or 1,4-naphthoquinone, if formed, does not contribute to a large extent to toxicity in rat nasal tissue.
The CitationCichocki et al. (2014) results are consistent with several studies (albeit ones not conducted in target respiratory tissue), showing that naphthalene metabolites (naphthalene epoxide, 1,2- naphthoquinone, and 1,4-naphthoquinone) react readily with cellular proteins in rats and mice in a dose-dependent manner, such that protein binding may be removing metabolites from the opportunity to redox cycle (CitationZheng et al. 1997; CitationWaidyanatha et al. 2002; CitationWaidyanatha and Rappaport, 2008; CitationBuckpitt et al. 2002). In other words, cytotoxicity would occur well before any 1,2-naphthoquinone is available to undergo redox cycling to generate ROS that could react with DNA.
Toxicogenomics
A recent study by CitationClewell et al. (2014) evaluated genomic responses in the male and female rat nasal epithelium following naphthalene exposures to 0.1, 1, 10, or 30 ppm (6 h/day, 5 days/week) for 90 days. There were a few significant gene expression changes in the female (but not male) olfactory and respiratory epithelium at 0.1 ppm. At the 1 ppm exposure concentration, signaling pathway enrichment was altered only in the male respiratory epithelium. A large number of significant gene expression changes and pathway responses were observed in both sexes and tissues at 10 and 30 ppm naphthalene exposure. The genomic responses were related to oxidative stress, GSH metabolism, cell cycle, inflammation, and proliferation. The responses reflect results from the CitationDodd et al. (2012) study, where animals were exposed to the same concentrations and duration and exhibited no histopathological findings at 0.1 ppm, only minimal effects (respiratory epithelial hyperplasia) at 1 ppm, and marked cytotoxicity and hyperplasia at 10 and 30 ppm.
A recent study by CitationCichocki et al. (2014) examined possible sex differences in the induction of antielectrophilic genes (glutamyl cysteine ligase [catalytic subunit] [gclc], nicotinamide adenine dinucleotide phosphate-oxidase [NADPH], quinone oxidase 1 [nqo1], and heme oxygenase-1 [hmox1]) in rats exposed via inhalation to 1, 3, 10, or 30 ppm naphthalene for 6 h. As discussed by the authors, these genes can be induced upon electrophilic stress in response to the antioxidant response element (ARE), and therefore can be used as biomarkers of cellular oxidative stress. The authors found that these genes were induced in the olfactory mucosa and respiratory epithelium of both male and female rats following all naphthalene exposure concentrations, and that gclc induction in particular was increased more in male than in female olfactory mucosa. No sex differences were observed in the respiratory epithelium. Gclc codes for the protein responsible for the rate-limiting step in GSH biosynthesis, and therefore could be important in naphthalene detoxification. These results point to a possible GSH-diminished adaptive response in female vs male rats that could explain the greater sensitivity of female rats to olfactory carcinogenicity following exposure to naphthalene. Clearly, more data would be necessary before being able to draw a firm conclusion.
Hypothesis-based weight-of-evidence evaluation
The first two steps of our HBWoE evaluation, as outlined in , are presented in the first five main sections of this paper for each realm of investigation. Step 2 is further considered and articulated in the context of the hypotheses in Steps 4 and 5 below. In this section, we present Steps 3–5 of the evaluation.
Hypotheses under consideration
The third step in the HBWoE approach is to identify and articulate the main alternative conceptions about how naphthalene carcinogenicity might operate. These overarching hypotheses constitute the opposing sides of the main question (i.e., how the body of relevant observations is informative about potential human risk) that one wants to address through a weight-of-evidence evaluation. The hypotheses can be newly proposed or may have already been put forth within the scientific community. The hypotheses are articulated so that one can evaluate, throughout the process of weighing all the evidence, how well each hypothesis is supported by the available data, how well each hypothesis explains patterns in the data, what additional assumptions or hypothesized influences are necessary to account for discordances, and whether other expected events or processes for a given hypothesis were, in fact, observed.
We consider two overarching hypotheses that have been put forth to explain the MoA for naphthalene carcinogenesis in animals, with consideration of their potential relevance to humans.
Mutagenic MoA
The first overarching hypothesis proposes a mutagenic MoA for naphthalene. This hypothesis assumes that naphthalene metabolites (e.g., naphthalene-1,2-epoxide, 1,2-naphthoquinone, and 1,4-naphthoquinone) are generated early in the carcinogenesis process and at subcytotoxic doses, and that one or more of these metabolites react with DNA, or generate ROS that can react with DNA, as an early, initiating genotoxic event leading to mutations and tumors in mouse lung and rat nasal tissue. It would presumably operate at some level in all tissues and at all doses that are able to generate the metabolites that are DNA-reactive. This overarching hypothesis does not preclude some contribution of high-dose cytotoxicity or other cellular dysfunction in further promoting the mutagenic initiations, but it sees the mutagenic effect as necessary and sufficient.
Non-mutagenic MoA
The second overarching MoA hypothesis proposes that the critical and necessary factor in naphthalene tumorigenesis is cytotoxicity, or other marked cellular dysfunctions that can manifest in tissue disruption, such as chronic inflammation or hyperplasia. It proposes that naphthalene can increase cancer risks only in those tissues, and at those doses, causing such cellular and tissue dysfunction. It does not preclude that, despite the absence of direct interaction of naphthalene or naphthalene metabolites with DNA at low doses, there may be processes that emerge from or along with cytotoxicity that increase the chance of somatic mutation.
These overarching hypotheses are expressed at the level of mode of action rather than specific mechanisms. A variety of particular underlying mechanisms could be considered, and it is worthwhile to trace through the degree to which available data can or cannot identify and characterize the potential role of such possible mechanisms, since such considerations constitute part of the assessment of biological plausibility, and of the explanatory power of the overarching MoA hypothesis. However, it is not necessary to identify and prove the contribution of any particular mechanistic detail in order to evaluate the support for the more general MoA proposal. As we have described them, the two contending overarching MoA proposals represent the main distinctions needed for the present analysis: (a) whether there is an impact on cancer risk from lower doses, or whether there is an exposure threshold for cancer risk corresponding to the levels needed to induce observable tissue toxicity; and (b) whether the presence and character of tissue toxicity is informative about tissue-, sex-, route-, and species-specificity of where naphthalene-associated tumors appear in animal bioassays, and how this specificity informs the existence and nature of potential human risk.
Evaluation of the logic of the hypotheses for each line of evidence and all evidence combined
For the fourth and fifth steps in the HBWoE approach, we considered the logic of the two hypotheses in the context of each line of evidence (epidemiology, animal studies, toxicokinetics, genotoxicity, and other mechanistic data), and also all evidence combined. We do this by summarizing results from Steps 1–2 for each realm of evidence (discussed above) and asking specific questions based on our understanding of the current data, and in the context of the proposed hypotheses.
Epidemiology
As discussed earlier in this paper, there are no epidemiology studies that show an association between naphthalene exposure and lung or nasal cancer in humans. In fact, there is very little evidence of any cancer in humans linked to naphthalene exposure. The data insufficiencies preclude any definitive conclusions, especially concerning lung cancer, given its high background in the population. Nasal cancers, however, are very rare in humans, and a number of chemical exposures have been identified that increase nasal cancer risk. It is probable that if naphthalene caused nasal cancers in humans, there would be case study reports in the literature suggesting such an association. The lack of any observation of effects when there are clear exposures (e.g., in asphalt workers), in combination with nasal cancers being very rare in humans, suggests no obvious association.
It is important to consider the epidemiology results in the context of the protein adducts observed in monkey nasal respiratory epithelial explants. Although the role of protein adducts in primate respiratory tissue is unclear, and could be an experimental artifact owing to high local metabolism with no clearance through the liver, the lack of cytotoxicity in nasal explants (CitationVan Winkle et al. 2014), following the same exposures that lead to protein adducts, is consistent with the lack of nasal tumors in the epidemiology studies. These results further call into question the relevance of the protein adduct results, suggesting that the existence of protein adducts in other explant tissues should be interpreted cautiously. Note that since CitationVan Winkle et al. (2014) is still a preliminary study, confirmation of these results is needed.
In order to address the potential for naphthalene to cause respiratory cancer in humans, we have compared naphthalene metabolism rates across species and tissues (rat, mouse, primate nose and lung), and have considered studies evaluating different CYP450s and other enzymes involved in metabolism of naphthalene in human respiratory tissues compared to rodents. We have also looked closely at tissue dosimetry in rats and applied a rat/human PBPK model to estimate nasal and lung tissue concentrations and metabolized doses in humans to try to predict whether typical human exposures could lead to respiratory tissue concentrations or metabolized doses that may lead to carcinogenesis.
We describe our analysis further below, in consideration of uncertainties and inconsistencies across studies and how they impact human relevance.
Animal studies
Based on our understanding of the animal toxicity data, as described earlier in this paper, we ask the following questions:
Is cytotoxicity sufficient to cause tumor formation in rodents? Why are there no tumors in the mouse nose although there is cytotoxicity?
Cytotoxicity appears to be necessary but not sufficient to cause tumors in rodents, based on the observation that there is cytotoxicity in the mouse nose at high exposure concentrations but no tumors. Although one could argue that mice simply need a higher dose of naphthalene before nasal tumors would be observed, the fact that the nasal respiratory epithelial hyperplasia in the mouse nose was quite extensive (67 out of 69 animals in the 10 ppm group, and 134 out of 135 animals in the 30 ppm group; in fact, a higher frequency of respiratory epithelial hyperplasia was observed in mouse vs rat nose), and yet, not one animal had a nasal tumor, suggests there is something different about how naphthalene is metabolized in the mouse vs the rat nose that results in tumors in the rat but not the mouse nose. The non-neoplastic effects in the rat and mouse nose from the 2-year NTP studies (2000, 2002) are very similar; that is, extensive chronic inflammation and regenerative hyperplasia occur in both the rat and mouse nose. The only clear difference appears to be atypical hyperplasia in the rat olfactory epithelium but not in the mouse olfactory epithelium. This difference may contribute to olfactory tumors in the rat but not the mouse nose. Hyperplasia is defined as an increase in the number of normal cells in a normal arrangement within a tissue; it exhibits a normal growth pattern and maturation sequence, and is not generally considered “preneoplastic” (CitationEustis 1989). Atypical hyperplasia (dysplasia), on the other hand, is defined as a proliferation of cells characterized by cellular atypia, alteration in the maturation sequence, or abnormal differentiation of cells within a tissue, and frequently indicates the emergence of a population of cells that may become cancerous (CitationEustis 1989). As discussed in CitationLong et al. (2003), atypical hyperplasia as observed in the rat tumor assay was considered “an unusual proliferative lesion… [and] may represent a precursor for nasal olfactory carcinogenesis.”
We have also compared naphthalene metabolism rates and involvement of different CYP450s and other metabolic enzymes (e.g., epoxide hydrolase) across species and tissues (rat, mouse, primate nose and lung) to try to address this question in the context of the proposed hypotheses. See further discussion in the toxicokinetics section below.
Is there co-occurrence of non-neoplastic lesions and tumors in the rat nose?
As part of our evaluation of an association between cytotoxicity, inflammation, and hyperplasia with tumor formation in the rat nose, we obtained individual animal pathology data from the NTP bioassay (CitationNTP 2000, Citation2012) to examine the co-occurrence of these lesions. Olfactory epithelial hyperplasia (atypical) occurred in almost 100% of male and female rats, with and without epithelial neuroblastomas, in all dose groups (10, 30, and 60 ppm). Although nasal respiratory epithelial adenomas were observed in male rats, nasal respiratory epithelial hyperplasia only occurred in 43–60% of male rats (and 37–45% of female rats), with a similar percentage of occurrence for respiratory epithelial degeneration and metaplasia. Therefore, we examined the co-occurrence of neoplastic and non-neoplastic nasal lesion data from individual male rats that developed respiratory epithelial adenomas. Interestingly, although the NTP assay did observe olfactory neuroblastomas amid non-neoplastic lesions (atypical hyperplasia) in female rats, NTP did not observe consistent co-occurrence of nasal respiratory epithelial degeneration, hyperplasia, and/or metaplasia in male rats that also had respiratory epithelial adenomas for all dose groups (10, 30, and 60 ppm). In the 10 ppm group, of the six animals that had adenomas, two had no reported respiratory epithelial degeneration, hyperplasia, and/or metaplasia. In the 30 ppm group, of the eight animals that had adenomas, two had no reported degeneration, hyperplasia, and/or metaplasia. In the 60 ppm group, of the 15 animals that had adenomas, three had no reported degeneration, hyperplasia, and/or metaplasia. The frequency of each of the lesions in animals that did not develop tumors is approximately the same or even slightly higher in some dose groups than in animals with respiratory epithelial adenomas. Furthermore, the frequency of an animal having a respiratory epithelial adenoma and at least one of these non-neoplastic lesions was 67%, 75%, and 87% in the 10, 30, and 60 ppm group, respectively. In the animals without tumors, the frequency of having at least one of these non-neoplastic lesions was not very different, and again even slightly higher than in the animals with tumors (79%, 88%, and 88% in the 10, 30, and 60 ppm group, respectively).
Consistent with our analysis, the NTP report stated that “In the present study, the cells involved in this lesion [olfactory neuroblastoma] and focal areas of intraepithelial hyperplasia/dysplasia appeared to be morphologically similar to and form a continuum with the neuroblastomas. In the respiratory epithelium, there was no clear association between the morphologies of the non-neoplastic proliferative changes and the development of respiratory epithelial adenomas” (CitationNTP 2000). CitationLong et al. (2003), referring to the NTP rat bioassay, stated that “a few animals had localized proliferative changes of the respiratory epithelium that were morphologically similar to respiratory epithelial adenomas.Citation” Harkema (2001) reviewed the histopathological findings from the NTP bioassay and reported consistent findings with NTP with regard to the morphologic character of the nasal tumors. CitationHarkema (2001) also noted that neoplastic lesions were often concurrent with inflammation, epithelial degeneration, epithelial hyperplasia, and glandular hyperplasia, and that this association was especially true for the neuroepithelial carcinomas.
How can the 90-day rat nasal study inform the apparent lack of consistent co-occurrence of non-neoplastic and neoplastic lesions in male rat respiratory epithelium in the NTP study?
The NTP (CitationNTP 2000) chronic naphthalene rat bioassay reported respiratory epithelial adenomas in Levels 1 and 2 of the nasal cavity. CitationDodd et al. (2012) observed transitional/respiratory epithelial hyperplasia in Level 2 of the nasal cavity (comparable to NTP Level 1 as per Dodd et al. [2012]) in 100% of rats exposed for 90 days to 1, 10, or 30 ppm naphthalene. Although the NTP data suggest that there is not a clear continuum from respiratory epithelial hyperplasia or other non-neoplastic respiratory epithelial lesions to tumors in the rat nose at the 2-year time point, the locations of hyperplasia in the 90-day study are the same locations where tumors were observed in the NTP study. These results suggest that there is a likely association between locations of respiratory epithelial hyperplasia and adenomas in the rat nose.
There is the possibility that the tumors obliterated the non-neoplastic lesions and therefore they were not observed in the tissues at the 2-year time point in the NTP study. Another possibility is that the non-neoplastic lesions that were consistently observed in the male rat respiratory epithelium at the 90-day time point in the CitationDodd et al. (2012) study, particularly respiratory epithelial hyperplasia (10/10 animals at 1, 10, and 30 ppm), resolved after 90 days before tumors were observed. CitationDodd et al. (2012) did show a dramatic decrease in respiratory epithelial hyperplasia and metaplasia in male and female rats following a 4-week recovery period from naphthalene exposure (e.g., 0/9, 1/9, and 0/9 hyperplasia at 1, 10, and 30 ppm, respectively, in male rats). In the same study, a 4-week recovery period did not result in as dramatic a decrease in olfactory hyperplasia, degeneration, or necrosis. Although the NTP assay involved continuous exposure, the CitationDodd et al. (2012) results suggest that the respiratory epithelium is capable of resolving respiratory epithelial hyperplasia and metaplasia.
Overall, although further studies would be helpful to elucidate an association between non-neoplastic and neoplastic lesions in male rat respiratory epithelia, the association of these lesions in female rat olfactory epithelia, in addition to the male rat nasal pathology data from the 90-day study, suggest important roles for cytotoxicity and regenerative hyperplasia with nasal olfactory and respiratory epithelial tumor formation.
What accounts for sex differences in tumor incidence in rats and mice?
It is not clear what accounts specifically for the sex differences in naphthalene-induced tumor incidence in mice and rats.
There is evidence that female mice are more susceptible to naphthalene-induced lung toxicity than male mice, and that airway epithelial repair occurs more slowly in males than in females (CitationVan Winkle et al. 2002; CitationOliver et al. 2009; CitationSutherland et al. 2012). It is also possible that the differences are related to differences in background incidences of alveolar/bronchiolar carcinomas in male (8%) and female (3%) B6C3F1 mice from the NTP historical control database (for years 1983–2007) (CitationMoore et al. 2013); lower background levels in females may allow for a more likely observed difference between control and exposed if there is a real effect. It is also worth considering the possibility that given the high background of lung tumors in B6C3F1 mice, and that only one statistically significant lung tumor was observed in one female mouse, naphthalene exposure is not related to lung tumors in mice.
There is little evidence for sex differences in the nasal toxicity response in rats. A recent study by CitationCichocki et al. (2014), discussed earlier, examined potential differences in GSH and GSSG levels in nasal respiratory and olfactory mucosa of male and female rats exposed to naphthalene vapor (1, 3, 10, or 30 ppm) for 4 or 6 h, and also analyzed expression of antioxidant response genes npo1, gclc, and hmox-1. The authors found that overall, there was no consistent sex difference observed in GSH levels between males and females in either tissue. All three genes examined, however, were significantly more induced in the olfactory epithelium of males compared to females at one or more exposure concentrations. These results are consistent with a greater sensitivity to naphthalene toxicity in female vs male olfactory epithelia. Males, however, also induced expression of npo1 and hmox-1 in the nasal respiratory epithelium to a greater extent compared to females, which is not consistent with increased nasal respiratory epithelial adenomas in males compared to females. Therefore, the reason for the sex difference in naphthalene induction of nasal respiratory epithelial adenomas remains unclear.
What do primate nasal explant cytotoxicity studies suggest about the potential for naphthalene-induced human respiratory toxicity/carcinogenesis?
As discussed earlier, a preliminary study in primate nasal explant tissue (CitationVan Winkle et al. 2014) observed little cytotoxicity even at exposure concentrations as high as 500 μM, likely equivalent to a greater than 10 ppm inhalation concentration in mice (CitationMorris 2013). These results suggest that human exposures as high as 10 ppm are unlikely to elicit more than minimal respiratory toxicity.
Conclusion from animal studies with respect to hypotheses
Overall, although additional studies could further elucidate associations between non-neoplastic and neoplastic lesions in respiratory epithelium, the animal data suggest that cytotoxicity, which is elicited only at comparatively high naphthalene concentrations, plays a key role in tumor formation. However, it is still not clear why cytotoxicity in the mouse nose does not lead to tumor formation. There is a need to look at differences in metabolism in mouse, rat, and primate, male and female, and target and non-target tissues to identify differences across species, sex, and tissue that may help explain observed differences in tumor formation and inform the mechanism of action.
Toxicokinetics
Based on our understanding of the current naphthalene toxicokinetic data described earlier in this paper, we ask the following questions:
Are there differences in naphthalene metabolism in the rat and mouse nose that could explain lack of tumors in the mouse nose?
As shown in , the naphthalene epoxide can be metabolized by epoxide hydrolase (EH) to the 1,2-dihydroxy-1,2-dihydronaphthalene (“dihydrodiol”), and then further by dihydrodiol dehydrogenase (DD) (also referred to as aldoketo-reductase or “AKR”) to the 1,2-naphthoquinone. Since there are studies indicating the involvement of the 1,2-napthoquinone in naphthalene cytotoxicity, and the potential for its genotoxic effects, understanding the tissue and species variability of the enzymes involved in formation of the 1,2-naphthoquinone, or other downstream naphthalene metabolites (such as the diol epoxides) is important for evaluating a mechanism of action for naphthalene carcinogenicity. Understanding species and tissue variability in the balance between the levels and activities of CYP2F (or other CYPs) and other enzymes (GSH S-transferase, EH, DD) involved in formation or removal of the 1,2-naphthoquinone or other downstream metabolites, or enzymes involved in repair of DNA damage induced by these metabolites, will help explain differences in species and tissue responses to naphthalene.
For example, as shown in a study by CitationGreen et al. (2001), the activity of EH toward metabolism of the styrene metabolite, styrene epoxide, is about 10-fold higher in the rat than in the mouse nose. The authors suggested that this could be why the rat nose is less susceptible to styrene toxicity than the mouse nose, because the rat is able to more rapidly detoxify the epoxide. A higher activity of EH toward naphthalene would likely have a different outcome and could suggest formation of the dihydrodiol and 1,2-naphthoquinone at a higher rate in the rat vs the mouse nose, and could also explain the difference in response to naphthalene injury between the mouse and rat nose. That is, upon high levels of naphthalene exposure, GSH depletion, and cytotoxicity, higher levels of EH activity in the rat nose could lead to higher levels of 1,2-naphthoquinone that could lead to genotoxic effects (either from direct reaction with DNA or redox cycling to generate DNA damage via ROS) on already hyperplastic tissue, potentially leading to regenerative hyperplasia and tumors. Further studies investigating the metabolic differences toward naphthalene in the rat and mouse nose, particularly involving EH, could possibly provide data to support this hypothesis.
More recent studies suggest that involvement of different CYPs in naphthalene metabolism in the rat and mouse nose may explain differences in toxicity. As shown in the CYP2F2 knockout mouse study by CitationLi et al. (2011), null mice were protected against lung toxicity but not against OM toxicity, suggesting that bioactivation of naphthalene by CYP2F2 is not necessary for OM toxicity in mice. A recent study by CitationHu et al. (2014) showed that CYP2A5 null mice were protected against OM toxicity but not lung toxicity, suggesting that bioactivation of naphthalene by CYP2A5 is necessary for OM toxicity in mice. It is not clear, however, why activation of naphthalene to the epoxide by a CYP other than CYP2F would lead to cytotoxicity but not tumor formation. Perhaps CYP2F is involved in further metabolism to a downstream toxic metabolite of naphthalene (e.g., 1,2-naphthoquinone, or the diol epoxides). Although this is a possibility, there is no direct evidence to suggest that this is the case for naphthalene.
However, a recent study of CYP2F2 involvement in styrene toxicity in mouse lung (CitationCruzan et al. 2012, Citation2013) suggests that styrene 7,8-epoxide does not cause lung toxicity in CYP2F2 knockout mice. The authors discussed that the results suggest CYP2F2 metabolism is responsible for styrene-induced mouse lung toxicity possibly via metabolism of styrene to ring- oxidized metabolites and further to 4-hydroxy styrene and then to 3,4-vinyl catechol or quinone metabolites. CYP-mediated metabolism of 1-naphthol to the 1,2-naphthoquinone, or to metabolites further downstream, has been proposed (CitationBrusick et al. 2008). If CYP2F2 is not involved in naphthalene-induced nasal toxicity in mice, as suggested by CitationHu et al. (2014), this may explain why toxicity, but not tumors, occurs in the mouse nose; that is, downstream toxic metabolites of naphthalene that may lead to tumorigenesis (such as the 1,2-naphthoquinone) may not be formed to a significant extent. By comparison, involvement of CYP2F4 in naphthalene-induced nasal toxicity in rats may be via generation of the epoxide and the downstream toxic 1,2-naphthoquinone.
The formation of the 1,2-naphthoquinone in different species depends on the balance of activities of CYP2F (or other CYPs), GSH S-transferase, EH, and DD (or AKR), and possibly other metabolic enzymes. Since GSH depletion has been shown to precede naphthalene-induced cytotoxicity (CitationPhimister et al. 2004), it is plausible that formation of the epoxide followed by GSH depletion are the first (and obligatory) steps in naphthalene-induced injury, but that the potential tumorigenic outcome of the injury will vary depending on the extent of activities of downstream enzymes. An understanding of the balance of activities of these enzymes in different target tissues (e.g., mouse lung and rat nose), and in individual cells, will help explain the differences in species and tissue responses to naphthalene injury. Further, understanding the balance in humans will allow for an extrapolation of the mechanism from animals to humans (discussed below).
Finally, it is worth considering that given the high rate of naphthalene metabolism and cytotoxicity in the mouse nose compared to the rat nose, it is possible that enhanced cytotoxicity in the mouse nose compared to that of the rat leads to diminished progression of initiated cells to tumors. The effect of cytotoxicity on tumor formation, however, cannot be assessed from the NTP studies, given that all doses in the studies are close to 100% cytotoxic.
How does naphthalene metabolism in rodents compare to monkey and human metabolism in the nose and lung? How might this comparison inform the mechanism of action and potential human relevance? Are rodents a good model for human metabolism and toxicity?
As discussed earlier, the current available data indicate that CYP2F1 is expressed in human respiratory tissue to a much lower extent compared to CYP2F4 in the rat nose, and also that the rate of human CYP2F1 metabolism of naphthalene is much lower than that of mouse CYP2F2. Other CYPs may be involved in naphthalene metabolism in human lung tissue (such as CYP2E1, CYP1A2, CYP3A4, CYP2A6, and CYP2A13, discussed earlier). To our knowledge, experiments examining activities of these CYPs toward naphthalene in human respiratory tissue, or in individual human lung or nasal cells, have not been conducted.
Moreover, since more recent studies suggest that CYP2A5 is responsible for naphthalene-induced toxicity in the mouse nose (CitationHu et al. 2014), it will be important to consider how the human homolog (CYP2A6), in addition to CYPA13 and CYP2F1, is involved in metabolism of naphthalene in human respiratory tissue. A recent CYP2A13/2F1-humanized and CYP2A13(only)-humanized mouse study by CitationDing et al. (2014) suggests that although both CYP2A13 and CYP2F1 are active toward naphthalene in the humanized mouse nose and lung, CYP2A13 is involved in metabolism primarily in the nose and CYP2F1 primarily in the lung. CYP2A13 and CYP2A6 are part of the same CYP2A subfamily and have 95.4% sequence homology (CitationLeclerc et al. 2010). These preliminary results could suggest that the human nose may be more similar to the mouse than the rat nose in the suite of metabolic processes in action, and thus perhaps in response to naphthalene exposure (i.e., cytotoxicity but no carcinogenicity). Beyond this possible qualitative difference affecting the sufficiency of cytotoxicity to prompt tumorigenesis, it remains true that humans are unlikely to reach concentrations high enough to lead to toxicity, given the much higher naphthalene metabolism in the mouse compared to human nose.
Although we are beginning to better understand the different CYPs involved in the initial metabolism of naphthalene in the human respiratory tract, compared to rodent tissues, there is not enough information on what the human/rodent differences may mean in terms of informing a specific mechanism of action in humans. There are sufficient data, however, to inform a MoA. The current data indicate that naphthalene metabolism to the epoxide (the obligatory first step) is considerably lower in human respiratory tissue compared to rat nose or mouse lung, suggesting that regardless of the CYP involved, the overall metabolism is lower, and therefore toxicity in human tissue is expected to be lower. These results are supported by the lack of cytotoxicity in primate nasal explants exposed to high concentrations of naphthalene (CitationVan Winkle et al. 2014).
As predicted by the CFD–PBPK model (CitationCampbell et al. 2014), the amount of naphthalene metabolized in the human nose and lung is much lower than that metabolized in the rat nose. The human/rat RGDRs for the NOAEL exposure in rats (0.1 ppm for 6 h/day, 5 days/week), based on the amount of naphthalene metabolized, are 5.56 for dorsal olfactory, 21 for ventral respiratory, and 20.5 for lung tissue, reflecting the much lower metabolic activity toward naphthalene in the human upper respiratory tract compared to rats.
Levels of EH and DD (AKR) in human tissues are known to be highly active (CitationBuckpitt et al. 2002), raising the concern that the protection against naphthalene-induced injury that human respiratory tissue gets from low levels of initial metabolism to naphthalene epoxide could be eroded to some degree by more efficient further metabolic activation of the epoxide that is formed. Since naphthalene metabolism in human respiratory tissue is much lower than in rodents, however, and therefore the extent of naphthalene epoxide formation is low enough that detoxification processes (i.e., GSH conjugation) are not likely to be overwhelmed, the majority of epoxide formed in human respiratory tissue is expected to be detoxified before it can be further metabolized by EH and DD (AKR) to more toxic naphthalene metabolites (quinones or diol epoxides).
Recent studies suggest that GSH is depleted in the rat nose at concentrations as low as 1 ppm naphthalene (CitationCichocki et al. 2014), but only begins to deplete in primate nasal explants at very high exposure concentrations (equivalent to approximately 10 ppm inhalation exposure in mice) (CitationVan Winkle et al. 2014).
One cannot rule out the possibility, however, that a small amount of epoxide in primates is further metabolized to more toxic metabolites. Since the levels of these metabolites would likely be very low such that they would be managed by cellular detoxification processes (GSH conjugation or DNA repair processes), they would be expected to contribute minimally to toxicity. See further discussion below.
Differences between humans and rodents in the balance of expression and rates of activities of enzymes involved in formation of naphthalene metabolites (e.g., epoxide and 1,2-naphthoquinone) and cellular detoxification mechanisms are important in evaluating potential human relevance. The very low rate of naphthalene metabolism in humans compared to rodents, as shown quantitatively in the dose-response section later in this paper, suggests that the mechanism for naphthalene-induced carcinogenesis in rodents is not likely to occur in human respiratory tissue at typical human exposure concentrations.
If naphthalene metabolism is very low in primate respiratory tissue, why are naphthalene protein adducts generated at the same rate in rhesus monkey lung and nasal explant tissue compared to mouse lung and rat nasal explant tissue?
As discussed, CitationDeStefano-Shields et al. (2010) and CitationBoland et al. (2004) observed covalent protein binding in rhesus monkey nasal epithelial and lung tissue, respectively, at rates similar to those in the rat nose and the mouse lung following naphthalene exposure. This is in contrast to the much lower rates of naphthalene metabolism in the rhesus monkey than in rodent airway tissues (CitationBuckpitt et al. 1992; CitationBaldwin et al. 2004). Given that explant studies involve incubation of naphthalene in vitro in dissected monkey airways and nasal epithelium, only local metabolism can be measured in the assays. As was observed in the PBPK model, however, the majority of inhaled naphthalene in humans is metabolized in the liver (approximately 90%). Therefore, without clearance from the liver, there is no process competing for removal of naphthalene, which can remain in the culture medium until it is eventually (even if slowly) metabolized locally in the respiratory explants. Given the high exposure concentrations in the explant studies (250 μM), metabolism is likely high enough to cause a buildup of naphthalene metabolites that can react with proteins, but this may not reflect what is happening in vivo. Although this is a reasonable explanation, further studies would be necessary to determine whether this is the explanation. Studies of protein adducts in the rat lung and mouse nose, where tumors are not observed, would be helpful to try to understand the potential for adduct formation in vitro and how it may or may not be related to toxicity in vivo.
As discussed earlier, although protein adducts have been observed in monkey nasal and lung explants following naphthalene exposure, a preliminary study (CitationVan Winkle et al. 2014) observed minimal cytotoxicity in nasal explants following the same or higher exposures (500 μM). These results suggest that even if protein adducts do occur in vivo in humans, they will not lead to cytotoxicity and, therefore, carcinogenicity. Further, as shown by CitationLin et al. (2006), differences in protein targets among species may contribute to the differences in species susceptibility to naphthalene toxicity.
Interestingly, the same study (CitationVan Winkle et al. 2014) also observed little GSH depletion until high concentrations (500 μM), suggesting little naphthalene metabolism at these concentrations. Therefore, it is not clear why protein adducts were observed at concentrations of 250 μM in explants. Although CitationVan Winkle et al. (2014), in their study, only looked at 100 and 500 μM, had they looked at 250 μM, perhaps some GSH depletion would have been observed. Or, it is possible that GSH depletion is still very low at all doses tested, but that without clearance by the liver, any amount of reactive naphthalene metabolites that escapes GSH conjugation, even if very low, is available to react with proteins, and although they may react with proteins in explants, this may not be the case in vivo. In any case, according to CitationVan Winkle et al. (2014), both 250 and 500 μM explant concentrations are likely equivalent to very high in vivo exposure concentrations (greater than 10 ppm) (CitationMorris 2013) that are not likely to be experienced in the general human population.
If naphthalene is being metabolized predominantly in the human liver, is there a possibility it could be getting back to the respiratory tract to cause toxicity?
As predicted by the CFD–PBPK model (CitationCampbell et al. 2014), discussed earlier in this paper, the majority of inhaled naphthalene is metabolized in the liver in both rats (69%) and humans (89%), with approximately 20% and 0.4% metabolized in the rat and human nose, respectively, and 0.3% metabolized in the rat and human lung. The remainder is likely exhaled or metabolized in other areas of the respiratory tract.
Given the low metabolism of naphthalene in human respiratory tissue and high metabolism in the liver, there is a question regarding the possibility of systemic circulation of naphthalene metabolites back to the respiratory tissue to cause toxicity and possibly carcinogenesis. There is more of a concern for human lung than nasal tissue, since, although human epidemiology studies have found no association between naphthalene exposure and oral, oropharyngeal, and nasal cancer, the existing human data are not sufficient to determine whether there is an association between naphthalene exposure and lung cancer (CitationLewis 2012).
Studies in the mouse lung and liver in vivo and in hepatocytes (CitationBuckpitt and Warren, 1983; CitationRichieri and Buckpitt, 1987) provide evidence to support a mechanism by which the naphthalene epoxide is delivered systemically to the lung. Using CYP inducers and tissue-selective modulators of lung and liver/kidney metabolism, CitationBuckpitt and Warren (1983) observed increased binding of naphthalene metabolites in the lung that could only have been caused by metabolism in the liver, suggesting systemic circulation from the liver. CitationRichieri and Buckpitt (1987) looked at efflux of naphthalene epoxide and other naphthalene-reactive metabolites from isolated mouse hepatocytes using radiolabeled naphthalene (14C) and extracellular glutathione (3H). The authors found that 30–60% of the GSH conjugates were produced extracellularly. These studies suggest that the naphthalene epoxide migrates outside of hepatocytes in mice and is available for systemic circulation and delivery to other tissues. It is not clear, however, how much systemic distribution from the liver contributes to naphthalene-induced toxicity in mouse lung.
Studies of non-cancer effects (cataracts and hemolytic anemia) are also useful to consider with respect to these questions because these effects are induced by oral or IP exposure, and therefore are the result of systemic distribution of naphthalene metabolites from the liver. Although cataracts are unlikely to be a significant health effect in humans following exposure to naphthalene (CitationATSDR 2005), naphthalene-induced cataracts are well studied in rats and rabbits and appear to occur at acute- or intermediate-duration oral exposure levels > 500 mg/kg-day (CitationVan Heyningen and Pirie 1967; CitationXu et al. 1992a,Citationb). These studies suggest that naphthalene is metabolized to the epoxide in the liver, and is then converted to the more stable 1,2-dihydrodiol that then circulates to the eye. CitationXu et al. (1992a,Citationb) provide evidence to suggest that the 1,2-dihydrodiol is metabolized by aldose reductase to the 1,2-DHN and the toxic 1,2-naphthoquinone in the lens, and that the 1,2-naphthoquinone is responsible for cataract formation. It is worth noting that no treatment-related gross or histopathological lesions of the eyes were observed in mice (CitationNTP 1992) or rats (CitationAbdo et al. 2001; CitationNTP 2000) exposed for two years to naphthalene concentrations as high as 30 or 60 ppm, respectively.
Hemolytic anemia has been observed in people, particularly children, after eating naphthalene-containing mothballs, but the data are inadequate to describe a dose–response relationship because the studies are typically case reports and lack quantitative information on naphthalene exposure levels (CitationATSDR 2005). Naphthoquinones are known hemolytic agents (CitationMunday et al. 2007). In mice and rats, however, no treatment-related effects on hematologic parameters were observed from exposure to 10 and 30 ppm naphthalene (mice) (CitationNTP 1992), and 10, 30, and 60 ppm naphthalene (rats) (CitationNTP 2000). Depletion of thymic lymphocytes, however, was observed in two female rats following oral exposure to 400 mg/kg for 90 days (CitationBattelle 1980b).
Cataract studies suggest that 1,2-dihydrodiol is the key metabolite circulated from the liver to the eye in rabbits and rats. Epoxide hydrolase is found at much higher levels in the human liver compared to livers of rats and mice, resulting in higher levels of hydrolysis of the naphthalene epoxide to naphthalene 1,2-dihydrodiol in humans compared to rats and mice (CitationKitteringham et al. 1996), and suggesting that if 1,2-dihydrodiol is the metabolite most likely distributed systemically in rats, it is the most likely metabolite distributed systemically in humans.
Therefore, in considering the relevance of systemic distribution of naphthalene metabolites in humans, we focused our attention on circulation of the 1,2-dihydrodiol from the liver to other tissues. One of the metabolites thought to be involved in naphthalene toxicity is 1,2-naphthoquinone, which can be generated from DD activity on 1,2-dihydrodiol to generate the 1,2-DHN that can then rearrange to 1,2-naphthoquinone, which may react with proteins and DNA and may also lead to oxidative stress. If we assume that 1,2-naphthoquinone is involved in naphthalene carcinogenesis in rodents, what is the potential that the quinone will be formed in other tissues (including the lung) in humans as a result of systemic distribution of the diol to these tissues and DD activity to generate the quinone?
To try to answer this question, we looked at the levels of aldoketo-reductase (AKR) activity and at the activities of these enzymes toward naphthalene 1,2-dihydrodiol in various tissues. AKR has DD activity as well as quinone reductase activity. The human AKR superfamily consists of many isoforms (AKR1A1, AKR1B1, AKR1B10, AKR1C1, AKR1C2, AKR1C3, AKR1C4, AKR1D1, AKR6A3, AKR6A5, AKR6A9, AKR7A2, and AKR7A3) that are expressed in different tissues and have different activities toward different chemical substrates (CitationO’Connor et al. 1999; CitationPenning and Drury 2007; CitationJin and Penning 2007). CitationJin and Penning (2007), in their review of human AKRs, indicated that AKR1A1 and AKR1C1-AKR1C4 are implicated in metabolic activation of PAHs to o-quinones, and that this metabolic activation may be involved in PAH-induced lung carcinogenesis. A more recent review by CitationShultz et al. (2011) suggests that AKR1A1 levels in normal human bronchoalveolar cells are very low and are unlikely to play a major role in PAH lung carcinogenesis.
Several of the AKR isoforms have been shown to have activity toward 1,2-naphthoquinone and naphthalene 1,2- dihydrodiol (CitationO’Connor et al. 1999; CitationPalackal et al. 2002; CitationShultz et al. 2011). As summarized in , AKR1A1, which is ubiquitously and constitutively expressed in human tissue (CitationJin and Penning 2007) with relatively lower expression in the lung, has been shown to have activity toward 1,2-naphthoquinone (CitationO’Connor et al. 1999), but no study was identified that evaluated AKR1A1 activity toward the 1,2-dihydrodiol. CitationPalackal et al. (2002) evaluated the DD activity of human AKR1C1–AKR1C4 (overexpressed as recombinant proteins in Escherichia coli) toward naphthalene 1,2-dihydrodiol in human lung carcinoma (A549) cells and found that all four AKR1Cs were active toward the 1,2-dihydrodiol. As observed by CitationShultz et al. (2011), the specific activities of NADPH-dependent quinone reduction are often 100–1000 times greater than the ability of the same AKR to oxidize the PAH dihydrodiol. Consequently, it is likely that AKR1A1 is also active toward the 1,2- dihydrodiol, but perhaps not to the extent that it is toward the 1,2-naphthoquinone. CitationShultz et al. (2011) indicated that AKR1A1 has a relatively high diol-to-quinone oxidation activity for PAHs in general, compared to AKR1C1–AKR1C3. Therefore, assuming the naphthalene 1,2-dihydrodiol is delivered systemically from the liver in humans to extrahepatic tissues, these data suggest that AKR1A1 in a number of tissues should be at least as active toward the diol as AKR1C1– AKR1C3 is in the lung to generate the 1,2-naphthoquinone.
Table 1. Human AKRs and activity toward naphthalene metabolites.
Further, if systemic delivery of the diol from the liver to the lung contributes to lung cancer in humans, owing to activity of AKR1C1–AKR1C3 on the diol, one would expect to see effects in other tissues as well (e.g., in the kidney, liver, and salivary gland), attributable to activity of AKR1A1 on the diol, yet naphthalene-induced cancers in these organs have not been identified in humans. One could argue that the levels of GSH transferase activity in these tissues may be higher than in the lung, thereby eliminating the toxic naphthalene metabolites before toxicity can occur. Although glutathione S-transferase (GST) expression levels are higher in the liver and kidney than in the lung, levels in other tissues are comparable to or lower than those in the lung; for example, GST expression is lower in salivary glands (CitationNishimura and Naito 2006). Salivary gland cancers are rare in the human population, with an incidence rate of approximately one in 100 000 persons in the US (NCI 2014), and therefore likely would have been observed in the naphthalene epidemiology studies had salivary gland cancers been elevated. Yet, naphthalene-induced tumors in these tissues are not observed.
Results from the CFD–PBPK model (CitationCampbell et al. 2014) can be used to more directly predict contribution from the liver. First, one can consider that there is also high naphthalene metabolism in the liver in rats (i.e., approximately 70% in rat liver following 1 ppm naphthalene exposure, as discussed earlier in this paper), where there were no lung tumors following inhalation exposures to naphthalene as high as 60 ppm in the CitationNTP (2000) bioassay. Further, there is a higher rate of metabolism in the rat liver compared to humans at a given exposure concentration. These results suggest that the lower overall rate of metabolism in the human liver compared to rats would also not contribute to lung tumors in humans at concentrations as high as 10.7 ppm (time-weighted continuous exposure equivalent to 60 ppm in rats for 6 h/day, 5 days/week). With respect to systemic distribution to the nose, one can compare the metabolic rate in the rat liver at exposure concentrations that do not lead to nasal lesions in the CitationDodd et al. (2012) 90-day study (5.5 × 10− 3 nmol/min-g liver tissue at 0.1 ppm in the rat) to the exposure concentration that it would take to reach the same metabolic rate in human liver (5.4 × 10− 3 nmol/min-g in human liver at 0.036 ppm with continuous exposure). A naphthalene exposure concentration of 0.036 ppm is well above (approximately 200-fold) typical human exposure concentrations of 0.95 μg/m3 (0.00017 ppm) naphthalene. See discussion of human equivalent concentrations (HECs) later in this paper.
Overall, based on the available data, it appears that systemic delivery of naphthalene metabolites from the liver back to the human respiratory tract would not contribute significantly to respiratory carcinogenesis in humans. This conclusion is supported by a lack of lung tumors in rats following inhalation exposures to naphthalene where there is also high metabolism in the liver (approximately 70% in rat liver at 1 ppm naphthalene), and an overall higher rate of metabolism than in the human liver, as predicted by the CFD–PBPK rat/human model. Further, if this were a plausible mechanism for naphthalene-induced lung carcinogenesis in humans, systemic delivery of naphthalene metabolites (particularly the naphthalene 1,2-dihydrodiol due to high levels of EH in human liver) would likely cause cancer in other tissues where AKRs are broadly expressed, and this is not what is observed in the naphthalene epidemiology studies.
Based on excretion studies in humans, what is the relevance of 1-naphthol, 2-naphthol, and 1,2-DHN in urine of occupationally exposed humans and in urban children?
Several studies have found elevated levels of 1-naphthol, 2-naphthol, and 1,2-DHN in urine of occupationally exposed humans (CitationBieniek 1994, Citation1997; CitationKlotz et al. 2011; CitationWu et al. 2005; CitationMarczynski et al. 2011). CitationKlotz et al. (2011) is the only study that reported 1,2-DHN. One- and 2-naphthol have also been detected in the urine of urban children (CitationOrjuela et al. 2012). Given that the majority of naphthalene is metabolized in the liver, it is likely that these levels in the urine reflect liver metabolism and not metabolism in respiratory tissue. As shown in , 1- and 2-naphthol are spontaneously formed from the epoxide and may be one of the first metabolites to be formed during GSH depletion and regeneration. At higher concentrations, with greater GSH depletion, EH in the liver likely begins to generate the dihydrodiol and 1,2-DHN, which are then released into the blood, where they are either excreted or react with proteins (such as hemoglobin). Given the very low levels of naphthalene metabolism in human respiratory tissues, these urinary metabolites likely reflect markers of exposure and are unlikely to be markers of respiratory toxicity in humans. This point is further supported through our dose–response analysis and derivation of HECs, discussed in later sections.
Conclusion from toxicokinetic studies with respect to hypotheses
Overall, the toxicokinetic data suggest that naphthalene toxicity in the mouse nose and lung and rat nose is dependent on GSH depletion. The current data suggest that CYP2F2 may be involved in metabolism of naphthalene to the epoxide in the mouse lung, and by comparison to results from styrene, possibly also in metabolism to more toxic downstream metabolites. CYP2A5 appears to be involved in naphthalene metabolism in the mouse nose where toxicity, but no tumors, are observed. Differences in CYP2A5 and CYP2F2 may explain the lack of tumors in the mouse nose; perhaps CYP2A5 does not further metabolize the epoxide to more toxic naphthalene metabolites. If CYP2F4 is involved in naphthalene metabolism in the rat nose, this could also explain why tumors are observed in the rat nose following naphthalene exposure. The difference in tumor formation and toxicity across species and tissues, however, ultimately depends on the balance of the rate of initial metabolism of naphthalene to the epoxide (from whatever CYPs are involved), in addition to activities of GST, EH, and DD, and possibly other metabolic enzymes. Additional data on the involvement of CYP2A13 and CYP2F1 in naphthalene metabolism in the primate nose and lung would be helpful to try to understand a possible mechanism of action in humans, given an exposure concentration high enough to be relevant to adverse effects in humans.
Although it is not clear which CYP is involved in naphthalene metabolism in human respiratory tissue, overall, the current data, including predictions from a recent rat–human CFD–PBPK model for inhaled naphthalene, suggest that metabolism is much lower in human lung and nasal tissue compared to the rat nose. At typical naphthalene exposure concentrations for humans, the low metabolism in human respiratory tissue likely results in little depletion of detoxification capacity (GSH), with little to no generation of downstream genotoxic metabolites of naphthalene, consistent with a non-mutagenic MoA. A preliminary study by CitationVan Winkle et al. (2014) observed little GSH depletion in primate nasal explants following high naphthalene exposures.
The CFD–PBPK model suggests that in humans, a significant portion of inhaled naphthalene is metabolized in the liver. Our analysis, however, suggests that systemic delivery of naphthalene metabolites from the liver back to human respiratory tissue is unlikely to contribute to respiratory toxicity and cancer in humans.
There is a question as to the relevance of protein adducts observed in primate nose and lung explants, since naphthalene metabolism is low in these tissues. As discussed, the results could be an artifact of explant assays that do not include clearance by and metabolism in the liver. The recent primate nasal explant study by CitationVan Winkle et al. (2014), however, suggests that if protein adducts do occur in vivo in primate respiratory tissue, they do not result in toxicity in these tissues, and presumably in carcinogenicity, since cytotoxicity is a likely precursor to regenerative hyperplasia and tumor formation.
Overall, the toxicokinetic data, in combination with the animal and human studies, suggest a mechanism of action in rodents that involves initial (and obligatory) metabolism of naphthalene to its epoxide intermediate followed by GSH depletion and toxicity. The fact that GSH depletion appears necessary for naphthalene-induced toxicity suggests a non-mutagenic MoA and a threshold for toxic effects. It also suggests that since there is very low naphthalene metabolism in human respiratory tissue, GSH is not likely to be depleted to a sufficient extent (as supported by the recent primate explant study) to lead to toxicity and tumors at typical human exposure concentrations.
These data further suggest that exposure to subcytotoxic naphthalene concentrations can produce no more than very low levels of genotoxic metabolites, strongly suggesting that a mutagenic MoA at low doses is not likely. See further discussion below.
Genotoxicity data
Based on our understanding of the naphthalene genotoxicity data, as described earlier in this paper, we ask the following question:
How should the genotoxicity results be interpreted with respect to the proposed MoAs? What is the dose–response relationship between naphthalene-induced cytotoxicity and genotoxicity? Do the genotoxicity results suggest a mutagenic MoA?
To try to answer these questions, we need to examine the key events in a mutagenic MoA and compare the dose–response for those steps to the dose–response for cytotoxicity. The following sections discuss these key events.
Key event 1: Metabolism to potentially genotoxic metabolites (e.g., 1,2-naphthoquinone, diol epoxides, others) at subcytotoxic concentrations.
There is no evidence that genotoxic metabolites of naphthalene are generated at subcytotoxic concentrations. As discussed above, 80% of the genotoxicity studies presented for naphthalene are negative and most of the positive genotoxicity studies were accompanied by cytotoxicity (CitationATSDR 2005). These results suggest that any potentially genotoxic metabolites are not readily generated at low naphthalene exposure concentrations. If that were the case, more of the in vitro assays would have been positive. The results are consistent with the fact that the potentially genotoxic metabolites are well downstream in the metabolism pathway for naphthalene () and are therefore likely not generated in sufficient quantities to induce mutations until GSH is depleted (a concentration that is likely too cytotoxic to be able to observe a significant mutation frequency experimentally). In fact, the only bacterial assay that was positive was an assay that treated cells directly with the downstream metabolite 1,2-naphthoquinone (CitationFlowers-Geary 1996). Since it would require a very large quantity of naphthalene to generate enough 1,2-naphthoquinone to be able to react with DNA, or to redox cycle to generate DNA damage via oxygen radicals, generation of this metabolite (or further downstream metabolites) at sufficient quantities to induce mutations at subcytotoxic concentrations is unlikely.
There are no studies that look specifically at the formation of potentially genotoxic metabolites at low concentrations in target tissues. More recent studies, however, as discussed earlier, have specifically evaluated the temporal and dose relationships between naphthalene exposure and genotoxicity and cytotoxicity, and have found that genotoxicity is observed only at naphthalene concentrations that also induce cytotoxicity (CitationRecio et al. 2012), or after cytotoxicity has been observed (CitationKaragiannis et al. 2012).
Taken together, these results suggest that genotoxic metabolites of naphthalene are not generated at sufficient quantities at subcytotoxic concentrations to suggest a mutagenic MoA.
Key event 2: DNA adduct formation from naphthalene metabolites at sub-cytotoxic concentrations.
Few studies have looked at DNA adduct formation from naphthalene, and none have looked in target tissue (rat nose or mouse lung). Although one in vivo (mouse skin) study found that DNA adducts can form following treatment with naphthalene or its metabolites (CitationSaeed et al. 2007, 2009), it is not clear whether these concentrations (0.5 and 1.2 μM) were also cytotoxic to mouse skin cells. Further, DNA adducts can often be repaired before DNA replication occurs and therefore will not always lead to mutations. Observations of DNA adducts, therefore, cannot be taken to equal observations of mutagenicity. First, it depends on the type of adduct; some DNA adducts are far more mutagenic than others. Further, in the cells that survive, only some of the DNA adducts formed will escape DNA repair and result in mutations, and an even smaller number might result in mutations in key genes that could lead to tumor formation. As discussed in the next section, subcytotoxic and cytotoxic concentrations of naphthalene in target tissue (rat nose) do not result in an increased level of mutations.
Key event 3: Naphthalene-induced mutations at sub-cytotoxic concentrations.
As discussed in CitationMeng et al. (2011), mutations in codon 271 of the p53 gene have been observed in rat nasal tumors induced by formaldehyde, and p53 mutations are also the most common mutation found in human nasal cavity tumors. Therefore, it is reasonable to expect an increase in this mutation in rat nasal epithelium following naphthalene exposure if it is part of the carcinogenic mechanism. The authors, however, observed no increase in p53 mutations at subcytotoxic or cytotoxic exposure concentrations of naphthalene (0.1, 1.0, 10, and 30 ppm for 90 days; see CitationDodd et al. 2012) in the male respiratory epithelium, but they did observe a significant decreasing trend in mutations with increasing dose. These results indicate no significant increase in mutations at the subcytotoxic concentration of 0.1 ppm and a loss of mutations due to chronic cytotoxicity at higher exposure concentrations (10 and 30 ppm), suggesting that naphthalene-induced carcinogenesis in the rat nose does not operate through a mutagenic MoA.
How can we interpret the genotoxicity results with respect to a potential MoA for carcinogenesis in rodents?
The genotoxicity/mutagenicity data suggest that the carcinogenic mechanism of action for the male rat nasal epithelium may involve high (cytotoxic) doses of naphthalene to generate sufficient levels of 1,2-naphthoquinone, or other downstream metabolites, and that these metabolites may react with DNA to cause mutations. These mutations, in combination with high-dose cytotoxicity, may then contribute to cell proliferation and tumor formation.
The CitationMeng et al. (2011) study suggests, however, that even at high doses of naphthalene, the frequency of p53 mutation is not increased. As discussed by the authors, there could be several reasons for this observation: 1) the p53 mutation was induced, but not to a sufficient degree to overcome loss of mutation due to cell death; 2) the p53 mutation is not a sensitive marker for naphthalene-induced nasal carcinogenesis; or 3) naphthalene-induced carcinogenesis is the result of long-term effects from chronic (longer than 90 days) cytotoxicity and regenerative cell proliferation, in which case mutations would be secondary to cytotoxicity. Although further studies evaluating potential mutagenesis in other genes are needed, as discussed by the authors, it is reasonable to expect the p53 mutation to be a sensitive marker for naphthalene-induced nasal tumors in rats.
Overall, the weight of evidence supports a mechanism of action for rat nasal carcinogenesis involving increased cytotoxicity with increasing exposure concentrations, but where cytotoxicity is sufficiently high, so that even if there is enough metabolism to generate downstream genotoxic metabolites, any mutations they may induce likely exist in dying cells and therefore are not relevant to tumor formation. Therefore, it is more likely that the carcinogenic mechanism of action involves genotoxicity and mutagenesis that is secondary to cytotoxicity.
A more specific genotoxic pathway has been proposed in rodents (CitationPiccirillo et al. 2012), whereby at high cytotoxic doses, 1,2-naphthoquinone forms an amino acid conjugate which in turn is metabolized by aryl amidase to produce a reactive naphthoquinone imine which is genotoxic. The authors indicate that this process is not likely to occur in humans. Although it is possible, evidence for this mechanism is limited.
How can we interpret the genotoxicity results with respect to human relevance?
There are no studies that examine genotoxic and mutagenic potential of naphthalene in human respiratory tissues or cells. There are several studies in human lymphoblasts, however, that observed no increase in DNA damage in workers exposed to naphthalene in jet fuel or bitumen (CitationMarczynski et al. 2011; CitationKrieg et al. 2012). In addition, an in vitro study by CitationRecio et al. (2012) in TK6 human lymphoblast cells observed genotoxicity (micronuclei induction), but only at concentrations that were already cytotoxic.
These results, although not in human respiratory tissue, suggest a consistent non-mutagenic MoA across rodent and human tissues that is associated with high doses of naphthalene. Although the involvement of genotoxicity in the MoA at high cytotoxic doses is unclear, importantly, these results suggest that low exposures to naphthalene (i.e., concentrations that do not induce cytotoxicity) do not induce genotoxicity or mutagenesis in humans.
How should we account for possible “lingering” genotoxicity at low exposure concentrations? That is, there will likely be some (albeit very low) potential for the generation of genotoxic metabolites at low exposure concentrations. Could this low level of genotoxicity lead to mutations and carcinogenesis?
As discussed in the previous sections, there is essentially no observable genotoxicity or mutagenicity at low (less than cytotoxic) exposures to naphthalene. Since all pathways in the proposed metabolic scheme for naphthalene are in equilibrium, it is possible that very low levels of downstream naphthalene metabolites are generated. The question is whether this level of metabolites could lead to genotoxicity, mutations, and carcinogenesis. Given that the levels will be very low, they will not likely overwhelm cellular detoxification mechanisms and will most likely be excreted. Any metabolites that are not detoxified, and further react with DNA, are likely to be removed by DNA repair mechanisms that should not be overwhelmed at these low concentrations. Further, if there are some that escape repair, the likelihood that the damage and possible subsequent mutation is in a critical gene that could lead to carcinogenesis is very small. Therefore, it is reasonable to assume, given the low genotoxic potential of naphthalene even at high concentrations that any lingering genotoxicity at very low exposure concentrations is highly unlikely to lead to mutagenesis and carcinogenesis. It is likely that any mutations would be consistent with or lower than background levels of mutations. It is also important to remember that there is no positive evidence for the existence of such hypothesized low-dose genotoxicity, nor is there any observation of tumorigenicity at sub-cytotoxic doses that such hypothesized low-dose genotoxicity might be invoked to explain.
Conclusions from genotoxicity/mutagenicity data with respect to hypotheses
Additional studies in target tissues, looking at temporal and dose–response relationships for DNA adduct formation and mutagenesis, in addition to cytotoxicity, would provide useful information with regard to the potential for naphthalene-induced mutagenesis at low exposure concentrations. The available data, however, strongly suggest that an initiating genotoxic event, and therefore a mutagenic MoA, is not likely for naphthalene.
Conclusions from mechanistic data
Although it is likely that a mutagenic MoA is not involved in naphthalene-induced carcinogenesis in rats, the specific mechanisms of action for naphthalene-induced carcinogenesis are not completely understood. The ultimate toxic metabolites are not entirely clear. Therefore, we need to focus on comparing overall metabolism and effects in animal tissues to those in humans/primates to try to understand differences across species that might inform a potential MoA. It is likely that cytotoxicity is involved in the carcinogenic mechanism in rodents, since it occurs in both the rat nose and mouse lung, and there are no elevations of animal bioassay tumors in tissues that lack cytotoxicity. However, cytotoxicity alone may not be sufficient, since there are target tissues where, following naphthalene exposure, toxicity occurs but tumors do not (i.e., mouse nasal tissue). It is also likely that if genotoxicity is involved, it is only at doses that are already cytotoxic. Cytotoxic doses of naphthalene, where inflammation also occurs, will likely result in the generation of ROS that may contribute to genotoxicity and mutations upon saturation of cellular protective mechanisms (i.e., GST and DNA repair pathways). Recent data, however, suggest that ROS from naphthalene metabolites (i.e., redox cycling of 1,2- or 1,4-naphthoquinone) are not likely involved in the mechanism for naphthalene carcinogenicity (CitationCichocki et al. 2014).
As discussed earlier, the 90-day rat study (CitationDodd et al. 2012), in addition to the re-evaluation of the NTP pathology data (CitationHarkema 2001), suggests that non-neoplastic lesions, specifically epithelial hyperplasia, are precursors to respiratory epithelial adenomas in male rats and olfactory neuroblastomas in female rats. The close association of these non-neoplastic lesions with nasal tumors suggests important roles for cytotoxicity and hyperplasia in the mechanism of action for nasal tumors in rats.
Toxicogenomic data (CitationClewell et al. 2014; CitationCichocki et al. 2014) suggest genomic responses related to oxidative stress, GSH metabolism, cell cycle, inflammation, and proliferation, but only at exposure concentrations of 1 ppm or higher in rats, with significantly higher responses at 10 and 30 ppm. These results are consistent with the histopathological dose–response in the rat nose (CitationDodd et al. 2012), suggesting a NOAEL of 0.1 ppm naphthalene and a non-mutagenic threshold MoA.
The role of protein adducts in primate respiratory tissue is unclear. Although protein adducts appear to correlate with sites of high metabolism and toxicity in rodents, it is not clear why protein adducts are observed in primate lung and nasal tissue where metabolism is low (although, as discussed earlier, these observations were in vitro in explants and at high exposure concentrations). The formation of protein adducts in primate respiratory explants indicates that these tissues have the potential to metabolize naphthalene to metabolites that are capable of reacting with cellular macromolecules. This leads to a question regarding the potential for these metabolites to react with DNA as well as proteins. This question needs to be considered in the context of other relevant data, including the following: 1) PBPK results suggest very low metabolism of naphthalene in human respiratory tissue with approximately 90% of inhaled naphthalene metabolized in the liver, and therefore, as discussed already, it may be that the explant results do not reflect what is happening in vivo, due to very high exposure concentrations (likely higher than what is attainable in humans) and lack of clearance from the liver; 2) there is no increase in mutations following naphthalene inhalation exposure in the rat nose (CitationMeng et al. 2011), where naphthalene metabolism is much higher than in human respiratory tissue; and 3) the recent primate nasal explant study by CitationVan Winkle et al. (2014) observed little cytotoxicity even at high exposure concentrations, suggesting that if protein adducts do occur in vivo in primate respiratory tissue, they do not result in toxicity in these tissues, and presumably carcinogenicity, since cytotoxicity is a likely precursor to regenerative hyperplasia and tumor formation.
Based on our understanding of the mechanistic data, we ask the following question:
If protein adducts can be formed in vivo in humans, but are not involved in cytotoxicity, are they toxicologically relevant to humans?
First, it has not been established that protein adducts are even plausible in vivo in human respiratory tissues at typical naphthalene exposure concentrations. Given that the results are from high exposures in explants (absent any clearance from the liver), and given the very low metabolism in human respiratory tissues, it is very likely that protein adducts do not form in humans at typical naphthalene exposure concentrations. The data suggest, however, that if protein adducts do form to any significant extent in vivo, they do not lead to cytotoxicity. If protein adducts can be formed in human respiratory tissue from naphthalene exposure, the possibility that these adducts could be related to diseases other than lung or nasal cancers should be explored. There is some evidence that naphthalene and other PAH exposures are associated with asthma incidence in children (CitationKim et al. 2005; CitationAl-Daghri 2008; CitationDeStefano-Shields et al. 2010). Further, while comparable levels of covalent binding have been demonstrated in target and non-target tissues, there is some evidence that differences in targeted proteins may contribute to differences in toxicity (CitationDeStefano-Shields et al. 2010; CitationBoland et al. 2004; CitationCho et al. 1994; CitationLin et al. 2006). The understanding of proteins targeted by naphthalene metabolites and the relationships to different effects across species will improve our understanding of possible risks the protein adducts may have in humans.
Further, it is important to point out that even if protein adducts are involved in naphthalene toxicity, the mechanism would involve a threshold, since modification of a single protein would not have a significant biological effect due to constant protein turnover in cells.
Finally, it is possible that the protein adducts are not associated with any adverse effects, and are only markers of exposure to naphthalene.
Summary of data integration
summarizes the results of the various lines of evidence we have evaluated, organized by how each key event in the proposed MoAs is supported by the available data; this table can be used to illustrate data integration across lines of evidence. Note that there are several cells labeled as “no data.” This does not necessarily mean these are data gaps. It is possible to have enough data to support a biologically plausible MoA without having data for every possible mechanistic key event in each tissue and species, as long as critical pieces of information are available and integrated into the weight-of-evidence analysis to support the MoA. In evaluating the weight of evidence with respect to a particular overarching hypothesized MoA, the importance of elements without much data to support them directly needs to be considered in light of how plausible it is to suppose that the elements act as hypothesized, and also how surprising it would be to find that they acted otherwise, in view of our wider understanding of biology and metabolism.
Table 2. Key Events in the proposed modes of action for naphthalene-induced carcinogenesis.
As shown in , although high-dose genotoxicity cannot be entirely ruled out, the in vitro and in vivo genotoxicity/mutagenicity data, consistently across all species and tissues, do not support a mechanism that includes even high-dose genotoxicity/mutagenicity, and therefore strongly suggest that a mutagenic MoA for naphthalene is not supportable. Again, note that although the mechanism of action may not be entirely clear, the results of our analysis strongly support a non-mutagenic threshold mode of action, involving high-dose GSH depletion, cytotoxicity, chronic inflammation, and regenerative hyperplasia, and importantly, whereby any mechanism relevant to carcinogenesis is not active until high cytotoxic doses.
Although there are still some questions and uncertainties in the data set, the current data strongly suggest that typical human exposures to naphthalene will not result in an increased risk of respiratory cancer. The plausibility of the explanations (or “accounts”) for these uncertainties within the data for one overarching hypothesis needs to be weighed in comparison to the alternate set of explanations (or “accounts”) of the data. This is described in the next section.
Evaluation of alternative accounts
An HBWoE evaluation comes down to an evaluation of alternative “accounts,” which are proposed sets of explanations for the observed phenomena across the body of relevant lines of evidence. These competing accounts should be evaluated to determine how the evidence supports them, what is necessary to assume for their support, and how the overall weight of the evidence for each suggests how compelling the account should be taken to be. An account is most compelling when it is not only supported by the factual record, but when it also helps explain the data by finding common reasons for sets of observations, and moreover, achieves this ability much more readily than any competing account. This approach—which is at the center of the HBWoE method—recognizes that the array of information available will seldom be able to prove or disprove any interpretation absolutely, especially when the candidate interpretations may include accommodations to justify why results, that on the face of things might seem inconsistent, might nonetheless be reconciled using further assumptions. Moreover, results that are explained in one account as consequences of a hypothesized causal process by the agent in question must still be explained (at least tentatively) under any alternative account that denies the same causal process as having other plausible reasons for having occurred. This approach is explained more fully in previous publications (CitationRhomberg et al. 2010, Citation2011; CitationRhomberg 2014; CitationPrueitt et al. 2011).
It is useful to distinguish between the large overarching hypotheses that propose alternative general conclusions about the toxicological properties of the agent (in our case, the alternative MoA proposals) and the more numerous subsidiary or contributing subset of hypotheses that each proposes how particular study results might be explained, and how such explanations do, or do not, support the larger overarching contention.
In this section, we evaluate how the apparent discrepancies in the naphthalene data set are explained and how plausible these explanations are for each account, asking whether the explanations require ad hoc assumptions, and how likely it is that additional data will support the explanation of the discrepancies.
summarizes our interpretation of the alternative accounts for each hypothesis (i.e., either a non-mutagenic or a mutagenic MoA). The first three columns of summarize our main arguments in support of an overarching hypothesis that the mechanism for naphthalene-induced nasal tumors in rats involves local high-dose GSH depletion and cytotoxicity, and that naphthalene-induced genotoxicity, if it occurs, does not happen until cytotoxic exposure concentrations, resulting in a threshold MoA and little to no human relevance for lung or nasal carcinogenesis at typical human exposure levels (Account #1). For each of our main arguments, we present the counter argument for another account of the data that proposes a mutagenic MoA that involves genotoxicity/mutagenicity from naphthalene metabolites that are generated at less than cytotoxic doses, with potential human relevance at typical human exposure levels (Account #2).
Table 3. Comparative Reasoning for Accounts of Naphthalene Carcinogenesis in Rodents and Human Relevance.
Fundamentally, Account #2 does not have a credible explanation for why, despite its proposed key role of direct DNA interaction and mutagenesis, there is no clear and consistent genotoxicity manifested for naphthalene, nor why there are apparently no somatic mutations in target tissues even at exposures prompting cytotoxicity. It treats the apparent confinement of target tissues for carcinogenesis in animal bioassays, to those tissues showing marked toxicity, as either an uninformative coincidence, or, at most, as a reflection of localization of metabolic activation. Though it cannot readily explain why tissues such as the liver (that produce the supposed genotoxic metabolites at high levels) are not targets for carcinogenesis, Account #1 attributes the consistent pattern of collocation of tissue toxicity and carcinogenic effect (and the lack of carcinogenic effect in tissues lacking toxicity), even though the affected tissues vary from rats to mice, as indicative of the causal role of such toxicity in the tumorigenic process. The main ad hoc element in Account #1 is that the lack of tumors in mouse nose, despite cytotoxicity, is attributed to some inherent difference in mouse nasal tissue that is yet to be fully identified, its cytotoxic response, or its metabolic pathways compared to rat nose and to other tissues. Account #1 addresses the possibility of tissue toxicity in humans exposed to naphthalene by evaluating human metabolic activity and by evaluation of the lack of relevant effects in primate explants.
Although Account #1 relies on a few ad hoc explanations of the available data in order to support the proposed hypothesis, the overall data suggest that these explanations are plausible, and if tested, could very well be supported. Account #2 has many more ad hoc explanations, and overall, lacks plausibility and is not supported by the available data. Summing the ad hoc explanations and instances where plausibility can be reasonably excluded for each account results in fewer of these (shaded cells) for Account #1. Overall, Account #1 is better supported by the available data. Note that even if less weight is given to Account #1 for nasal tumors, given that the CitationVan Winkle et al. (2014) study is still preliminary, Account #1 is still stronger than Account #2, based on comparisons of all remaining data in the table.
Potentially vulnerable subpopulations
There may be certain subpopulations that are more susceptible to potential carcinogenicity from exposure to naphthalene than the general human population. Children are more susceptible to many toxicants due to differences in their anatomy and physiology compared to adults. With regard to naphthalene, neonates may be especially vulnerable because their enzyme systems for conjugation and excretion are not well developed (CitationATSDR 2005). For example, neonates have smaller stores of reduced GSH, making them more susceptible to oxidative stress (CitationSudakin et al. 2011). CitationOrjuela et al. (2012) reported an association between chromosomal aberrations in the lymphocytes of 5-year-old urban children and urinary levels of 1- and 2-naphthol. It is unclear however, what role naphthalene may have had since there was no measurement of total PAHs or other possible genotoxic agents.
Genetic variation in metabolism may also affect susceptibility to naphthalene toxicity. Polymorphisms that affect activity levels have been observed for several enzymes that are responsible for naphthalene metabolism. Increased bioactivation and/or decreased conjugation and excretion may lead to higher concentrations of downstream reactive metabolites.
CYP enzymes
Among the CYP enzymes found in human respiratory tissue that are capable of metabolizing naphthalene, polymorphisms have been reported for CYP1A1, CYP2E1, CYP2A6, CYP2S1, CYP2A13, and CYP2F1 genes (CitationSaarikoski et al. 2005; CitationNan et al. 2001; CitationYang et al. 1999; CitationWang et al. 2003; CitationCauffiez et al. 2004; CitationTournel et al. 2007).
The *2C polymorphism in exon 7 of the CYP1A1 gene and the *5A/B polymorphisms in the 5’ flanking region of the CYP2E1 gene are well established in the human population. The frequency of the CYP1A1*2C polymorphism is reported to be 22% in Caucasians and 5% in Japanese populations (CitationRoco et al. 2012). The frequency of CYP2E1*5A/B polymorphisms (combined) is reported to be 2–4% in the US population and higher in Asian populations (30–50%) (CitationNeafsey et al. 2009).
CitationNan et al. (2001) studied the effects of CYP1A1 and CYP2E1 polymorphisms on naphthalene metabolism in two different populations: occupationally exposed coke oven workers and students with no occupational exposure. The authors observed significantly higher urinary concentrations of 2-naphthol, indicating higher enzyme activity, only in workers who were either heterozygous or homozygous for the CYP2E1 Rsa1 polymorphism (c1/c2 or c2/c2) compared to the homozygous wild-type genotype (c1/c1). No effect was evident in the students, and no effect of the CYP1A1 polymorphism was observed in either subject group. Similar results were obtained in a study by CitationYang et al. (1999). The authors found that among Japanese men (not occupationally exposed to naphthalene), the CYP2E1 RsaI polymorphism (c1/c2 and c2/c2) was significantly associated with higher urinary levels of 1- and 2-naphthols compared to the wild-type genotype. The CYP1A1 polymorphism had no effect on naphthol levels. These studies suggest that CYP2E1 may be involved in naphthalene metabolism in humans.
Polymorphisms of the naphthalene-metabolizing enzymes CYP2A13 and CYP2F1 have also been observed in human populations. While the effects of these variants on naphthalene toxicity are unknown, certain CYP2A13 variants have been associated with specific types of lung cancer and with changes in enzyme activity toward other carcinogens (CitationFukami et al. 2008; CitationWang et al. 2003; CitationCauffiez et al. 2004). Several variants of CYP2F1 were also recently identified, but no evidence of associations with lung cancer was observed (CitationTournel et al. 2007).
It is unclear as of yet what effect, if any, these polymorphisms might have on susceptibility to naphthalene-induced respiratory cancer.
Epoxide hydrolase
There are two well-established variants of the microsomal epoxide hydrolase gene. The first is a tyrosine to histidine at codon 113 in exon 3 that reduces in vitro enzyme activity by approximately 39–50%; the second is a histidine to arginine at codon 139 in exon 4 that increases in vitro enzyme activity by approximately 25% (CitationSalam et al. 2007; CitationSeidegard and Ekstrom 1997; CitationKitteringham et al. 1996). These variations may affect the stability of the enzyme, which may account, in part, for the interindividual differences in activity among human populations (CitationSeidegard and Ekstrom 1997). The epoxide hydrolase polymorphisms may affect individual susceptibility to cancer that is associated with environmental carcinogens (CitationSeidegard and Ekstrom 1997). CitationWu et al. (2001) noted that the exon 4 polymorphism had different effects on susceptibility to lung cancer in different human populations. This polymorphism was associated with an increased risk for lung cancer among a population of Mexican Americans, but not among a population of African Americans. The exon 3 polymorphism had no effect on lung cancer in either group (CitationWu et al. 2001). Little is known about the effects of epoxide hydrolase polymorphisms on naphthalene metabolism. An in vitro assay of human liver enzyme activity revealed no correlation between these two epoxide hydrolase genotypes and rate of naphthalene metabolism (CitationKitteringham et al. 1996). CitationSalam et al. (2007) reported that in children, higher epoxide hydrolase activity was associated with increased incidence of asthma, possibly due to increased bioactivation of PAHs. Whether this observation is relevant to naphthalene bioactivation and cancer risk is unknown.
Glutathione-S-transferases
Individuals with reduced GST activity may be at higher risk for naphthalene toxicity since GSH conjugation is a major detoxification pathway. There are three types of GST enzymes that have well-characterized polymorphisms. Individuals who are homozygous for the GSTT1 null or GSTM1 null genotypes have no activity of those respective enzymes (CitationSeidegard and Ekstrom 1997). GSTM1 has high activity towards many epoxide metabolites of PAHs (CitationSeidegard and Ekstrom 1997) and may be important in the metabolism of naphthalene. In the study of coke oven workers described above (CitationNan et al. 2001), workers who smoked and had the null GSTM1 genotype had higher urinary concentrations of 2-naphthol compared to non-null workers. There was no association between the GSTT1 null genotype and 2-naphthol concentrations. Similar results were noted in the study of Japanese male workers (CitationYang et al. 1999). In this study, smokers who had only the GSTM1 null genotype had higher concentrations of urinary naphthols than did workers with the normal GSTM1 genotype. The GSTP1 variant is a substitution of valine for isoleucine at the 105 position, which may result in decreased enzyme activity (CitationMcCarty et al. 2007). In the study of asthmatic children by CitationSalam et al. (2007), low-activity GSTP1 phenotype was associated with asthma. While this study did not focus on naphthalene metabolism, PAHs are known to be associated with asthma and wheeze (CitationMiller et al. 2004; CitationJedrychowski et al. 2005), therefore suggesting an increased susceptibility to PAHs for children with this phenotype.
Conclusions related to susceptibility
Although we did not identify any investigations of naphthalene toxicity in association with enzyme polymorphisms, the above studies demonstrate that metabolism of naphthalene may differ with different phenotypes of CYP, epoxide hydrolase, and GST enzymes. This may indicate that people with increased CYP and/or epoxide hydrolase activity, or with deficiencies in GST enzymes, may be more susceptible to naphthalene toxicity than the general population.
Dose–response assessment
The most recent finalized assessment of naphthalene carcinogenicity by the CitationUS EPA (1998) recognizes that “[given that] in most genotoxicity tests with naphthalene, negative results have been obtained, it appears unlikely that naphthalene represents a genotoxic carcinogen” (p. 34). CitationBogen (2008) presented an analysis of a dual MoA for induction of respiratory tract tumors by inhaled naphthalene, in a model that assumed cytotoxicity and mutagenicity are each increased independently by exposure but can interact in their impact on tumor risk. He employed pharmacokinetic modeling to estimate target tissue-metabolized doses and used in vitro data on human lymphocytes to estimate the comparative patterns of dose-dependence of cell-killing and genotoxic effects. The joint action of these influences on tumor generation was described using an Moolgavkar–Venzon–Knudson (MVK)-model that expresses the development of dose-specific tumor risk over time as a function of underlying birth, death, and mutation among the population of target-tissue cells, with increasing exposures acting progressively to elevate the rates of these processes over those in unexposed tissue. A number of assumptions are needed to achieve this modeling, and Bogen's aim was to set these so as to find a conservative estimate of the factor by which a purely mutagenic MoA model (and its low-dose linear extrapolation) could overestimate the risks by failing to account for the role of cytotoxicity and its disproportionate role in tumorigenesis at the exposure levels in animal bioassays at which tumor responses were observed.
In view of newer data, described above, that suggest little role for mutagenicity in target tissues (and that any such role would occur, if at all, only secondarily to cytotoxicity), and because we have the added information about the dose-dependence of the relevant tissue toxicity at lower doses, we have taken an approach to dose–response analysis that focuses on characterizing the dependency of tissue toxicity on tissue metabolized dose.
The outcome of our HBWoE evaluation (summarized in ) is that the current data more strongly support a MoA that is non-mutagenic, and that there is a much larger degree of ad hoc argument (with little plausibility that additional data will support those arguments) in the hypothesis that accounts for the data as supporting a mutagenic MoA.
The data, therefore, support a MoA that likely involves a threshold for cytotoxicity; that is, a dose below which cytotoxicity, and therefore tumors, are not expected to occur. The dose– response evaluation needs to attempt to identify this threshold in rodent tissues, and to identify the lowest doses associated with non-neoplastic lesions that are likely precursors to tumors. Our dose–response evaluation attempts to align exposure– response relationships of key precursor non-neoplastic lesions to exposure–response relationships for tumors in an attempt to develop a sequence of key events for tumor formation in the rat nose that is consistent with the tissue dose–response and the biologically plausible MoA supported by the HBWoE evaluation. Part of our evaluation involved, as discussed already, examining what non-neoplastic lesions occur in the individual rats with tumors. In this section, we try to understand the dose– response relationship for those precursors compared to the dose–response for tumors, and the tissue concentrations and metabolized doses associated with those precursor effects.
The challenge in this approach is that the NTP bioassays themselves include only high doses—high enough that the target tissues for tumorigenicity had widespread tissue toxicity that was little diminished between the higher exposure and the lower one. As the PBPK model of CitationCampbell et al. (2014) shows, the tissue-specific metabolized doses in rat nose are not markedly different for the NTP exposure levels, owing to saturation of metabolic capacity at the high air concentrations used. Thus, the ability to describe the dose–response for precursor lesions that are deemed to be key necessary events requires other information, for which we use the 90-day study of CitationDodd et al. (2012) that aimed to extend observation of noncancer nasal respiratory lesions in rats to lower exposure levels, and also the PBPK model results of CitationCampbell et al. (2014) that enable estimation of how tissue-metabolized doses diminish at lower exposure concentrations.
As discussed, CitationDodd et al. (2012) conducted a 90-day low-dose (0.1, 1, 10, 30 ppm naphthalene) rat inhalation study that identified doses at which non-neoplastic lesions (e.g., inflammation, hyperplasia, degeneration/necrosis, squamous metaplasia, and goblet-cell hyperplasia) in nasal respiratory and olfactory epithelial tissue did not occur (0.1–1 ppm), thereby suggesting a naphthalene-inhalation threshold exposure concentration in rats for lesions that are precursors of nasal cancer. The genomics data (CitationClewell et al. 2014) are consistent with this NOAEL, and although GSH depletion was not evaluated at 0.1 ppm in the rat nose, and does occur at concentrations as low as 1 ppm (CitationCichocki et al. 2014), the GSH-depletion data are not inconsistent with a NOAEL at 0.1 ppm; further investigation of GSH-depletion at lower concentrations for longer exposure durations would be useful. Our approach is based on the presumption that a NOEL for these apparent precursor lesions in rats will also be NOEL for tumors; this is also consistent with the results of our HBWoE analysis, within which we conclude that the data suggest important roles for cytotoxicity and regenerative hyperplasia with nasal tumor formation, and that low doses of naphthalene are not likely to lead to tumor-initiating genotoxic and mutagenic events.
Therefore, we do not use the NTP tumor data for our main dose–response analysis, and instead use the non-neoplastic data from the CitationNTP (2000) and CitationDodd et al. (2012) bioassays.
Selection of lesion type
We obtained the raw data through CitationNTP (2012) and from the authors of the CitationDodd et al. (2012) study for the dose–response modeling. As discussed in the CitationNTP (2000) rat bioassay, a number of non-neoplastic lesions in nasal tissue were observed in animals that developed tumors, some that are more likely to be associated with tumor formation, and others that are not. As NTP discussed, hyaline degeneration and goblet-cell hyperplasia of the nasal respiratory and olfactory epithelium are considered non-specific protective or adaptive responses to chronic inhalation of irritants. Therefore, these lesions are not considered further in our dose–response analysis.
The CitationDodd et al. (2012) 90-day study examined rat nasal tissues using several sections through the nose, beginning at the tip and continuing through progressively posterior sections, designated Levels 1 through 5. Inflammation occurs in the nasal tip and the Level 1 section of the male and female rat nose at all doses including the controls, with no dose response (i.e., 50–100% variable response across control and dose groups). Tumors have not been observed in the nasal tip, and although the CitationNTP (2000) bioassay reports tumors in the Level 1 section, the NTP Level 1 section appears to correlate with the CitationDodd et al. (2012) Level 2 section. Therefore, we did not conduct dose–response modeling for inflammation in the nasal tip and Level 1 nasal section from CitationDodd et al. (2012).
Dose–response modeling was conducted on non-neoplastic lesions determined to be key based on results of our HBWoE evaluation (respiratory and olfactory epithelial hyperplasia, degeneration [if not specified as hyaline], and respiratory epithelial inflammation and squamous metaplasia), observed in both male and female rats in the CitationDodd et al. (2012) 90-day and the NTP 2-year bioassay (CitationNTP 2000). [Note that squamous metaplasias are considered benign lesions but are maintained nonetheless in our dose–response analysis].
Selection of lesion location
We modeled respiratory epithelial lesions reported in Level 2 of the CitationDodd et al. (2012) study, and not Level 1, since no tumors and no non-neoplastic lesions (that were not also in the controls) were observed there. Although olfactory tumors occurred predominantly in Level 3 of the CitationNTP (2000) bioassay, one could argue that a tumor could arise from a relevant non-neoplastic lesion wherever that lesion may occur. Therefore, although the NTP Level 3 section of the nose is comparable to the CitationDodd et al. (2012) Level 4 section, we modeled all relevant olfactory lesions from the CitationDodd et al. (2012) study that occurred in Levels 2–5. We also checked to see how the dose–response estimates would vary had we just considered animals with lesions in Level 4. For all but two dose/lesion combinations, there was no difference. For the two that were different, the 95% lower BMDLs went up slightly if only Level 4 was included. The CitationNTP (2000) bioassay reported non-neoplastic lesions in the nasal respiratory and olfactory epithelium, but did not specify whether the lesions were located in Level 1, 2, or 3. Therefore, we modeled all relevant nasal lesions as reported by NTP (respiratory and olfactory epithelial hyperplasia and respiratory epithelial squamous metaplasia).
Dose–response modeling approach
The noncancer lesions (in both the 90-day and the 2-year bioassay) are described in terms of severity by scoring into one of several ranked categories. CitationDodd et al. (2012) scored lesions as 1 = minimal, 2 = slight/mild, 3 = moderate, 4 = moderately severe, or 5 = severe; average severity of all relevant nasal lesions from all exposure groups ranged from 0.6 to 2.7 for male rats, and 0.8 to 2.5 for female rats. CitationNTP (2000) scored lesions as 1 = minimal, 2 = mild, 3 = moderate, or 4 = marked; average severity of all relevant nasal lesions from all exposure groups ranged from 1.8 to 3.5 in male rats, and from 1.6 to 3.2 in female rats. It is possible that lesions of severity greater than the mildest category might be more informative about the ensuing carcinogenic process. As a conservative estimate, however, we conducted dose–response modeling considering all lesions present at some severity level (i.e., severity 1 or higher) for 90-day and 2-year data.
Responses based on the metabolized dose were derived through application of the recent CFD–PBPK model (CitationCampbell et al. 2014) that incorporates metabolic rate constants for naphthalene from nasal and lung tissue of rat and rhesus monkey, and has allowed for an understanding of metabolized doses in specific rat nasal tissue locations. As shown in , the shape of the relationship between inhaled air concentration vs the tissue-specific metabolized dose is quite non-linear. Given that naphthalene toxicity is clearly dependent on initial metabolism to the epoxide, as discussed here in earlier sections, we derived dose–response curves and points of departure (PODs) based only on metabolized dose, and not inhaled dose.
Results of dose–response modeling
All supporting modeling files are available in the Supplementary Materials to be found online at http://informahealthcare.com/doi/abs/10.3109/10408444.2015.1061477, including best-fit model selection criteria and averaging methods, along with the dose–response curves for all endpoints evaluated. A summary of BMDs and PODs based on the best-fit model, and on the average BMDs from all models that adequately fit the data, are shown in . The average and best-fit model BMDs for all of the 90-day non-neoplastic endpoints are very similar. Since the average considers all models with adequate fit, we used the average model BMDLs (PODs) to estimate HECs, discussed in the next section.
Table 4. Points of departure for lesions in naphthalene-exposed rats from NTP (CitationNTP 2000) and CitationDodd et al. (2012)*.
We compared the modeling results (BMDs in and dose–response curves in the Supplementary Materials to be found online at http://informahealthcare.com/doi/abs/10.3109/10408444.2015.1061477) for the same endpoint from the NTP 2-year bioassay and the CitationDodd et al. (2012) 90-day study, to determine which results were most appropriate for use as PODs to derive HECs. As shown in (for male respiratory and female olfactory epithelial hyperplasia as examples), the 90-day dose–response model provides information for lower doses (0.1 and 1 ppm), including doses where effects were not observed. These low doses were not evaluated in the 2-year NTP bioassay, and therefore the 90-day modeling results provide a better dose–response estimate in the lower dose range for these endpoints. As shown in (for female olfactory epithelial hyperplasia) and in the Supplementary Materials to be found online at http://informahealthcare.com/doi/abs/10.3109/10408444.2015.1061477, the BMD software was not able to fit the NTP data for a number of the NTP non-neoplastic endpoints, owing largely to the similarity in tissue-metabolized dose levels—a consequence of saturating metabolism—at the available NTP dose levels. Also, as shown in , the ranges of viable BMDs from the NTP data are the same or larger (e.g., hyperplasia) than the ranges from the 90-day data, suggesting that the 90-day data are less variable in the dose–response output. Therefore, we used the 90-day BMDs to estimate PODs and HECs.
Figure 4. Two-year (CitationNTP 2000) vs 90-day (CitationDodd et al. 2012) metabolized dose–response in male and female rats following naphthalene exposure via inhalation (BMR of 10% extra risk for the 0.95 lower confidence limit on the BMD). Figures show the best-fit curve. See supplemental material for all modeling results. (A) Male rat respiratory epithelial hyperplasia dose–response from 2-year study (Logistic); (B) male rat respiratory epithelial hyperplasia dose–response from 90-day study (Weibull); (C) female rat olfactory epithelial hyperplasia dose–response from 2-year study (Log-Logistic with NO FIT); (D) female olfactory epithelial hyperplasia dose–response from 90-day study (Log-Logistic).
We compared our BMDL10 (POD) values to the BMDL values presented in the recent study of genomic responses in the rat nasal epithelium following inhalation of naphthalene (CitationClewell et al. 2014). The BMDL values from the genomics study are very similar to our values shown in . The lowest BMDL (based on metabolized dose) in the female olfactory epithelium in CitationClewell et al. (2014) is 2.9 nmol/min-g tissue, compared to our 1.25 nmol/min-g tissue for olfactory epithelial hyperplasia. The lowest BMDL for the male respiratory epithelium in the same study is 1.4 nmol/min-g tissue, compared to our 0.78 nmol/min-g tissue for respiratory epithelial hyperplasia. Therefore, there is consistency between BMDs from non-neoplastic endpoints and gene expression endpoints, providing support for the range of BMDs across studies.
Human equivalent concentrations
In this section, we apply the CFD–PBPK model (CitationCampbell et al. 2014) to derive HECs based on the rat PODs described in the previous section. The questions that we are trying to address in this section are:
Given the differences in naphthalene metabolism across species and tissues, are the nasal and lung respiratory tissue metabolized doses modeled in rodents achievable in humans?
If achievable, what exposure concentrations in humans would be necessary and are they typical?
The low dose evaluation in the recent 90-day rat bioassay by CitationDodd et al. (2012), in addition to the recent rat/human CFD–PBPK model (CitationCampbell et al. 2014), has allowed for incorporation of species differences in tissue dosimetry to evaluate whether parallel tissues, or other tissues in the respiratory tract in humans will be subject to tissue doses that could prompt the key events of the apparent MoA and associated adverse effects in humans at typical human exposures.
The PBPK model is linked to computational fluid dynamic models of human air flows and metabolic capacities so that the model can predict metabolically activated doses in humans in the nose and other locations in the respiratory tract. Since our approach is based on the presumption that a no-effect level for the apparent precursor lesions in rats will also be a no-effect level for tumors, we can use this assumption, in combination with an understanding about what tissue doses are not likely to lead to precursor lesions, to answer the question about the possibility of human responses in tissues other than the nose (e.g., lung). We apply the PBPK model to ask whether humans have sufficient metabolic activation in nasal or non-nasal tissues to be near levels needed to produce the non-neoplastic lesions seen in the rat nose.
Overall, our approach is to look at the dose–response for different components of the apparent MoA in the rat nose (non-neoplastic lesions) as they depend on tissue-metabolized dose, and then, through application of the CFD–PBPK model, try to understand the relevance of those associations to adverse effects in nasal and non-nasal human tissues.
Human equivalent concentrations for nasal tissue
Table 7 in CitationCampbell et al. (2014) describes the amount of naphthalene metabolized in dorsal olfactory, ventral respiratory, lung, and liver tissues of rats and humans for naphthalene exposure concentrations of 0.1, 1, 3, 10, 30, and 60 ppm (6 h/day, 5 days/week), equivalent to time-weighted continuous exposures for humans of 0.018, 0.18, 0.54, 1.8, 5.4, and 10.7 ppm (24 h/day, 7 days/week), respectively. We applied a linear interpolation between each metabolized dose point in Table 7 in CitationCampbell et al. (2014), with the metabolized dose proportional to the log of the air concentration, to estimate the inhalation concentrations for humans that would result in tissue-specific metabolized doses equivalent to metabolized doses for each PODRAT (HEC). Interestingly, as shown in Table 7 of CitationCampbell et al. (2014), the metabolized doses in the low-dose inhalation range for rats (0.2 ppm for ventral respiratory and approximately 3.6 ppm for olfactory for 6 h/day, 5 days/week, resulting in metabolized doses of 1.7 and 4.9 nmol/min-g tissue, respectively) are not achievable in humans at even the highest dose evaluated in the PBPK model (60 ppm, or 10.7 ppm continuous exposure), suggesting saturation of metabolism in human nasal tissue at doses that are less than cytotoxic in the rat nose.
summarizes the rat BMDs and PODs (average from all adequately fitting models, AM_BMDs and AM_PODS, respectively), and corresponding HEC values. As shown, the rates of metabolism associated with the PODRAT for male squamous metaplasia, female hyperplasia, and female squamous metaplasia in the ventral respiratory epithelium (approximately 4 nmol/min-g tissue) are higher than the highest metabolized dose estimated in the human ventral respiratory epithelium in the PBPK model (i.e., 1.5 nmol/min-g tissue in the ventral respiratory tissue at 10.7 ppm continuous exposure) (CitationCampbell et al. 2014). Therefore, we were not able to estimate HEC values for these lesions. The metabolic rate is clearly not achievable in humans until exposure concentrations much higher than 10.7 ppm, which is much higher than typical human exposures (0.00017 ppm [0.95 μg/m3]; CitationATSDR 2005), or possibly not achievable at all due to saturation of metabolism.
Table 5. Summary of BMDs (AM_BMDs), BMDLs (AM_PODs), HECs, and MOEs.
We also calculated margins of exposure (MOEs) by dividing the HEC values, where we were able to calculate them, by the typical residential exposure level of 0.00017 ppm (0.95 μg/m3). An MOE greater than 30 is considered to be without appreciable risk based on a combined uncertainty factor of 3 for rat–human pharmacodynamic difference, 3 for human–human pharmacodynamic differences, and 3 for human–human pharmacokinetic differences (i.e., the PBPK model accounts for rat–human pharmacokinetic differences). All MOEs shown in are much greater than 30, suggesting that naphthalene does not represent an unacceptable cancer risk to humans at typical residential exposures. Further, as discussed by CitationGriego et al. (2008), potential high-end residential naphthalene exposures of 10–100 μg/m3, including on-label use of mothballs, would still result in MOEs greater than 30.
Human equivalent concentrations for lung tissue
Lung tumors occurred in mice but not rats in the NTP bioassays. As we have set out above, the mouse lung tumors also appear to be secondary to noncancer toxicity. The challenge, however, is that we have no mouse data on dose–response of lung noncancer toxicity observations below the range of the high NTP bioassay doses. Furthermore, we have no mouse PBPK model to estimate the tissue-metabolized doses in the lung at bioassay exposures and at lower levels, though the expectation is that nonlinearities similar to those seen in the rat nasal-tissue-metabolized doses would apply. As discussed, humans are expected to have markedly lower lung-tissue metabolized doses than the mice (owing to their known lower metabolic activity in lung, as measured in vitro by microsomes). In view of these limitations in available information, we have taken two approaches to address whether human exposures could plausibly reach levels that would engender risks, based on the evidence from rats and taking into account relevant systematic differences between rats and mice, as discussed below.
We first attempted to estimate naphthalene HECs protective of lung cancer in humans by assuming that tissue-level metabolized doses that were unable to cause tissue toxicity in the rat nose would also be unable to cause such toxicity in the human lung—that is, that human lung epithelium is no more sensitive than rat nasal epithelium. The nasal-tissue-metabolized doses equivalent to the PODRAT values in are therefore taken to represent the BMDL-metabolized doses in human lung tissue. Then, the inhalation concentration for humans that would be required to result in that metabolized dose in human lung tissue is estimated through application of the PBPK model (Table 7 in CitationCampbell et al. 2014).
The second approach is to note that the NTP bioassay rats did not have lung tumors, even at the maximum bioassay exposure of 60 ppm, and so the PBPK model's estimate of rat lung- metabolized dose at the 60 ppm exposure gives a level that represents a NOAEL for the rat lung epithelium. We can then use the PBPK model to calculate the inhalation concentration in humans that would result in the same metabolized dose in human lung tissue that was shown to be a NOAEL for rat lung.
For both of these estimates, the metabolized doses in human lung tissue that are equivalent to the lowest rat nasal BMDL (0.78 nmol/min-g tissue from our ) or the rat lung NOAEL (0.38 nmol/min-g tissue, from Table 7 in Campbell et al. [2014] for rat lung at 60 ppm) are 78- to 38-fold above the metabolized dose associated with the largest air concentration that has been examined for human lung tissue in the PBPK model (0.01 nmol/min-g tissue, from Table 7 in Campbell et al. [2014] for human lung at 60 ppm, or 10.7 ppm continuous exposure). Therefore, an inhalation concentration for humans that would achieve these metabolized doses cannot be estimated without extrapolation far beyond the range of exposures over which the human model is deemed meaningful, but it can be assumed that the level would be above the maximum naphthalene time-weighted continuous exposure of 10.7 ppm, which is 63 000-fold higher than typical residential exposures to naphthalene (0.00017 ppm), or perhaps not achievable due to saturation of metabolism. These results are consistent with the very low rates of naphthalene metabolism in the primate lung compared to those in the rat lung, both of which are much lower than rates of metabolism in the mouse lung where tumors were observed.
Based on these results, the very low level of naphthalene metabolism in human lung tissue is not likely to lead to lung cancer in humans, even at high exposure concentrations; that is, 10.7 ppm naphthalene is about 60-fold higher than the upper-end of the range of occupational exposure levels (0.17 ppm, or 1000 μg/m3) as described in CitationGriego et al. (2008). Although our estimates here only account for metabolism in the human lung, our analysis above suggests that systemic delivery of metabolized naphthalene from the liver back to the human lung would also not contribute to increased risk of human lung cancer.
Discussion
Overall, the results of our HBWoE evaluation of naphthalene-induced nasal carcinogenicity in rodents, and its potential relevance to human respiratory cancers, suggest a threshold MoA for carcinogenesis in the rat nose and mouse lung that involves high-dose GSH depletion, followed by cytotoxicity, chronic inflammation, regenerative hyperplasia, and tumor formation. Our results, based on a weight-of-evidence evaluation of the genotoxicity data, including limited evidence of genotoxicity in rat nasal tissue where tumors are observed (CitationMeng et al. 2011), also suggest that if genotoxicity from naphthalene metabolites is involved in the MoA, it is only at high cytotoxic concentrations and therefore is not consistent with a mutagenic MoA. Our evaluation of human relevance suggests that low naphthalene metabolism in human respiratory tissue is most consistent with little to no toxicity or carcinogenic risk at typical naphthalene environmental exposures.
We formulated an account of the available naphthalene data to assess how well the evidence supports our proposed MoA, including explanations for the observed apparent discrepancies and uncertainties in the data set. The logic and reasoning necessary to assume support for our account was compared to the logic and reasoning necessary to assume support for an alternative account that suggests a mutagenic MoA for naphthalene. The comparison of the two accounts suggests that a non-mutagenic threshold MoA is better supported by the available data than a low-dose mutagenic MoA. It is important to keep in mind that the comparison of accounts, as presented in our , is not intended to prove one account over the other, but to show the weights of both accounts and to show which is better supported by the available data.
It is possible that the alternative account presented here may be presented slightly differently by others who may view the data differently. The general goal of account comparisons in any HBWoE analysis is that the comparisons can be used as a basis of discussion for those who have a difference of opinion with respect to how the accounts for each overarching hypothesis are supported by the available data. It can also be used to guide future experiments, if necessary, to continue to better understand how well both accounts are supported by the data overall.
Our dose–response analysis further supports the conclusions regarding human relevance of our HBWoE evaluation. As shown in our summary of naphthalene HEC values in , the MOEs for the non-neoplastic lesions, based on a comparison to typical residential naphthalene exposure concentrations of 0.95 μg/m3, are all much greater than 30, suggesting no significant risk. These values are based on BMDLs very close to those derived recently from toxicogenomics data (CitationClewell et al. 2014).
With respect to potential human lung cancer risk from exposure to naphthalene, based on results from the rat–human PBPK model (CitationCampbell et al. 2014), our analysis suggests that there is little risk even for high exposures to naphthalene, consistent with the very low rate of naphthalene metabolism in the human lung compared to that in the mouse lung where tumors are observed. Given the greater uncertainty in observations of elevated lung cancer risk in the human population, and the fact that our conclusions are based on observations in rat respiratory tissue, when lung tumors were observed in mice following naphthalene exposure, an analysis that considers more specifically the human relevance of mouse lung tumors is important. The US EPA is currently trying to determine whether chemically induced mouse lung tumors are relevant to tumor formation in humans, using data for naphthalene, ethylbenzene, and styrene, all of which lead to lung tumors in mice via a mechanism of action that involves CYP2F2. Recent studies by CitationCruzan et al. (2009, Citation2012, Citation2013) suggest that mouse lung tumors are not relevant to humans. The US EPA recently held a workshop on this topic (CitationUS EPA 2014).
Although our analysis strongly supports a non-mutagenic MoA whereby any mechanism relevant to naphthalene-induced carcinogenesis is not active until high cytotoxic doses, the available data do not sufficiently support a specific mechanism of action. Given the complicated nature of naphthalene metabolism, the many enzymes that are likely involved, and the number of potentially toxic metabolites, it may be difficult to entirely sort out what combination of activities and what specific naphthalene metabolites lead to tumors in rodents, and the relevance for humans. Additional studies could provide more information about the mechanism of carcinogenesis in rodents and relevance to humans. These might include analyses designed to further investigate 1) the relevance of protein adducts in explants and whether they occur in vivo in rodents and humans; 2) the extent of GSH depletion at inhalation concentrations of less than 1 ppm naphthalene in rats, followed by derivation of a GSH-depletion BMDL; 3) the mechanistic difference between involvement of CYP2A13 and CYP2A5 vs CYP2F in naphthalene metabolism; 4) a better understanding of the mechanism of mouse lung tumors; and 5) better characterization of more susceptible/sensitive populations (e.g., genetic polymorphisms) so that chemical-specific UFs, rather than default UFs, can be applied to the MOE estimates.
Importantly, although the mechanism of action may not be entirely clear, a non-mutagenic threshold MoA for naphthalene-induced rat nasal tumors should be considered to determine human relevance and to guide regulatory and risk-management decisions.
Supplementary material available online
Supplementary Tables and plots to be found online at http://informahealthcare.com/doi/abs/10.3109/10408444.2015.1061477.
itxc_a_1061477_sm5412.pdf
Download PDF (7.2 MB)Acknowledgements
The authors would like to thank Daniel Skall, a Gradient employee, for his efforts in finalizing the dose–response modeling analysis. The authors gratefully acknowledge the extensive comments from five anonymous reviewers selected by the Editor. These comments were most helpful in revising the manuscript.
Declaration of interest
The authors’ affiliation during the course of this work is as shown on the first page. All authors, other than Dr. Marc Nascarella, are currently affiliated with Gradient, a private environmental consulting firm. Dr. Nascarella's current affiliation is with the Massachusetts Department of Public Health. This paper was prepared with financial support to Gradient from the Naphthalene Research Committee (NRC). The NRC is a group of companies and organizations that produce or use naphthalene-containing materials and/or products. The work reported in the paper was conducted during the normal course of employment by Gradient. The NRC was given the opportunity to comment on the manuscript. The authors retained final decision-making, and have the sole responsibility for the writing and contents of this paper. The views and opinions expressed are not necessarily those of the NRC.
References
- Abdo KM, Eustis SL, McDonald M, Jokinen MP, Adkins B, Haseman JK. (1992). Naphthalene: A respiratory tract toxicant and carcinogen for mice. Inhal Toxicol, 4, 393–409.
- Abdo KM, Grumbein S, Chou BJ, Herbert R. (2001). Toxicity and carcinogenicity study in F344 rats following 2 years of whole–body exposure to naphthalene vapors. Inhal Toxicol, 13, 931–50.
- Adkins B, Van Stee EW, Simmons JE, Eustis SL. (1986). Oncogenic response of strain A/J mice to inhaled chemicals. J Toxicol Environ Health, 17, 311–22.
- Agency for Toxic Substances and Disease Registry (ATSDR). (2005). Toxicological Profile for Naphthalene 1–Methylnaphthalene and 2–Methylnaphthalene.
- Al-Daghri NM. (2008). Serum polycyclic aromatic hydrocarbons among children with and without asthma: Correlation to environmental and dietary factors. Int J Occup Med Environ Health, 21, 211–17.
- Bagchi D, Bagchi M, Balmoori J, Vuchetich PJ, Stohs SJ. (1998a). Induction of oxidative stress and DNA damage by chronic administration of naphthalene to rats. Res Commun Mol Pathol Pharmacol, 101, 249–57.
- Bagchi D, Balmoori J, Bagchi M, Ye X, Williams CB, Stohs SJ. (2000). Role of p53 tumor suppressor gene in the toxicity of TCDD endrin naphthalene and chromium (VI) in liver and brain tissues of mice. Free Radic Biol Med, 28, 895–903.
- Bagchi D, Balmoori J, Bagchi M, Ye X, Williams CB, Stohs SJ. (2002). Comparative effects of TCDD endrin naphthalene and chromium (VI) on oxidative stress and tissue damage in the liver and brain tissues of mice. Toxicology, 175, 73–82.
- Bagchi M, Bagchi D, Balmoori J, Ye X, Stohs SJ. (1998b). Naphthalene–induced oxidative stress and DNA damage in cultured macrophage J774A.1 cells. Free Radic Biol Med, 25, 137–43.
- Bailey LA, Prueitt RL, Rhomberg LR. (2012). Hypothesis-based weight-of-evidence evaluation of methanol as a human carcinogen. Regul Toxicol Pharmacol, 62, 278–91.
- Baldwin RM, Jewell WT, Fanucci MV, Plopper CG, Buckpitt AR. (2004). Comparison of pulmonary/nasal CYP2F expression levels in rodents and rhesus macaque. J Pharmacol Exp Ther, 309, 127–36.
- Battelle Columbus Laboratories (Batelle). (1980a). Subchronic Toxicity Study: Naphthalene (C52904), B6C3F1 Mice. Report to National Toxicology Program (NTP) 25p, March 4.
- Battelle Columbus Laboratories (Batelle). (1980b). Subchronic Toxicity Study: Naphthalene (C52904), Fischer 344 Rats. Report to National Toxicology Program (NTP) Docket No. EPA-HQ-OW-2002-0021-0075. 56p, March 4.
- Bergamini CM, Gambetti S, Dondi A, Cervellati C. (2004). Oxygen reactive oxygen species and tissue damage. Curr Pharm Des, 10, 1611–26.
- Bieniek G. (1994). The presence of 1–naphthol in the urine of industrial workers exposed to naphthalene. Occup Environ Med, 51, 357–9.
- Bieniek G. (1997). Urinary naphthols as an indicator of exposure to naphthalene. Scand J Work Environ Health, 23, 414–20.
- Boffetta P, Burstyn I, Partanen T, Kromhout H, Svane O, Langard S, et al. (2003b). Cancer mortality among European asphalt workers: An international epidemiological study. I. Results of the analysis based on job titles. Am J Ind Med, 43, 18–27.
- Boffetta P, Burstyn I, Partanen T, Kromhout H, Svane O, Langård S, et al. (2003a). Cancer mortality among European asphalt workers: An international epidemiological study. II. Exposure to bitumen fume and other agents. Am J Ind Med, 43, 28–39.
- Bogen KT, Benson JM, Yost GS, Morris JB, Dahl AR, Clewell HJ, et al. (2008). Naphthalene metabolism in relation to target tissue anatomy physiology cytotoxicity and tumorigenic mechanism of action. Regul Toxicol Pharmacol, 51, 27–36.
- Bogen KT. (2008). An adjustment factor for mode-of-action uncertainty with dual-mode carcinogens: The case of naphthalene-induced nasal tumors in rats. Risk Anal, 28, 1033–51.
- Boland B, Lin CY, Morin D, Miller L, Plopper C, Buckpitt A. (2004). Site-specific metabolism of naphthalene and 1-nitronaphthalene in dissected airways of rhesus macaques. J Pharmacol Exp Ther, 310, 546–54.
- Bosetti C, Garavello W, Gallus S, La Vecchia C. (2006). Effects of smoking cessation on the risk of laryngeal cancer: An overview of published studies. Oral Oncol, 42, 866–72.
- Boysen G, Pachkowski BF, Nakamura J, Swenberg JA. (2009). The formation and biological significance of N7-guanine adducts. Mutat Res, 678, 76–94.
- Brusick D, Small MS, Cavalieri EL, Chakravarti D, Ding X, Longfellow DG, et al. (2008). Possible genotoxic modes of action for naphthalene. Regul Toxicol Pharmacol, 51, 43–50.
- Brusick D. (2008). Critical assessment of the genetic toxicity of naphthalene. Regul Toxicol Pharmacol, 51, 37–42.
- Buckpitt A, Boland B, Isbell M, Morin D, Shultz M, Baldwin R, et al. (2002). Naphthalene-induced respiratory tract toxicity: Metabolic mechanisms of toxicity. Drug Metab Rev, 34, 791–820.
- Buckpitt A, Buonarati M, Avey LB, Chang AM, Morin D, Plopper CG. (1992). Relationship of cytochrome P450 activity to Clara cell cytotoxicity. II. Comparison of stereoselectivity of naphthalene epoxidation in lung and nasal mucosa of mouse hamster rat and rhesus monkey. J Pharmacol Exp Ther, 261, 364–72.
- Buckpitt A, Morin D, Murphy S, Edwards P, Van Winkle L. (2013). Kinetics of naphthalene metabolism in target and non-target tissues of rodents and in nasal and airway microsomes from the Rhesus monkey. Toxicol Appl Pharmacol, 270, 97–105.
- Buckpitt AR, Bahnson LS. (1986). Naphthalene metabolism by human lung microsomal enzymes. Toxicology, 41, 333–41.
- Buckpitt AR, Warren DL. (1983). Evidence for hepatic formation export and covalent binding of reactive naphthalene metabolites in extrahepatic tissues in vivo. J Pharmacol Exp Ther, 225, 8–16.
- Campbell JL, Andersen ME, Clewell HJ. (2014). A hybrid CFD–PBPK model for naphthalene in rat and human with IVIVE for nasal tissue metabolism and cross–species dosimetry. Inhal Toxicol, 26, 333–44.
- Cauffiez C, Lo–Guidice JM, Quaranta S, Allorge D, Chevalier D, Cenne S, et al. (2004). Genetic polymorphism of the human cytochrome CYP2A13 in a French population: Implication in lung cancer susceptibility. Biochem Biophys Res Commun, 317, 662–9.
- Chang JH, Kochansky CJ, Shou M. (2006). The role of P-glycoprotein in the bioactivation of raloxifene. Drug Metab Dispos, 34, 2073–8.
- Cho M, Chichester C, Morin D, Plopper C, Buckpitt A. (1994). Covalent interactions of reactive naphthalene metabolites with proteins. J Pharmacol Exp Ther, 269, 881–9.
- Cho TM, Rose RL, Hodgson E. (2006). In vitro metabolism of naphthalene by human liver microsomal cytochrome P450 enzymes. Drug Metab Dispos, 34, 176–83.
- Cichocki JA, Smith GJ, Mendoza R, Buckpitt AR, Van Winkle LS, Morris JB. (2014). Sex differences in the acute nasal antioxidant/antielectrophilic response of the rat to inhaled naphthalene. Toxicol Sci, 139, 234–44.
- Clewell HJ, Efremenko A, Campbell JL, Dodd DE, Thomas RS. (2014). Transcriptional responses in the rat nasal epithelium following subchronic inhalation of naphthalene vapor. Toxicol Appl Pharmacol, 280, 78–85.
- Consonni D, Pesatori AC, Tironi A, Bernucci I, Zocchetti C, Bertazzi PA. (1999). Mortality study in an Italian oil refinery: Extension of the follow–up. Am J Ind Med, 35, 287–94.
- Cruzan G, Bus J, Banton M, Gingell R, Carlson G. (2009). Mouse specific lung tumors from CYP2F2-mediated cytotoxic metabolism: An endpoint/toxic response where data from multiple chemicals converge to support a mode of action. Regul Toxicol Pharmacol, 55, 205–18.
- Cruzan G, Bus J, Hotchkiss J, Harkema J, Banton M, Sarang S. (2012). CYP2F2-generated metabolites not styrene oxide are a key event mediating the mode of action of styrene-induced mouse lung tumors. Regul Toxicol Pharmacol, 62, 214–20.
- Cruzan G, Bus J, Hotchkiss J, Sura R, Moore C, Yost G, et al. (2013). Studies of styrene oxide and 4-hydroxystyrene toxicity in CYP2F2 knockout and CYP2F1 humanized mice support lack of human relevance for mouse lung tumors. Regul Toxicol Pharmacol, 66, 24–9.
- DeStefano-Shields C, Morin D, Buckpitt A. (2010). Formation of covalently bound protein adducts from the cytotoxicant naphthalene in nasal epithelium: Species comparisons. Environ Health Perspect, 118, 647–52.
- Ding X, Kaminsky LS. (2003). Human extrahepatic cytochromes P450: Function in xenobiotic metabolism and tissue–selective chemical toxicity in the respiratory and gastrointestinal tracts. Annu Rev Pharmacol Toxicol, 43, 149–73.
- Ding X, Li L, Hartog MA, Wang Y, Van Winkle LS. (2014). Role of human CYP2A13 and CYP2F1 in naphthalene bioactivation and toxicity in the lung and nasal mucosa of a CYP2A13/2F1–humanized mouse. Toxicologist, 138, 137.
- D’Mello TA, Yamane GK. (2007). Occupational Jet Fuel Exposure and Invasive Cancer Occurrence in the United States Air Force 1989–2003. Air Force Institute for Operational Health: Brooks City–Base TX. (IOH–RS–BR–TR–2007–0001).
- Dodd DE, Gross EA, Miller RA, Wong BA. (2010). Nasal olfactory epithelial lesions in F344 and SD rats following 1– and 5–day inhalation exposure to naphthalene vapor. Int J Toxicol, 29, 175–84.
- Dodd DE, Wong BA, Gross EA, Miller RA. (2012). Nasal epithelial lesions in F344 rats following a 90-day inhalation exposure to naphthalene. Inhal Toxicol, 24, 70–9.
- European Union European Commission Joint Research Centre European Chemicals Bureau (EUR). (2003). European Union Risk Assessment Report: Naphthalene (CAS NO. 91–20–3) (EINECS No. 202–049–5). Office for Official Publications of the European Communities. Luxembourg. (EUR 20763 EN, 1 st Priority List Volume 33).
- Eustis SL. (1989). The sequential development of cancer: A morphological perspective. Toxicol Lett, 49, 267–81.
- Fayerweather WE. (2007). Meta-analysis of lung cancer in asphalt roofing and paving workers with external adjustment for confounding by coal tar. J Occup Environ Hyg, 4, 175–200.
- Flowers-Geary L, Bleczinki W, Harvey RG, Penning TM. (1996). Cytotoxicity and mutagenicity of polycyclic aromatic hydrocarbon ortho–quinones produced by dihydrodiol dehydrogenase. Chem Biol Interact, 99, 55–72.
- Fukami T, Katoh M, Yamazaki H, Yokoi T, Nakajima M. (2008). Human cytochrome P450 2A13 efficiently metabolizes chemicals in air pollutants: Naphthalene styrene and toluene. Chem Res Toxicol, 21, 720–5.
- Genter MB, Marlowe J, Kerzee JK, Dragin N, Puga A, Dalton TP, Nebert DW. (2006). Naphthalene toxicity in mice and aryl hydrocarbon receptor-mediated CYPs. Biochem Biophys Res Commun, 348, 120–3.
- Green T, Lee R, Toghill A, Meadowcraft S, Lund V, Foster J. (2001). The toxicity of styrene to the nasal epithelium of mice and rats: Studies on the mode of action and relevance to humans. Chem Biol Interact, 137, 185–202.
- Griego FY, Bogen KT, Price PS, Weed DL. (2008). Exposure epidemiology and human cancer incidence of naphthalene. Regul Toxicol Pharmacol, 51, 22–6.
- Harkema J. (2001). Attachment F: Pathology review of NTP chronic study of naphthalene in rats for the American Chemistry Council. Report to American Chemistry Council (ACC) Naphthalene Panel. Submitted to National Toxicology Program (NTP). In Comments of the Naphthalene Panel of the American Chemistry Council on Draft Report on Carcinogens Background Document for Naphthalene. August 26, 2002.
- Hu J, Sheng L, Li L, Zhou X, Xie F, D’Agostino J, et al. (2014). Essential role of the cytochrome P450 enzyme CYP2A5 in olfactory mucosal toxicity of naphthalene. Drug Metab Dispos, 42, 23–7.
- International Agency for Research on Cancer (IARC). (2002). Some traditional herbal medicines some mycotoxins naphthalene and styrene: Section on naphthalene. In IARC Monographs on the Evaluation of Carcinogenic Risks to Humans (Volume 82). Lyon: International Agency for Research on Cancer (IARC). Geneva: World Health Organization (WHO).
- Jedrychowski W, Galas A, Pac A, Flak E, Camman D, Rauh V, Perera F. (2005). Prenatal ambient air exposure to polycyclic aromatic hydrocarbons and the occurrence of respiratory symptoms over the first year of life. Eur J Epidemiol, 20, 775–82.
- Jin Y, Penning TM. (2007). Aldo–keto reductases and bioactivation/detoxication. Annu Rev Pharmacol Toxicology, 47, 263–92.
- Kapuci M, Ulker Z, Gurkan S, Alpsoy L. (2012). Determination of cytotoxic and genotoxic effects of naphthalene 1–naphthol and 2–naphthol on human lymphocyte culture. Toxicol Ind Health, 30, 82–9.
- Karagiannis TC, Li X, Tang MM, Orlowski C, El-Osta A, Tang ML, Royce SG. (2012). Molecular model of naphthalene-induced DNA damage in the murine lung. Hum Exp Toxicol, 31, 42–50.
- Karlgren M, Miura S, Ingelman–Sundberg M. (2005). Novel extrahepatic cytochrome P450s. Toxicol Appl Pharmacol, 207, 57–61.
- Kedderis GL, Shepard KG, Recio L. (2014). Cytotoxicity of naphthalene toward cells from target and non-target organs in vitro. Chem Biol Interact, 209, 85–95.
- Kim HY, Finneman JI, Harris CM, Harris TM. (2000). Studies of the mechanisms of adduction of 2′-deoxyadenosine with styrene oxide and polycyclic aromatic hydrocarbon dihydrodiol epoxides. Chem Res Toxicol, 13, 625–37.
- Kim JH, Kim JK, Son BK, Oh JE, Lim DH, Lee KH, et al. (2005). Effects of air pollutants on childhood asthma. Yonsei Med J, 46, 239–44.
- Kim SJ, Jajoo HK, Kim HY, Zhou L, Horton P, Harris CM, Harris TM. (1995). An efficient route to N6 deoxyadenosine adducts of diol epoxides of carcinogenic polycyclic aromatic hydrocarbons. Bioorg Med Chem, 3, 811–22.
- Kitteringham NR, Davis C, Howard N, Pirmohamed M, Park BK. (1996). Interindividual and interspecies variation in hepatic microsomal epoxide hydrolase activity: Studies with cis-stilbene oxide carbamazepine 10 11-epoxide and naphthalene. J-Pharmacol Exp Ther, 278, 1018–27.
- Klotz K, Schindler BK, Angerer J. (2011). 1,2-Dihydroxynaphthalene as biomarker for a naphthalene exposure in humans. Int J Hyg Environ Health, 214, 110–14.
- Krieg EF Jr, Mathias PI, Toennis CA, Clark JC, Marlow KL, B'hymer C, et al. (2012). Detection of DNA damage in workers exposed to JP-8 jet fuel. Mutat Res, 747, 218–27.
- Lanza DL, Code E, Crespi CL, Gonzalez FJ, Yost GS. (1999). Specific dehydrogenation of 3-methylindole and epoxidation of naphthalene by recombinant human CYP2F1 expressed in lymphoblastoid cells. Drug Metab-Dispos, 27, 798–803.
- Leclerc J, Tournel G, Courcot-Ngoubo Ngangue E, Pottier N, Lafitte JJ, Jaillard S, et al. (2010). Profiling gene expression of whole cytochrome P450 superfamily in human bronchial and peripheral lung tissues: Differential expression in non-small cell lung cancers. Biochimie, 92, 292–306.
- Lee MG, Phimister A, Morin D, Buckpitt A, Plopper C. (2005). In situ naphthalene bioactivation and nasal airflow cause region-specific injury patterns in the nasal mucosa of rats exposed to naphthalene by inhalation. J-Pharmacol Exp Ther, 314, 103–10.
- Lewis DF, Ito Y, Lake BG. (2009). Molecular modelling of CYP2F substrates: Comparison of naphthalene metabolism by human rat and mouse CYP2F subfamily enzymes. Drug Metab Drug Interact, 24, 229–57.
- Lewis RJ, Schnatter AR, Drummond I, Murray N, Thompson FS, Katz AM, et al. (2003). Mortality and cancer morbidity in a cohort of Canadian petroleum workers. Occup Environ Med, 60, 918–28.
- Lewis RJ. (2012). Naphthalene animal carcinogenicity and human relevancy: Overview of industries with naphthalene-containing streams. Regul Toxicol Pharmacol, 62, 131–7.
- Li L, Wei Y, Van Winkle L, Zhang QY, Zhou X, Hu J, et al. (2011). Generation and characterization of a Cyp2f2-Null mouse and studies on the role of CYP2F2 in naphthalene-induced toxicity in the lung and nasal olfactory mucosa. J Pharmacol Exp Ther, 339, 62–71.
- Lin CY, Boland BC, Lee YJ, Salemi MR, Morin D, Miller LA, et al. (2006). Identification of proteins adducted by reactive metabolites of naphthalene and 1-nitronaphthalene in dissected airways of rhesus macaques. Proteomics, 6, 972–82.
- Lin CY, Isbell MA, Morin D, Boland BC, Salemi MR, Jewell WT, et al. (2005). Characterization of a structurally intact in situ lung model and comparison of naphthalene protein adducts generated in this model vs lung microsomes. Chem Res Toxicol, 18, 802–13.
- Lin PH, Chen DR, Wang TW, Lin CH, Chuang MC. (2009). Investigation of the cumulative tissue doses of naphthoquinones in human serum using protein adducts as biomarker of exposure. Chem Biol Interact, 181, 107–14.
- Long PH, Herbert RA, Peckham JC, Grumbein SL, Shackelford CC, Abdo K. (2003). Morphology of nasal lesions in F344/N rats following chronic inhalation exposure to naphthalene vapors. Toxicol Pathol, 31, 655–64.
- Marczynski B, Raulf-Heimsoth M, Spickenheuer A, Pesch B, Kendzia B, Mensing T, et al. (2011). DNA adducts and strand breaks in workers exposed to vapours and aerosols of bitumen: Associations between exposure and effect. Arch Toxicol, 85, S53–S64.
- McCarty KM, Ryan L, Houseman EA, Williams PL, Miller DP, Quamruzzaman Q, et al. (2007). A case-control study of GST polymorphisms and arsenic related skin lesions. Environ Health, 6, 5.
- Meng F, Wang Y, Myers MB, Wong BA, Gross EA, Clewell HJ III, et al. (2011). P53 codon 271 CGT to CAT mutant fraction does not increase in nasal respiratory and olfactory epithelia of rats exposed to inhaled naphthalene. Mutat Res, 721, 199–205.
- Merletti F, Boffetta P, Ferro G, Pisani P, Terracini B. (1991). Occupation and cancer of the oral cavity or oropharynx in Turin Italy. Scand J Work Environ Health, 17, 248–54.
- Miller RL, Garfinkel R, Horton M, Camann D, Perera FP, Whyatt RM, Kinney PL. (2004). Polycyclic aromatic hydrocarbons environmental tobacco smoke and respiratory symptoms in an inner-city birth cohort. Chest, 126, 1071–8.
- Moore NP, McFadden LG, Landenberger BD, Thomas J. (2013). Gender differences in the incidence of background and chemically induced primary pulmonary neoplasms in B6C3F1 mice: A retrospective analysis of the National Toxicology Program (NTP) carcinogenicity bioassays. Exp Toxicol Pathol, 65, 1109–15.
- Morley N, Rapp A, Dittmar H, Salter L, Gould D, Greulich KO, Curnow A. (2006). UVA-induced apoptosis studied by the new apo/necro-Comet-assay which distinguishes viable apoptotic and necrotic cells. Mutagenesis, 21, 105–14.
- Morris JB, Buckpitt AR. (2009). Upper respiratory tract uptake of naphthalene. Toxicol Sci, 111, 383–91.
- Morris JB. (2013). Nasal dosimetry of inspired naphthalene vapor in the male and female B6C3F1 mouse. Toxicology, 309, 66–72.
- Munday R, Smith BL, Munday CM. (2007). Structure-activity relationships in the haemolytic activity and nephrotoxicity of derivatives of 1,2- and 1,4-naphthoquinone. J Appl Toxicol, 27, 262–9.
- Nagata K, Martin BM, Gillette JR, Sasame HA. (1990). Isozymes of cytochrome P-450 that metabolize naphthalene in liver and lung of untreated mice. Drug Metab Dispos, 18, 557–64.
- Nan HM, Kim H, Lim HS, Choi JK, Kawamoto T, Kang JW, et al. (2001). Effects of occupation lifestyle and genetic polymorphisms of CYP1A1 CYP2E1 GSTM1 and GSTT1 on urinary 1-hydroxypyrene and 2-naphthol concentrations. Carcinogenesis, 22, 787–93.
- National Cancer Institute (NCI). (2014). “SEER Fast Stats: Compare statisics by data type, 1975-–2011.” Surveillance, Epidemiology, and End Results (SEER) Program. Accessed at http://seer.cancer.gov/faststats/selections.php.
- National Institute for Occupational Safety and Health (NIOSH). (2000). Hazard Review: Health Effects of Occupational Exposures to Asphalt. (United States Dept. of Health and Human Services NIOSH Publication No. 2001–110).
- National Toxicology Program (NTP). (2011). Naphthalene (CAS No. 91–20–3). In Report on Carcinogens (12th Edition). US Dept. of Health and Human Services Public Health Service: National Toxicology Program.
- National Toxicology Program (NTP). (1992).Toxicology and Carcinogenesis Studies of Naphthalene (CAS No. 91–20–3) in B6C3F1 Mice (Inhalation Studies). US Public Health Service: National Institutes of Health. (NTP–TR–410, NIH Publication No. 92–3141).
- National Toxicology Program (NTP). (December 2000). NTP Technical Report on the Toxicology and Carcinogenesis Studies of Naphthalene (CAS No. 91–20–3) in F344/N Rats (Inhalation Studies). US Public Health Service: National Institutes of Health. (NTP–TR–500, NIH Publication No. 01–4434).
- National Toxicology Program (NTP). 2012. Long–term bioassay study search options for C52904C. [Online] Available from: http://tools.niehs.nih.gov/ntp_tox/index.cfm?fuseaction = longtermbioassaydata.datasearch&&study_no = C52904C&&study_length = 2%20Years. [Accessed August 2012].
- Neafsey P, Ginsberg G, Hattis D, Johns DO, Guyton KZ, Sonawane B. (2009). Genetic polymorphism in CYP2E1: Population distribution of CYP2E1 activity. J Toxicol Environ Health B Crit Rev, 12, 362–88.
- Nishimura M, Naito S. (2006). Tissue-specific mRNA expression profiles of human phase I metabolizing enzymes except for cytochrome P450 and phase II metabolizing enzymes. Drug Metab Pharmacokinet, 21, 357–74.
- North DW, Abdo KM, Benson JM, Dahl AR, Morris JB, Renne R, Witschi H. (2008). A review of whole animal bioassays of the carcinogenic potential of naphthalene. Regul Toxicol Pharmacol, 51, 6–14.
- O’Connor T, Ireland LS, Harrison DJ, Hayes JD. (1999). Major differences exist in the function and tissue-specific expression of human aflatoxin B1 aldehyde reductase and the principal human aldo-keto reductase AKR1 family members. Biochem J, 343, 487–504.
- Oliver JR, Kushwah R, Wu J, Cutz E, Yeger H, Waddell TK, Hu J. (2009). Gender differences in pulmonary regenerative response to naphthalene-induced bronchiolar epithelial cell injury. Cell Prolif, 42, 672–87.
- Olsson A, Kromhout H, Agostini M, Hansen J, Lassen CF, Johansen C, et al. (2010). A case-control study of lung cancer nested in a cohort of European asphalt workers. Environ Health Perspect, 118, 1418–24.
- Orjuela M, Liu X, Miller RL, Warburton D, Tang DL, Jobanputra V, et al. (2012). Urinary naphthol metabolites and chromosomal aberrations in 5 yr old children. Cancer Epidemiol Biomarkers Prev, 21, 1191–202.
- Palackal NT, Lee SH, Harvey RG, Blair IA, Penning TM. (2002). Activation of polycyclic aromatic hydrocarbon trans-dihydrodiol proximate carcinogens by human aldo-keto reductase (AKR1C) enzymes and their functional overexpression in human lung carcinoma (A549) cells. J Biol Chem, 277, 24799–808.
- Penning TM, Drury JE. (2007). Human aldo-keto reductases: Function gene regulation and single nucleotide polymorphisms. Arch Biochem Biophys, 464, 241–50.
- Pfuhler S, Fellows M, van Benthem J, Corvi R, Curren R, Dearfield K, et al. (2011). In vitro genotoxicity test approaches with better predictivity: Summary of an IWGT workshop. Mutat Res, 723, 101–7.
- Pham NT, Jewell WT, Morin D, Buckpitt AR. (2012a). Analysis of naphthalene adduct binding sites in model proteins by tandem mass spectrometry. Chem Biol Interact, 199, 120–8.
- Pham NT, Jewell WT, Morin D, Jones AD, Buckpitt AR. (2012b). Characterization of model Peptide adducts with reactive metabolites of naphthalene by mass spectrometry. PLoS ONE, 7, e42053.
- Phimister AJ, Lee MG, Morin D, Buckpitt AR, Plopper CG. (2004). Glutathione depletion is a major determinant of inhaled naphthalene respiratory toxicity and naphthalene metabolism in mice. Toxicol Sci, 82, 268–78.
- Piccirillo VJ, Bird MG, Lewis RJ, Bover WJ. (2012). Preliminary evaluation of the human relevance of respiratory tumors observed in rodents exposed to naphthalene. Regul Toxicol Pharmacol, 62, 433–40.
- Plopper CG, Macklin J, Nishio SJ, Hyde DM, Buckpitt AR. (1992a). Relationship of cytochrome P-450 activity to Clara cell cytotoxicity. III. Morphometric comparison of changes in the epithelial populations of terminal bronchioles and lobar bronchi in mice hamsters and rats after parenteral administration of naphthalene. Lab Invest, 67, 553–65.
- Plopper CG, Suverkropp C, Morin D, Nishio S, Buckpitt A. (1992b). Relationship of cytochrome P-450 activity to Clara cell cytotoxicity. I. Histopathologic comparison of the respiratory tract of mice rats and hamsters after parenteral administration of naphthalene. J Pharmacol Exp Ther, 261, 353–63.
- Prueitt RL, Goodman JE, Bailey LA, Rhomberg LR. (2011). Hypothesis-based weight-of-evidence evaluation of the neurodevelopmental effects of chlorpyrifos. Crit Rev Toxicol, 41, 822–903.
- Raunio H, Hakkola J, Hukkanen J, Lassila A, Paivarinta L, Pelkonen O, et al. (1999). Expression of xenobiotic-metabolizing CYPs in human pulmonary tissue. Exp Toxicol Pathol, 51, 412–17.
- Recio L, Shepard KG, Hernandez LG, Kedderis GL. (2012). Dose-response assessment of naphthalene-induced genotoxicity and glutathione detoxication in human TK6 lymphoblasts. Toxicol Sci, 126, 405–12.
- Rhomberg L. (2014). Hypothesis-based weight-of-evidence: An approach to assessing causation and its application to regulatory toxicology. Risk Anal, doi:10.1111/risa.12206.
- Rhomberg LR, Bailey LA, Goodman JE, Hamade A, Mayfield D. (2011). Is exposure to formaldehyde in air causally associated with leukemia? – A hypothesis-based weight-of-evidence analysis. Crit Rev Toxicol, 41, 555–621.
- Rhomberg LR, Bailey LA, Goodman JE. (2010). Hypothesis-based weight of evidence: A tool for evaluating and communicating uncertainties and inconsistencies in the large body of evidence in proposing a carcinogenic mode of action – Naphthalene as an example. Crit Rev Toxicol, 40, 671–96.
- Ribble D, Goldstein NB, Norris DA, Shellman YG. (2005). A simple technique for quantifying apoptosis in 96-well plates. BMC Biotechnol, 5, 12.
- Richieri PR, Buckpitt AR. (1987). Efflux of naphthalene oxide and reactive naphthalene metabolites from isolated hepatocytes. J Pharmacol Exp Ther, 242, 485–92.
- Roberts ES, Alworth WL, Hollenberg PF. (1998). Mechanism-based inactivation of cytochromes P450 2E1 and 2B1 by 5-phenyl-1-pentyne. Arch Biochem Biophys, 354, 295–302.
- Roco A, Quinones L, Agundez JA, Garcia-Martin E, Squicciarini V, Miranda C, et al. (2012). Frequencies of 23 functionally significant variant alleles related with metabolism of antineoplastic drugs in the Chilean population: Comparison with Caucasian and Asian populations. Front Genet, 3, 229.
- Rushton L. (1993). A 39-year follow-up of the U.K. oil refinery and distribution center studies: Results for kidney cancer and leukemia. Environ Health Perspect, 101, 77–84.
- Saarikoski ST, Rivera SP, Hankinson O, Husgafvel-Pursiainen K. (2005). CYP2S1: A short review. Toxicol Appl Pharmacol, 207, 62–9.
- Saeed M, Higginbotham S, Gaikwad N, Chakravarti D, Rogan E, Cavalieri E. (2009). Depurinating naphthalene-DNA adducts in mouse skin related to cancer initiation. Free Radic Biol Med, 47, 1075–81.
- Saeed M, Higginbotham S, Rogan E, Cavalieri E. (2007). Formation of depurinating N3adenine and N7guanine adducts after reaction of 12-naphthoquinone or enzyme-activated 12-dihydroxynaphthalene with DNA. Implications for the mechanism of tumor initiation by naphthalene. Chem Biol Interact, 165, 175–88.
- Salam MT, Lin PC, Avol EL, Gauderman WJ, Gilliland FD. (2007). Microsomal epoxide hydrolase glutathione S-transferase P1 traffic and childhood asthma. Thorax, 62, 1050–7.
- Schmahl D. (1955). Testing of naphthalene and anthracene for carcinogenic effect in rats. Z Krebsforsch, 60, 697–710.
- Schreiner CA. (2003). Genetic toxicity of naphthalene: A review. J Toxicol Environ Health B Crit Rev, 6, 161–83.
- Seidegard J, Ekstrom G. (1997). The role of human glutathione transferases and epoxide hydrolases in the metabolism of xenobiotics. Environ Health Perspect, 105, 791–9.
- Shultz CA, Quinn AM, Park JH, Harvey RG, Bolton JL, Maser E, Penning TM. (2011). Specificity of human aldo-keto reductases NAD(P)H:quinone oxidoreductase and carbonyl reductases to redox-cycle polycyclic aromatic hydrocarbon diones and 4-hydroxyequilenin-o-quinone. Chem Res Toxicol, 24, 2153–66.
- Simmonds AC, Ghanayem BI, Sharma A, Reilly CA, Millen B, Yost GS, Forkert PG. (2004). Bioactivation of 11-dichloroethylene by CYP2E1 and CYP2F2 in murine lung. J Pharmacol Exp Ther, 310, 855–64.
- Su T, Bao Z, Zhang QY, Smith TJ, Hong JY, Ding X. (2000). Human cytochrome P450 CYP2A13: Predominant expression in the respiratory tract and its high efficiency metabolic activation of a tobacco-specific carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone. Cancer Res, 60, 5074–9.
- Sudakin DL, Stone DL, Power L. (2011). Naphthalene mothballs: Emerging and recurring issues and their relevance to environmental health. Curr Top Toxicol, 7, 13–19.
- Sutherland KM, Edwards PC, Combs TJ, Van Winkle LS. (2012). Sex differences in the development of airway epithelial tolerance to naphthalene. Am J Physiol Lung Cell Mol Physiol, 302, L68–L91.
- Talamini R, Bosetti C, La Vecchia C, Dal Maso L, Levi F, Bidoli E, et al. (2002). Combined effect of tobacco and alcohol on laryngeal cancer risk: A case-control study. Cancer Causes Control, 13, 957–64.
- Thornton-Manning JR, Dahl AR. (1997). Metabolic capacity of nasal tissue interspecies comparisons of xenobiotic-metabolizing enzymes. Mutat Res, 380, 43–59.
- Tournel G, Cauffiez C, Billaut-Laden I, Allorge D, Chevalier D, Bonnifet F, et al. (2007). Molecular analysis of the CYP2F1 gene: Identification of a frequent non-functional allelic variant. Mutat Res, 617, 79–89.
- United States Environmental Protection Agency (US EPA). (2004). Toxicological Review of Naphthalene (CAS No. 91–20–3) (External review draft). (NCEA–S–1707).
- United States Environmental Protection Agency (US EPA). (2012a). Benchmark Dose Technical Guidance. Risk Assessment Forum. (EPA/100/R–12/001).
- United States Environmental Protection Agency (US EPA). (2012b). Benchmark Dose Software (BMDS) (Version 2.3.1). National Center for Environmental Assessment (NCEA). [Online] Available from: http://www.epa.gov/ncea/bmds/.
- United States Environmental Protection Agency (US EPA). (2014). Mouse Lung Tumor Workshop. [Online]Available from: http://www.epa.gov/iris/irisworkshops/mltw/.
- United States Environmental Protection Agency (US EPA). (1998). “Toxicological Review of Naphthalene (CAS No. 91-–20-3).” 116p.
- Van Heyningen R, Pirie A. (1967). The metabolism of naphthalene and its toxic effect on the eye. Biochem J, 102, 842–52.
- Van Winkle LS, Buckpitt AR, Nishio SJ, Isaac JM, Plopper CG. (1995). Cellular response in naphthalene-induced Clara cell injury and bronchiolar epithelial repair in mice. Am J Physiol, 269, L800–L818.
- Van Winkle LS, Buckpitt AR, Plopper CG. (1996). Maintenance of differentiated murine Clara cells in microdissected airway cultures. Am J Respir Cell Mol Biol, 14, 586–98.
- Van Winkle LS, Cichocki J, Mendoza R, Edwards P, Morris JB. (2014). Sex differences in glutathione levels and cytotoxicity following naphthalene or acrolein exposure in nasal epithelium. Toxicologist, 138, 322.
- Van Winkle LS, Gunderson AD, Shimizu JA, Baker GL, Brown CD. (2002). Gender differences in naphthalene metabolism and naphthalene-induced acute lung injury. Am J Physiol Lung Cell Mol Physiol, 282, L1122–L1134.
- Van Winkle LS, Isaac JM, Plopper CG. (1997). Distribution of epidermal growth factor receptor and ligands during bronchiolar epithelial repair from naphthalene–induced Clara cell injury in the mouse. Am J Pathol, 151, 443–59.
- Vineis P, Schatzkin A, Potter JD. (2010). Models of carcinogenesis: An overview. Carcinogenesis, 31, 1703–9.
- Waidyanatha S, Rappaport SM. (2008). Hemoglobin and albumin adducts of naphthalene-12-oxide 12-naphthoquinone and 14-naphthoquinone in Swiss Webster mice. Chem Biol Interact, 172, 105–14.
- Waidyanatha S, Troester MA, Lindstrom AB, Rappaport SM. (2002). Measurement of hemoglobin and albumin adducts of naphthalene-12-oxide 12-naphthoquinone and 14-naphthoquinone after administration of naphthalene to F344 rats. Chem Biol Interact, 141, 189–210.
- Wang H, Tan W, Hao B, Miao X, Zhou G, et al. (2003). Substantial reduction in risk of lung adenocarcinoma associated with genetic polymorphism in CYP2A13 the most active cytochrome P450 for the metabolic activation of tobacco-specific carcinogen NNK. Cancer Res, 63, 8057–61.
- Warren DL, Brown DL Jr., Buckpitt AR. (1982). Evidence for cytochrome P-450 mediated metabolism in the bronchiolar damage by naphthalene. Chem Biol Interact, 40, 287–303.
- West JA, Buckpitt AR, Plopper CG. (2000). Elevated airway GSH resynthesis confers protection to Clara cells from naphthalene injury in mice made tolerant by repeated exposures. J Pharmacol Exp Ther, 294, 516–23.
- West JA, Pakehham G, Morin D, Fleschneer CA, Buckpitt AR, Plopper CG. (2001). Inhaled naphthalene causes dose dependent Clara cell cytotoxicity in mice but not in rats. Toxicol Appl Pharmacol, 173, 114–19.
- Willems BA, Melnick RL, Kohn MC, Portier CJ. (2001). A physiologically based pharmacokinetic model for inhalation and intravenous administration of naphthalene in rats and mice. Toxicol Appl Pharmacol, 176, 81–91.
- Wilson AS, Davis CD, Williams DP, Buckpitt AR, Pirmohamed M, Park BK. (1996). Characterisation of the toxic metabolite(s) of naphthalene. Toxicology, 114, 233–42.
- Wong O, Harris F. (2005). Retrospective cohort mortality study and nested case-control study of workers exposed to creosote at 11 wood-treating plants in the United States. J Occup Environ Med, 47, 683–97.
- Wong O, Raabe GK. (2000). A critical review of cancer epidemiology in the petroleum industry with a meta–analysis of a combined database of more than 350000 workers. Regul Toxicol Pharmacol, 32, 78–98.
- Wu R, Waidyanatha S, Henderson AP, Serdar B, Zheng Y, Rappaport SM. (2005). Determination of dihydroxynaphthalenes in human urine by gas chromatography–mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci, 826, 206–13.
- Wu X, Gwyn K, Amos CI, Makan N, Hong WK, Spitz MR. (2001). The association of microsomal epoxide hydrolase polymorphisms and lung cancer risk in African-Americans and Mexican-Americans. Carcinogenesis, 22, 923–8.
- Xu GT, Zigler JS Jr., Lou MF. (1992a). Establishment of a naphthalene cataract model in vitro. Exp Eye Res, 54, 73–81.
- Xu GT, Zigler JS Jr., Lou MF. (1992b). The possible mechanism of naphthalene cataract in rat and its prevention by an aldose reductase inhibitor (ALO1576). Exp Eye Res, 54, 63–72.
- Yamane GK. (2006). Cancer incidence in the U.S. Air Force: 1989–2002. Aviat Space Environ Med, 77, 789–94.
- Yang M, Koga M, Katoh T, Kawamoto T. (1999). A study for the proper application of urinary naphthols new biomarkers for airborne polycyclic aromatic hydrocarbons. Arch Environ Contam Toxicol, 36, 99–108.
- Zheng J, Cho M, Jones AD, Hammock BD. (1997). Evidence of quinone metabolites of naphthalene covalently bound to sulfur nucleophiles of proteins of murine Clara cells after exposure to naphthalene. Chem Res Toxicol, 10, 1008–14.