
Abstract
Biosimilar development involves a target-directed iterative process to ensure a similar product to the originator. Here we report the preclinical development of the proposed biosimilar rituximab (GP2013). Post-translational modifications and bioactivities of GP2013 versus originator rituximab were engineered and monitored to ensure similar pharmacological profiles. Antibody-dependent cellular cytotoxicity (ADCC) was used to illustrate how different glycosylation patterns and structure–function relationships were controlled during process development. Pharmacological comparability between GP2013 and originator rituximab were confirmed in preclinical studies using clinical scale drug product. Similar in vitro ADCC potency was demonstrated when compared in a dose–response manner against two lymphoma cell lines using freshly purified human natural killer (NK) cells. In vivo efficacy was demonstrated in two well characterized mouse xenograft models, testing at sensitive sub-therapeutic dose levels. Pharmacokinetics and pharmacodynamics (CD20 cell depletion) were likewise comparable in cynomolgus monkeys. This preclinical comparability exercise confirms that GP2013 and originator rituximab are pharmacologically similar.
Introduction
Biologically derived therapeutics have revolutionized modern medicine, significantly improving health care outcomes for patients since they were first introduced in the 1980s [Citation1,Citation2]. However, the increasing use of therapeutic monoclonal antibodies (mAbs), especially in the areas of cancer and autoimmune disease, constitutes a major and increasing cost burden for health care systems. Biosimilars provide a comparable level of efficacy and safety with that of the originator product with the added advantage of being more affordable, thereby expanding patient access to therapies that otherwise may be restricted for cost reasons [Citation3].
Biosimilars are biologics that are approved in highly-regulated markets as being similar to existing agents for which patents have expired. Regulatory approval is provided on the basis of comparable quality, safety and efficacy to an originator product [Citation4,Citation5]. In Europe, the European Medicines Agency (EMA) has developed a specific regulatory pathway and recently issued guidelines that specifically describe non-clinical and clinical requirements for the development of biosimilar mAbs [Citation6]. Other countries have also adopted similar regulatory frameworks based on the European guidelines. In the USA, the Food and Drug Administration (FDA) released draft guidance for the regulatory review of biosimilars in early 2012 [Citation7]. Second-generation drugs, such as biobetters, cannot be approved via the biosimilar regulatory pathways [Citation4].
Therapeutic mAbs and immunoglobulin G (IgG)/Fc-fusion proteins are typically manufactured using mammalian cell expression systems, and as such are subject to post-translational modifications (also known as quality attributes). The overall profile of these quality attributes has been shown to exhibit minor batch-to-batch variability, as well as more significant variability due to manufacturing process changes [Citation8]. The variance in these target specifications provides a variability range within which the biosimilar product attributes need to be to achieve comparability [Citation4]. Thus, thorough physicochemical and in vitro biological characterization of the attributes in the originator product over an extended period (e.g. 5–7 years) is the first step in the development of a biosimilar.
Physicochemical similarity to the originator is essential to ensure a comparable clinical efficacy and safety profile of the end product. To bring key outlying attributes in line with the variability ranges of the originator product, specific steps in the manufacturing process are repeatedly modified. In this regard, the design of the entire manufacturing process quality is integral to the quality of a biosimilar product. Paramount to this exercise is the implementation of comprehensive and sensitive analytical tools [Citation9,Citation10].
Rituximab (Rituxan®/MabThera®) was the first mAb to show a clinically significant anticancer effect, and has been in clinical use for 15 years for the treatment of patients with non-Hodgkin lymphoma and chronic lymphocytic leukemia, as well as rheumatoid arthritis and other autoimmune conditions. It is a chimeric mouse/human mAb where the Fab domain of rituximab binds to the CD20 antigen and the Fc domain recruits immune effector functions to mediate B-cell lysis, as well as modulating exposure. Postulated mechanisms of effector-mediated cell lysis include antibody-dependent cellular cytotoxicity (ADCC) mediated by one or more of the Fcγ receptors on the surface of granulocytes, macrophages and natural killer (NK) cells, and complement-dependent cytotoxicity (CDC) resulting from C1q binding and the subsequent lytic cascade [Citation11–13]. Rituximab binding to CD20 has also been shown to activate signaling cascades that result in the induction of cell death via apoptosis [Citation14].
Several candidate rituximab biosimilars are in development [Citation15,Citation16]. GP2013 is a proposed rituximab biosimilar being developed according to the biosimilar regulatory guidance and by applying quality-by-design (QbD) principles [Citation17]. Using the example of ADCC activity, we illustrate the target-directed development and characterization of the proposed biosimilar GP2013 and show how structure– function relationships can be used to ensure comparability at the in vitro level. This phase is critical for regulatory approval of biosimilars; it ensures that critical post- translational modifications affecting effector functions (and other pharmacological properties of the biologic introduced during mammalian cell line expression) are in line with the reference product [Citation18], and also importantly avoids modifications that are recognized as foreign in human subjects and potentially induce adverse reactions in patients [Citation19]. Furthermore, a tailored program of preclinical studies using clinical-scale drug product comparing biosimilar rituximab with the originator product is reported, providing preclinical confirmatory evidence of in vivo similarity with regard to pharmacokinetics (PK), pharmacodynamics (PD) and efficacy.
Materials and methods
Glycan quantification
N-glycans were released from the Fc part of the mAbs using N-glycosidase F (Roche Diagnostics, Mannheim, Germany). Subsequent separation and concentration was performed by ultracentrifugal filtration and centrifugal evaporation. Excess 2-aminobenzamide (2-AB; Fluka, Sigma-Aldrich Chemie GmbH, Munich, Germany, and Steinheim, Germany) was used to label the glycans. Free 2-AB label glycan derivates were removed by gel filtration using Sephadex G10 (GE Healthcare, Munich, Germany). Normal phase chromatography, using an ACQUITY UPLC BEH glycan 1.7 μm 100 × 2.1 mm column (Waters, Saint-Quentin en Yvelines, France) was employed to separate the 2-AB labeled glycans. A fluorescence detector, set at an excitation wavelength of 250 nm and emission wavelength of 428 nm, recorded elution of the 2-AB labeled glycans.
In vitro ADCC development potency assay
The characterization assay to assess the in vitro ADCC activities of GP2013 and originator rituximab used the Raji B-cell line as target cell and the CD16 overexpressing NK3.3 cell line as effector cell. Raji B-cells were loaded with the fluorochrome calcein, and subsequently incubated with different concentrations of GP2013 or originator rituximab and an excess of NK3.3 cells (ratio of 1:10). Concentration-dependent killing of the Raji B-cells was analyzed by measuring the release of calcein at 515 nm. ADCC activity was calculated using a parallel line assay according to the European Pharmacopoeia 7th edition. The final result was expressed as the relative potency of a sample compared to a reference.
In vitro ADCC confirmatory potency assay
The relative ADCC activity of GP2013 and originator rituximab were evaluated in a dose–response manner across a wide concentration range against the SU-DHL-4 (diffuse large B-cell lymphoma) and Daudi (Burkitt lymphoma) cell lines using freshly purified human NK cells and an effector to target ratio of 10:1. As ADCC is sensitive to the phenylalanine (F) to valine (V) polymorphism at position 158 in FcγRIIIa (V/V having greater potency than V/F or F/F) [Citation11], freshly purified human NK cells of the V/V genotype from a healthy donor were used to increase the sensitivity of the assay to identify potential differences. This assay differed from that of the characterization ADCC assay described above as part of the development process in the source of effector (NK) cells used (freshly purified, V/V genotyped human NK cells versus cell line), use of a wider range of dose concentrations used in the more physiological setting, and use of two different target cell lines.
Mouse xenograft models
Anti-tumor activity was assessed in two xenograft SCID (severe combined immune deficiency) mouse models of non-Hodgkin lymphoma with the SU-DHL-4 (diffuse large B-cell lymphoma) and Jeko-1 (mantle cell lymphoma) cell lines. SCID mice were subcutaneously injected with 1 × 107 tumor cells into the flank, and those with tumor volumes of 100–150 mm3 (SU-DHL-4) or 200 mm3 (Jeko-1) after 22 days were randomized to a negative control group (n = 10) which received placebo (Privigen®; polyvalent human IgG 30 mg/kg) and multiple treatment groups which received GP2013 or originator rituximab over a range of sub-therapeutic doses defined according to a pre-established maximal effect dose. These were 3 mg/kg or 30 mg/kg for SU-DHL-4 (n = 50 per group) and 0.03 (n = 24), 0.1 (n = 40), 0.3 (n = 40) or 1 mg/kg (n = 24) for Jeko-1. Antibodies were administered intraperitoneally once weekly for 4 weeks. Tumors were calipered twice weekly and mice were euthanized when tumors attained the 2000 mm3 volume endpoint or at study end (60 days).
Pharmacokinetics and pharmacodynamics in cynomolgus monkeys
Comparative PK and PD of GP2013 and originator rituximab (MabThera®) were assessed in single and multiple dose studies in cynomolgus monkeys. These were chosen as the most appropriate pharmacologically relevant species, given the lack of cross-reactivity of rituximab to non-primate CD20 [Citation20]. In the single-dose study, 14 cynomolgus monkeys were administered intravenous (i.v.) 5 mg/kg doses of GP2013 or MabThera®. Serum samples were collected for up to 10 weeks after administration. In the multiple-dose study, 32 animals received either 20 mg/kg or 100 mg/kg doses of GP2013 or MabThera® (eight per group), administered via weekly i.v. bolus injection for 4 weeks (days 1, 8, 15 and 22). Animals were then followed for a 4-week dosing-free period, with a subset (n = 4 per group) followed by a 6-month recovery period. Serum samples were collected for up to 8 or 30 weeks.
Pharmacokinetics assay
A competitive enzyme-linked immunosorbent assay (ELISA) format was used to determine rituximab concentrations in cynomolgus monkey serum. A microtiter plate was coated with a monoclonal rat anti-rituximab antibody and non- specific binding sites were blocked. A mixture of serum containing rituximab and a defined concentration of biotinylated rituximab was then added to the coated microtiter plate. After a further incubation with streptavidin–horseradish peroxidase (HRP) which bound to the biotinylated rituximab, a chromogen was added into the wells and was oxidized by HRP to form a blue-colored complex. After stopping the reaction with acid, the optical density was measured at 450 nm using a microplate reader. The concentrations of the quality control and study samples were interpolated using a standard curve from known concentrations of rituximab that were processed similarly to the study samples.
Pharmacodynamics assay
B-cells were subcategorized to follow the relative and absolute counts of the two B-cell subsets found in cynomolgus monkeys (CD20high CD40low CD21− and CD20low CD40high CD21+) [Citation20]. Immunostaining analysis of relative cell numbers (percentage of lymphocytes) was performed using a FACSCalibur instrument (Becton Dickinson GmbH, Heidelberg, Germany). Total lymphocyte counts were determined on the same day. Absolute numbers of the lymphocyte subpopulations were calculated from relative and total numbers. Antigen saturation was reported as the mean fluorescence intensity (MFI) and anti-CD20 isotype antibody stained B-cells (gated on CD40).
Statistics
In vitro ADCC confirmatory assay
The fluorescence values for the triplicate wells representing the spontaneous release control were averaged, as were the values for the triplicate wells representing the maximum lysis control. For each of the wells representing the antibody dilution series, a “% specific lysis” value was calculated. The triplicate “% specific lysis” values for each step of each dilution series were then averaged, and the results graphed as antibody concentration (ng/mL) versus average % specific lysis using GraphPad Prism4 software. The GraphPad Prism4 software was also used to calculate the 50% effective concentration (EC50) in each case. To determine whether the effects of GP2013 were significantly different from those of MabThera® in the assays, the results at each antibody dilution level were compared using a one-way analysis of variance (ANOVA) with Tukey's post-test in the SigmaStat program.
Mouse xenograft models
Due to approximate log-normal distribution of tumor volumes, geometric means were derived for each study day. The geometric means for each assessment day (before the first drop-out) were then compared between GP2013 and MabThera® by deriving their ratio and 95% confidence interval (CI). All calculations and plots were done using R 2.8.1 run on MODESIM.
Pharmacokinetics
The area under the concentration–time curve (AUC) from 0 to tlast was calculated by trapezoidal integration.
Pharmacodynamics
The PD analyses were conducted for CD40high B-cells (CD20low CD40highCD21+) and CD40low B-cells (CD20high CD40low CD21−). The effect of rituximab on peripheral B-cells was assessed as percentage change from baseline. The area under the effect–time curve (AUEC) was calculated for 0–7 days for the above mentioned parameter by trapezoidal integration. This time interval was chosen as it reflects B-cell depletion after a single dose of rituximab in both studies. To account for some of the intraindividual variability, the mean of three pre-dose measurements was taken as the baseline value. These pre-dose values were taken at the day of dosing and 2 and 4 days prior to dosing.
Descriptive analysis
Descriptive statistics (geometric mean [for PK analysis], arithmetic mean [for PD analysis], co-efficient of variation) were calculated for each parameter and for each treatment group.
Inferential statistical analysis
Based on fundamental PK relationships, the multiplicative model was applied for the concentration related parameters AUC and AUEC for percentage change from baseline using log-transformed data. The test item group was compared to the reference group using 90% confidence intervals for the ratio of the means for the PK parameter AUC and using 95% confidence intervals for the ratio of the means of the PD parameter AUEC for percentage change from baseline. An ANOVA was performed to estimate the residual error, which was used to construct the confidence intervals. Treatment was the only effect considered in the ANOVA model. Arithmetic and geometric means used for the calculation of point estimators, such as differences or ratios between treatments, were derived from the ANOVA as least squares means or exponentially transformed least squares means, respectively. The analyses were performed using the SAS procedure PROC GLM. The corresponding confidence intervals were then calculated using the least squares means and the root of the residual mean squares from the ANOVA of the log-transformed data with subsequent exponential transformation. All statistical calculations were performed using the SAS software version 8.2.
Results
Target-directed biosimilar development: linking a-fucosylated glycans to in vitro ADCC activity
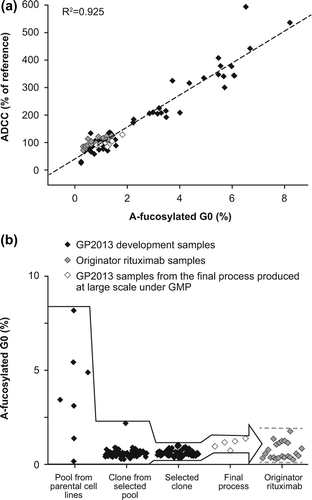
Glycosylation of the antibody Fc fragment is essential for Fc receptor-mediated activity and complement binding [Citation21]. Human IgG1s carry two N-linked complex-type glycans, which influence the conformation of the Fc domain via multiple non-covalent interactions with the CH2 domain. It has been shown that reduced levels of core-fucose increases the affinity to FcγRIIIa (CD16), which is expressed on ADCC effector cells, resulting in increased ADCC [Citation22,Citation23]. The importance of ADCC as a key mechanism of action for rituximab was shown in clinical settings, as the response rate of patients with follicular lymphoma (FL) was higher in a cohort carrying the higher affinity FcγRIIIa V158 variant when compared to the cohort carrying the lower affinity FcγRIIIa F158 variant, also resulting in the classification of rituximab as a class I therapeutic antibody [Citation11,Citation14]. As small changes in the low abundant a-fucosylated bG0 glycan species can influence ADCC, it was important to closely monitor this glycan during the development of GP2013. Using a sensitive and specific quantification method, the a-fucosylated bG0 glycan species was shown to occur in originator rituximab at levels between 0.3 and 1.8%. The target-directed biosimilar development approach includes a multiple-step cell line selection process that enables steering the biosimilar product toward the originator product quality. Using the variability in the a-fucosylated bG0 structures, which in these initial pools and clones ranged between 0.2 and 8.2% in the early development stages of the proposed biosimilar rituximab, a clear correlation (R2 = 0.925) could be shown between a-fucosylated bG0 and results from a quantitative in vitro ADCC potency assay []. A similar quantitative relationship between a-fucosylated glycans and ADCC has previously been reported [Citation24–26]. Thus, while starting with large a-fucosylated bG0 glycan variability, the multiple-step biosimilar cell line selection approach allowed targeted selection of a clone producing rituximab closely matching the a-fucosylated bG0 levels of the originator product []. The proposed biosimilar rituximab from the selected clone was subsequently shown to display the same in vitro ADCC potency activity as the originator rituximab.
Figure 1. (a) The wide range of a-fucosylated bG0 glycans in GP2013 development samples allowed the establishment of a quantitative structure–function relationship between a-fucosylated bG0 and ADCC. Black squares: GP2013 development samples; gray squares: rituximab originator samples; white squares: GP2013 samples from the final process produced on a large scale under good manufacturing practice (GMP). (b) Target-directed development of GP2013 to ensure that a-fucosylated bG0, and thus ADCC, is within the originator target range. The different steps entailed: (1) selection and subsequent cloning of a pool from a parental Chinese hamster ovary (CHO) cell line with a good overall quality and productivity profile and an a-fucosylated bG0 value closest to originator rituximab; (2) selection of the clone with the best overall quality profile, with a-fucosylated bG0 structure being close to the originator and showing little variation; (3) exposing the selected clone to different process conditions to optimize the overall quality and productivity profile resulting in the selection of a final process; (4) GMP production of GP2013 on a large scale using the final process, resulting in a-fucosylated bG0 values in the middle of the originator range.
In vitro ADCC confirmatory assay
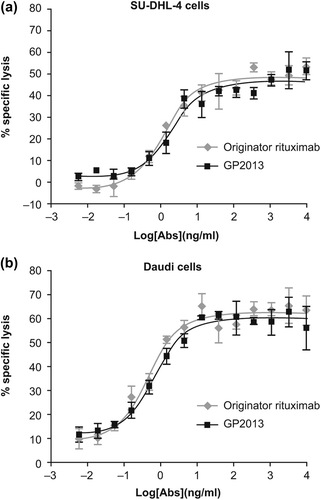
To further confirm that the final quality of GP2013 ultimately used in clinical trials displayed ADCC activity comparable to that of MabThera®, an alternative format of the ADCC assay was employed in which freshly purified NK cells carrying a CD16 (FcγRIII) isoform from a V/V genotyped healthy donor were used. This isoform of CD16 has a higher affinity to the Fc-domain of IgG and is highly sensitive to even very small changes in relative levels of fucose in the glycoform. The ADCC results obtained against both SU-DHL-4 and Daudi target cells are shown in , and demonstrate that both products have a similar potency across multiple concentrations tested, with an overlapping profile over the entire range. Statistical analysis of the individual concentrations as well as of the data set as a whole confirmed that there were no statistically significant differences between GP2013 and originator rituximab on the two cell lines tested (not shown).
Figure 2. Comparative assessment of ADCC potency against (a) SU-DHL-4 cells and (b) Daudi cells.
Mouse xenograft models
To confirm the successful engineering of the structure–function relationships impacting on efficacy at the in vivo level, two mouse xenograft models that have been extensively employed for the characterization of rituximab were used. These models more comprehensively assess the complex interactions between various attributes influencing the overall pharmacological profile of the therapeutic antibody in a single, enclosed system. To further increase the sensitivity of the models to identify any potential differences between GP2013 and MabThera®, suboptimal therapeutic doses were tested, since compensatory effects of one attribute versus another are less likely to occur at sub-maximal drug exposure levels. Both GP2013 and originator rituximab inhibited tumor growth to a similar extent, including at the sensitive mid-dose levels ().
Figure 3. In vivo comparability in two mouse xenograft models of non-Hodgkin lymphoma. (a) SU-DHL-4 model, (b) Jeko-1 model.
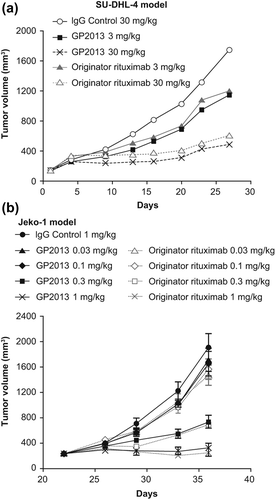
In the SU-DHL-4 model, both GP2013 and MabThera® at 3 and 30 mg/kg produced comparable dose-dependent changes of mean tumor volume of treated versus placebo []. The normalized data for tumor volume measurements up to day 23 resulted in ratios of geometric means (GP2013 vs. MabThera®) ranging from 1.00 to 1.19 at the 3 mg/kg and from 0.89 to 1.08 at the 30 mg/kg dose (). The 95% CI values increased with time, indicating the increasing intra-group heterogeneity associated with the xenograft model. No trends were observed in the ratios that would indicate deviation from standard therapeutic comparability parameters, notwithstanding the increasing intra-group heterogeneity at later time points.
Table I. Comparison of tumor volume in two mouse xenograft models of non-Hodgkin lymphoma.
In the Jeko-1 model, similar dose–response effects on efficacy were observed with GP2013 and MabThera®. The maximal anti-tumor response with both antibodies was stasis at the highest dose level of 1 mg/kg. The minimally effective dose for each of the antibodies was determined to be 0.3 mg/kg []. Ratios of geometric means of tumor volumes ranged from 1.00 to 1.06 at the 0.1 mg/kg and from 1.00 to 0.95 at the 0.3 mg/kg dose from day 21, which was the start of treatment, to day 36, which was the last day that all animals remained. Thus, inhibition of tumor growth was comparable for GP2013 and originator rituximab at both sensitive doses (0.1 and 0.3 mg/kg) ().
Pharmacokinetics and pharmacodynamics in cynomolgus monkeys
Pharmacokinetics
PK analyses were limited to the first 9 days and 14 days post-first i.v. administration in the single (5 mg/kg) and repeat dose (20 and 100 mg/kg) PK/PD studies, respectively. The reason for this was the development of anti-rituximab antibodies to both GP2013 and MabThera® between day 9 and day 14 (single dose) and from day 14 onwards (repeated dose).
Mean concentration–time profiles after single 5 mg/kg administration were fairly similar over the first 9 days post-administration []. Non-compartmental PK analysis confirmed bioequivalence between GP2013 and MabThera® with nearly identical AUCs and 90% CIs within the standard bioequivalence acceptance range of 0.8–1.25. For maximum concentration (Cmax) values, geometric means were approximately 13% lower with GP2013 compared with originator rituximab. This small difference is not considered relevant, and is most likely due to intrinsic heterogeneity in serum sampling collection after i.v. administration.
Figure 4. Pharmacokinetics comparison in cynomolgus monkeys after i.v. administration of (a) single 5 mg/kg dose (b) repeat 20 mg/kg dose, (c) repeat 100 mg/kg dose.
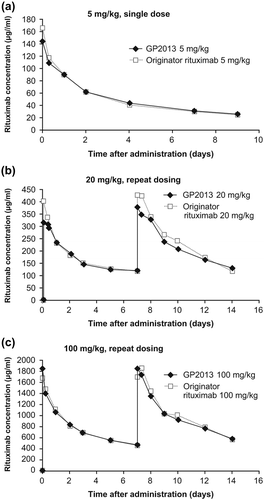
Mean concentration–time profiles after repeated dosing of 20 mg/kg and 100 mg/kg were also comparable between GP2013 and the reference product over the 14-day observation period []. A comparable AUC was observed under all treatments. GP2013 and MabThera® exhibited nearly identical AUCs and 90% CIs lying entirely within the standard bioequivalence acceptance range of 0.8–1.25. Comparable AUCs were observed following both dose levels, indicating that GP2013 and MabThera® are similar at the PK level preclinically.
Pharmacodynamics
A pronounced depletion within the first week and a slow return to baseline from week 6, which was sustained until the end of the study at week 10, were observed for the two peripheral B-cell subpopulations (CD20lowCD40highCD21+ and CD20highCD40lowCD21−) in the single-dose study []. These two B-cell subsets differ in their susceptibility to rituximab-induced depletion driven by the relatively different levels of target (CD20) density on the cell surface [Citation20]. In both GP2013 and MabThera®, the two subsets recovered with comparable kinetics. A slightly lower, but still comparable depletion (up to day 7) was seen for GP2013 in the CD20lowCD40highCD21+ B-cell subpopulation with an approximately 9% lower depletion when compared with MabThera® (i.e. a ratio of the AUECs for percentage change from baseline of 0.91 [95% CI 0. 82–1.01] for GP2013 vs. MabThera®). However, this small difference was diminished when the CD20highCD40lowCD21− B-cell subpopulation was analyzed (with an AUEC ratio for percentage change from baseline of 0.99 [95% CI 0.97–1.00]). In both subsets, bioequivalence between GP2013 and MabThera® was demonstrated, with the 95% CIs of the AUEC ratios falling within the acceptance range of 0.80–1.25.
Figure 5. Pharmacodynamics comparison in cynomolgus monkeys after i.v. administration of (a) single 5 mg/kg dose, (b) repeat 20 mg/kg dose, (c) repeat 100 mg/kg dose.
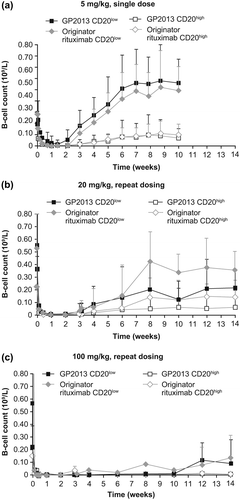
Similarly, a pronounced depletion and a slow return of the two peripheral B-cell subpopulations analyzed were observed in the multiple-dose study []. Both treatment groups showed a very similar course for B-cell depletion and recovery. For CD20lowCD40highCD21+ B-cells, recovery started from week 3 onward for the 20 mg/kg dose groups, while for the 100 mg/kg dose group none of the animals showed a B-cell recovery during the initial 10-week observation period (except for one animal in the MabThera® group). Of note, in this toxicokinetic study, data after 4 weeks following administration refer only to a subgroup of animals (n = 4/treatment group), which explains the heterogeneity in the data. Analyses of other immune cell subtypes, part of a standard preclinical toxicology assessment carried out in parallel, equally showed comparable distribution in both treatment groups and throughout the entire observation period (data not shown).
Discussion
Biologics have become increasingly integral to the treatment of various cancers and autoimmune diseases. However, the high costs associated with these products have led to reimbursement challenges which in turn have resulted in restricted knock-on patient access to many treatments. Biosimilars offer a solution to this downward spiral, providing similar medication at a more cost- effective price, and ultimately leading to expanded patient access.
Despite their complexity and heterogeneity, mAbs can be comprehensively characterized by using modern and sensitive analytical techniques [Citation9,Citation10]. Biosimilar development leverages this knowledge and, through a target-directed iterative development process, can deliver a product that is as similar to the reference product as the reference product is to itself.
Proof of similarity is then assessed by a comparability exercise based on comprehensive physicochemical and biological characterization, preclinical studies and focused clinical assessment. This totality of evidence approach ensures comparability between the proposed biosimilar and the reference product with regard to quality, safety and efficacy, and subsequent regulatory approval with the same label as the originator drug without the need for clinical trials in all indications.
The preclinical data presented here show the structure–function relationship between variations in glycan fucosylation and ADCC potency of the proposed biosimilar rituximab GP2013, and demonstrate how cell line selection process adjustments were made to engineer a product with similar attributes and comparable biological activity to that of the originator. This example illustrates how state-of-the-art analytical techniques can effectively detect minute variations in post-translational modifications. A range of assays, including affinity binding to a representative panel of Fcγ-receptors, FcRn and CD20 binding, as well as assessment of the two other postulated mechanisms-of-action of rituximab, e.g. CDC and apoptosis, further confirmed the functional similarity, preclinically, of the two products. The comprehensive physicochemical and functional characterization of GP2013, forming the basis for the proposed biosimilarity, has previously been reported [Citation17].
The comparability of GP2013 and originator rituximab was also demonstrated in in vivo preclinical models. In xenograft SCID mice, both GP2013 and originator rituximab inhibited tumor growth with similar potency. The use of a dose-scaling approach, which included administration of suboptimal and mid-dose levels which are more sensitive for detecting small differences in activity between products, did not show any differences in anti-tumor potency. Moreover, the relative anti-tumor efficacy of both products remained comparable throughout the observation period.
Finally, preclinical similarity between GP2013 and originator rituximab was also demonstrated in cynomolgus monkeys, the pharmacologically most relevant species, for both PK (AUC) and PD (B-cell depletion). Exposure at three different dose levels following either a single or repeated dose administration was comparable between GP2013 and MabThera®, showing that post-translational modifications that might affect interactions with FcRn and subsequent recycling and clearance of mAbs had been adequately engineered into the proposed biosimilar rituximab [Citation27–29]. Two different B-cell subsets have been identified in cynomolgus monkeys: CD20highCD40lowCD21 − and CD20lowCD40highCD21+ [Citation20]. These two B-cell subsets differ significantly in their relative in vitro and in vivo susceptibility to rituximab, with lower doses more readily depleting the CD20high B-cell subset and significantly higher doses being required for depletion of the CD20low set, reflecting the influence of the avidity of the target antigen (CD20) expression levels on B-cells on the pharmacological properties of the antibody. Importantly, the observation that both GP2013 and MabThera® resulted in similar levels of recovery of B-cell counts following discontinuation of treatment further suggests that no differences in the reconstitution of this immune cell compartment from bone marrow derived pre-B-cells are to be expected, nor that there will be differences in overall immune responses mediated by other compartments. This assumption is corroborated by clinically relevant analytical data from these cynomolgus studies showing comparable responses between the two treatment groups, including neutrophil counts (data not shown). The use of a wide range of doses indicates that equivalent exposure and PD response can be expected for GP2013 and originator rituximab in different disease treatment settings using varying dose regimens. Extrapolating from the findings here, the data also confirm that the approved dose for the clinical use of the originator rituximab can be used to conduct clinical trials to assess biosimilarity of the products. GP2013 is being assessed in two clinical trials: one initial phase II study in patients with rheumatoid arthritis to assess PK and PD similarity and an additional phase III study in patients with follicular lymphoma to assess similarity in efficacy and safety.
To conclude, biosimilar development involves a target-directed iterative process using state-of-the-art technologies to obtain a product with very similar attributes to the originator drug. The primary amino-acid sequence is identical to that of the reference, which is often not the case for so-called copy or generic biologics that do not follow the biosimilar regulatory principles [Citation10]. The engineered comprehensive physicochemical and functional characterization together with appropriate preclinical and clinical confirmatory programs aim to prove that biosimilars are comparable to the originator with regard to quality, safety and efficacy. Such biosimilars then have an important role in bringing high quality, and similarly efficacious and safe medications at a more affordable cost, to more patients around the world.
Supplementary Material
Download Zip (10 MB)Acknowledgements
Medical writing support was provided by Andy Bond of Spirit Medical Communications Ltd, supported by Sandoz Biopharmaceuticals/Hexal AG. The entire GP2013 project team is acknowledged for their contribution to the development of GP2013.
Potential conflict of interest:
Disclosure forms provided by the authors are available with the full text of this article at www.informahealthcare.com/lal.
References
- Brekke OH, Sandlie I. Therapeutic antibodies for human diseases at the dawn of the twenty-first century. Nat Rev Drug Discov 2003;2: 52–62.
- Reichert JM. Trends in US approvals: new biopharmaceuticals and vaccines. Trends Biotechnol 2006;24:293–298.
- Cornes P. The economic pressures for biosimilar drug use in cancer medicine. Target Oncol 2012;7(Suppl. 1):S57–S67.
- McCamish M, Woollett G. Worldwide experience with biosimilar development. MAbs 2011;3:209–217.
- McCamish M, Woollett G. The state of the art in the development of biosimilars. Clin Pharmacol Ther 2012;91:405–417.
- European Medicines Agency, Committee for Medicinal Products for Human Use (CHMP). Guideline on similar biological medicinal products containing monoclonal antibodies – non-clinical and clinical issues. London: European Medicines Agency; 2012. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/06/WC500128686.pdf
- Food and Drug Administration, Center for Drug Evaluation and Research (CDER). Guidance for Industry. Scientific considerations in demonstrating biosimilarity to a reference product. Rockville, MD, Food and Drug Administration; 2012. Available from: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM291128.pdf
- Schiestl M, Stangler T, Torella C, et al. Acceptable changes in quality attributes of glycosylated biopharmaceuticals. Nat Biotechnol 2011;29:310–312.
- Berkowitz SA, Engen JR, Mazzeo JR, et al. Analytical tools for characterizing biopharmaceuticals and the implications for biosimilars. Nat Rev Drug Discov 2012;11:527–540.
- Beck A, Diemer H, Ayoub D, et al. Analytical characterization of biosimilar antibodies and Fc-fusion proteins. Trends Anal Chem 2013;48:81–95.
- Cartron G, Dacheux L, Salles G, et al. Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcgammaRIIIa gene. Blood 2002;99:754–758.
- van Meerten T, van Rijn RS, Hol S, et al. Complement-induced cell death by rituximab depends on CD20 expression level and acts complementary to antibody-dependent cellular cytotoxicity. Clin Cancer Res 2006;12:4027–4035.
- Weiner GJ. Rituximab: mechanism of action. Semin Hematol 2010;47:115–123.
- Maloney DG, Smith B, Rose A. Rituximab: mechanism of action and resistance. Semin Oncol 2002;29(Suppl. 2):2–9.
- Vital EM, Kay J, Emery P. Rituximab biosimilars. Expert Opin Biol Ther 2013;13:1049–1062.
- Dorvignit D, Palacios JL, Merino M, et al. Expression and biological characterization of an anti-CD20 biosimilar candidate antibody. A case study. mAbs 2012;4:488–496.
- Visser J, Feuerstein I, Stangler T, et al. Physicochemical and functional comparability between the proposed biosimilar rituximab GP2013 and originator rituximab. BioDrugs2013;27:495–507.
- Jiang X-R, Song A, Bergelson S, et al. Advances in the assessment and control of the effector functions of therapeutic antibodies. Nat Rev Drug Discov 2011;10:101–111.
- Beck A, Sanglier-Cianferani S, Van Dorsselaer A. Biosimilar, biobetter, and next generation antibody characterization by mass spectroscopy. Anal Chem 2012;84:4637–4646.
- Vugmeyster Y, Howell K, Bakshl A, et al. Effect of anti-CD20 monoclonal antibody, Rituxan, on cynomolgus monkey and human B cells in a whole blood matrix. Cytometry 2003;52:101–109.
- Tao MH, Morrison SL. Studies of aglycosylated chimeric mouse-human IgG. Role of carbohydrate in the structure and effector functions mediated by the human IgG constant region. J Immunol 1989;143:2595–2601.
- Kubota T, Niwa R, Satoh M, et al. Engineered therapeutic antibodies with improved effector functions. Cancer Sci 2009;100: 1566–1572.
- Li C, Rossomando A, Wu S-L, et al. Comparability analysis of anti-CD20 commercial (rituximab) and RNAi-mediated fucosylated antibodies by two LC-MS approaches. mAbs 2013;5:565–575.
- Shields RL, Lai J, Keck R, et al. Lack of fucose on human IgG1 N-linked oligosaccharide improves binding to human Fcgamma RIII and antibody-dependent cellular toxicity. J Biol Chem 2002;277: 26733–26740.
- de Romeuf C, Dutertre CA, Le Garff-Tavernier M, et al. Chronic lymphocytic leukaemia cells are efficiently killed by an anti-CD20 monoclonal antibody selected for improved engagement of FcgammaRIIIA/CD16. Br J Haematol 2008;140: 635–643.
- Chung S, Quarmby V, Gao X, et al. Quantitative evaluation of fucose reducing effects in a humanized antibody on Fcγ receptor binding and antibody-dependent cell-mediated cytotoxicity activities. mAbs 2012;4:326–340.
- Ternant D, Paintaud G. Pharmacokinetics and concentration-effect relationships of therapeutic monoclonal antibodies and fusion proteins. Expert Opin Biol Ther 2005;5(Suppl. 1):S37–S47.
- Wang W, Lu P, Fang Y, et al. Monoclonal antibodies with identical Fc sequences can bind to FcRn differentially with pharmacokinetic consequences. Drug Metab Dispos 2011;39:1469–1477.
- Keizer RJ, Huitema AD, Schellens JH, et al. Clinical pharmacokinetics of therapeutic monoclonal antibodies. Clin Pharmacokinet 2010;49:493–507.