Abstract
The advanced glycoxidation end products (AGEs) and lipoxidation end products (ALEs) contribute to the development of diabetic complications and of other pathologies. The review discusses the possibilities of counteracting the formation and stimulating the degradation of these species by pharmaceuticals and natural compounds. The review discusses inhibitors of ALE and AGE formation, cross-link breakers, ALE/AGE elimination by enzymes and proteolytic systems, receptors for advanced glycation end products (RAGEs) and blockade of the ligand–RAGE axis.
| Abbreviations | ||
| Aβ | = | amyloid β-peptide |
| AD | = | Alzheimer's disease |
| AGEs | = | advanced glycoxidation end products |
| ALEs | = | advanced lipoxidation end products |
| CEL | = | N-ϵ-(carboxyethyl)lysine |
| CML | = | N-ϵ-(carboxymethyl)lysine |
| DAP | = | diaminopropionic acid |
| 3-DG | = | 3-deoxyglucosone |
| FN3K | = | fructosamine-3-kinase |
| GO | = | glyoxal |
| GOLD | = | glyoxal–lysine dimer |
| GSH | = | glutathione |
| HMGB1 | = | high-mobility group box 1proteins |
| HNE | = | 4-hydroxynonenal |
| LDL | = | low-density lipoprotein |
| oxLDL | = | oxidized LDL |
| LPA | = | lysophosphatidic acid |
| MDA | = | malondialdehyde |
| MG | = | methylglyoxal |
| MMP | = | metalloproteinase |
| MOLD | = | methylglyoxal–lysine dimer |
| ONE | = | 4-oxo-2-nonenal |
| PM | = | pyridoxamine |
| PTB | = | N-phenacylthiazolium |
| PMT | = | pyridinium (3-[[2-(methylsulfonyl) hydrazino] carbonyl]-1-[2-oxo-2-2-thienyl) ethyl]) |
| RAGE | = | receptor for advanced glycation end products |
| fRAGE | = | full-length RAGE |
| sRAGE | = | shortened RAGE |
| esRAGE | = | endogenous secretory RAGE |
| RCS | = | reactive carbonyl species |
| ROS | = | reactive oxygen species |
| STZ | = | streptozotocin |
| UPS | = | ubiquitin-proteasome system |
| VEGF | = | vascular endothelial growth factor |
AGEs, ALEs, and RAGE as drug target (GA)
Advanced glycoxidation [Citation1–3] end products (AGEs) and advanced lipoxidation end-products (ALEs) are widely studied as reporters of oxidative and glycoxidative damage [Citation4–8]. The most common analytical methods for their quantitative determination are based on ELISA or Western blot assays, using a variety of commercially available antibodies against AGEs, such as N-ϵ-(carboxymethyl)lysine (CML), N-ϵ-(carboxyethyl)lysine (CEL), and ALEs, the most popular being 4- hydroxynonenal (HNE)- and malondialdehyde (MDA)-adducted proteins. The content of AGEs and ALEs in tissue and body fluids has been correlated with different oxidative-stress based diseases. For instance, the serum levels of CML, one of the most studied AGEs, are associated with the severity of diabetes-related disease, including retinopathy [Citation9] and microangiopathy [Citation10], chronic kidney disease, as well as with the formation and acceleration of diabetic and non-diabetic atherosclerotic lesions [Citation11]. CML was also found to be elevated in cerebrospinal fluid of patients with amyotrophic lateral sclerosis [Citation12], and the tissue level of CML in cortical neurons and cerebral vessels was related to the severity of cognitive impairment in patients with cerebrovascular disease [Citation13]. Also, ALEs have been reported as useful biomarkers of oxidative damage. The Michael adduct of HNE to proteins was found to be significantly elevated in patients with a variety of oxidative stress-based diseases, such as Alzheimer's disease [Citation14], chronic pancreatitis [Citation15], obesity and insulin resistance [Citation16], and systemic lupus erythematosus [Citation17].
Besides being considered as reliable biomarkers of oxidative damage, as well as as predictors and prognostic factors, more recently, AGEs and ALEs have also been recognized as important pathogenetic factors of some oxidative-based diseases, as supported by the following facts: (1) as pointed out above, a strict correlation between the amount of AGEs/ALEs in tissues and fluids and disease states has been found, in both animal and human subjects; (2) a substantial amount of literature is now available reporting the molecular and cellular pathogenic mechanisms for the AGEs/ALEs involvement in the onset and progression of different diseases, including atherosclerosis, diabetes, and neurological disorders. The AGE/ALE damaging effect is mediated by different mechanisms, including the dysfunction of the proteins undergoing the oxidative modification, protein polymerization, signal transduction, immunoresponse, and RAGE activation; (3) compounds effective as inhibitors of AGEs/ALEs formation or able to block their biological effects have been found to significantly ameliorate different oxidative-based diseases.
Hence, AGEs/ALEs are now considered as promising drug targets, and a substantial effort is dedicated to delve into the molecular strategies aimed at preventing, reducing, or removing these protein oxidation products [Citation18,Citation19]. The different molecular approaches thus far reported can be grouped by considering at which level of the damaging AGE/ALE cascade they are effective and in particular if they act by inhibiting the AGE/ALE formation, accelerating their catabolism or blocking their biological effects.
The first level of action, the inhibition of AGE/ALE formation, also consists of different approaches, which target the different inducers (ROS, metal ions) and intermediate products (mainly reactive carbonyl species (RCS)) involved in the AGE/ALE formation. Antioxidants, metal ion chelators, and reactive carbonyl compound quenchers represent the most promising approaches so far reported for inhibiting AGE/ALE formation, in both in vitro and in vivo conditions. In some cases, as found for both natural and synthetic compounds, the inhibition of AGEs/ALEs formation does not proceed through a single specific mechanism but implicates multiple mechanisms, involving at least two of the following ones: antioxidant, metal ion chelation, and RCS quenching (reactions with RCS preventing their binding to vital macromolecules). Belonging to this first group of approaches, also the xenobiotics that act by potentiating the endogenous detoxification system devoted to the metabolization/detoxification of all the endogenous compounds involved in the AGE/ALE formation. Such a protective system consists of non-enzymatic and enzymatic antioxidants (compounds that prevent undesired oxidation by reacting with oxidants or oxidation intermediates), the enzymes including superoxide dismutase (SOD), catalase (CAT), and glutathione peroxidase (GPX), as well as of enzymes involved in the metabolic inactivation of reactive carbonyl derivatives, which include both phase I enzymes, namely aldehyde dehydrogenases (ALDHs), aldo-keto reductases (AKRs) and carbonyl reductase (CBR), and phase II enzymes, that is, glutathione S-transferases (GSTs) [Citation20].
The second level of intervention involves accelerating the catabolism of already-formed AGEs/ALEs; this can be achieved by potentiating the endogenous proteolytic system or by using xenobiotics that are able to catalytically degrade AGEs/ALEs.
The third level of intervention, which is also the most innovative, consists of blocking the biological response of AGEs/ALEs. Such an approach has emerged in parallel to the discovery of the receptor for AGEs and ALEs such as RAGE and galectin-3 [Citation21]. It should be noted that such an approach permits the blocking of the damaging effect induced not only by endogenously formed AGEs/ALEs but also by exogenously derived AGEs/ALEs.
In summary, AGEs/ALEs are involved as pathogenetic factors in some oxidative-based diseases including atherosclerosis, diabetes, and Alzheimer's disease (AD), and are now recognized as promising drug targets. On these grounds, several molecular approaches have been described with the aim of inhibiting the damaging response induced by AGE/ALE formation. The aim of the present review is to systemically describe the molecular approaches thus far reported with a particular view of the rational drug design approach used. Compounds and strategies will also be critically commented on and their limits highlighted.
Inhibitors of ALE/AGE formation (GA)
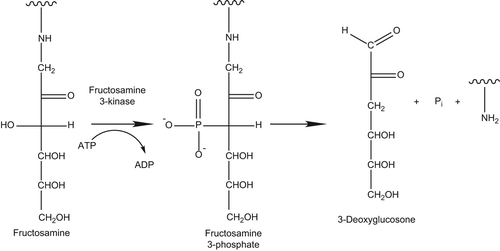
Several molecular approaches have been considered for inhibiting AGE/ALE formation, and such diversity is due to the complex reactions leading to the AGE/ALE formation, which involves different inducers, precursors, and intermediates. In particular, as better described in the paper entitled “Advanced glycoxidation and lipoxidation end products (AGEs and ALEs): an overview of their mechanisms of formation” and published in this series, two main pathways are involved in the AGE formation. The former is based on the reaction of reducing sugars (any sugar that either has an aldehyde group (aldoses) or is capable of forming one in solution through keto-enol isomerism (ketoses)) with the protein primary amino groups (the amino terminus and the ϵ-amino group of Lys), followed by metal ion catalyzed rearrangements. The latter involves the reaction of RCS such as glyoxal (GO), methylglyoxal (MG), and 3-deoxyglucosone (3-DG; ) with the nucleophilic protein sites, and in particular Arg, Lys, and Cys. The RCS acting as AGE precursors are generated by either sugar or AGEs decomposition/autoxidation, a series of reactions which are catalyzed by metal ions. ALEs are generated by only one pathway, based on the covalent adduction of lipid- derived RCS (HNE, GO, and MDA) with Arg, Lys, His, and Cys nucleophilic residues. The RCS acting as ALE precursors are generated by the lipid peroxidation cascade, and hence, several oxidative stress inducers and propagators impact their formation, including ROS and metal ions.
Figure 1. Main reactive carbonyl species: glyoxal, methylglyoxal, and 3-deoxyglucosone.
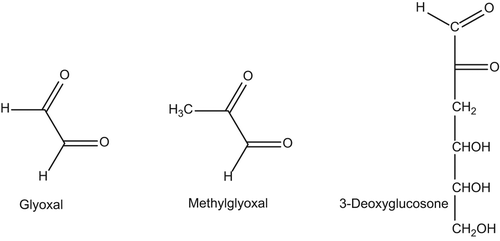
By considering the diverse pathways leading to AGE/ALE formation, the following molecular strategies for the inhibition of their formation can be considered, based on the use of the following: (1) antioxidants, able to inhibit the lipid peroxidation cascade as well as the radical-based reactions involved in AGE formation; (2) metal ion chelators, inhibiting different oxidative pathways such as those leading to ROS production, hydroperoxide decomposition, sugar and AGE decomposition to RCS; (3) compounds able to quench RCS which act as AGE and ALE precursors. Furthermore, the first two mentioned approaches can be seen as indirect strategies since they prevent the formation of AGE and ALE precursors, while the last one is a direct approach in which the compounds react with the already-formed RCS, deactivating them and promoting their rapid excretion. The above-mentioned approaches are grouped and described below ().
Figure 2. Molecular intervention strategies to inhibit and degrade AGEs and ALEs.
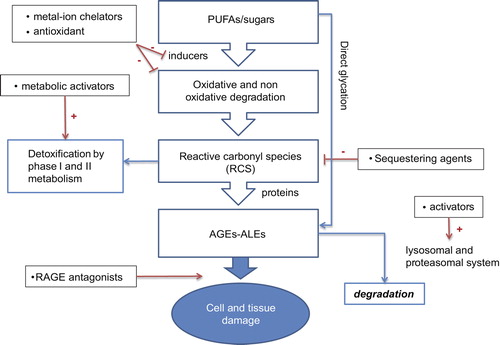
Multifactorial etiology of post-translational modifications of proteins/lipids would serve as a rationale for multi-target-oriented interventions in anti-AGE/ALE therapeutic strategy. Combination therapies that simultaneously target multiple pathways may obviously be more successful than those that modify a single pathway. This approach also decreases the risk of side effects and is economical.
Indirect inhibition mechanisms (MS)
Antioxidants
Irreversible AGEs were shown to be formed via a sequence of glycation and oxidation reactions [Citation1–3]. Under physiological conditions, glucose, like other α-hydroxyaldehydes, can enolize and reduce molecular oxygen. This process is catalyzed by transition metals yielding reactive α-ketoaldehydes and oxidizing free radical intermediates [Citation22]. The ketoamine Amadori products also undergo autoxidation, contributing to the oxidative damage of proteins exposed to hyperglycemia [Citation1,Citation2]. At an early stage in this research, the terms “autoxidative glycosylation” and “glycoxidation” were introduced for the above-mentioned processes [Citation1], to emphasize the importance of oxidation chemistry in AGE formation.
As shown in the classical experiments of [Citation22], glycoxidation and cross-linking of collagen as a model protein exposed to glucose in vitro was strongly inhibited under antioxidative conditions (nitrogen atmosphere and metal chelators). Antioxidative conditions had no effect on glycation per se measured by the degree of covalent attachment of the radiolabeled glucose.
In addition to the above-mentioned autoxidation processes, the oxidative stress in hyperglycemia would be enhanced by generation of ROS occurring at mitochondrial level as a consequence of the increased intracellular glucose metabolism [Citation23,Citation24].
Hyperglycemia does not only generate more ROS but also attenuates endogenous antioxidative mechanisms through glycation of scavenging enzymes. Excessive consumption of NADPH by the polyol pathway in hyperglycemia may result in the depletion of glutathione (GSH).
Taking into consideration the oxidation component(s) of advanced glycation, pharmacological intervention by antioxidants represents a reasonable therapeutic strategy aimed against AGE formation.
Protective effects of exogenously administered antioxidants have been extensively studied in chemical and animal models of glycoxidation, mostly in relation to the development of diabetic complications. Antioxidants and chelators were found to inhibit the formation of glycoxidation products, without a significant effect on the extent of the protein glycation. Antioxidants may thus dissociate glycoxidative changes caused by the exposure of protein to glucose from the incorporation of the monosaccharide into the protein itself.
There are countless experiments and studies of antioxidant action on processes of non-enzymatic glycation covered by excellent reviews (see, e.g., [Citation25]). Most of these studies are aimed at modeling the situation in a hyperglycemic milieu relevant to diabetes. The effect of a typical chain-breaking antioxidant on glycoxidation can be exemplified by the extensive studies performed on the pyridoindole antioxidant drug stobadine (). Under conditions of an experimental glycation model in vitro, comprising bovine serum albumin incubated in the presence of glucose, stobadine inhibited glycation-related absorbance and fluorescence changes of the protein as well as the yield of 2,4-dinitrophenyl-hydrazine-reactive carbonyls with an efficacy comparable with that of the reference antioxidant trolox, and more efficiently than did the glycation inhibitor aminoguanidine. Since stobadine did not affect the early steps of glycation measured as Amadori product formation and the covalent binding of glucose, the observed protective effects were explained by the ability of the drug to eliminate free radical intermediates of glycoxidation reactions, operative after the preceding glycation steps [Citation26,Citation27].
Figure 3. Relevant xenobiotics acting as antioxidants (first row) and metal chelators (second and third rows).
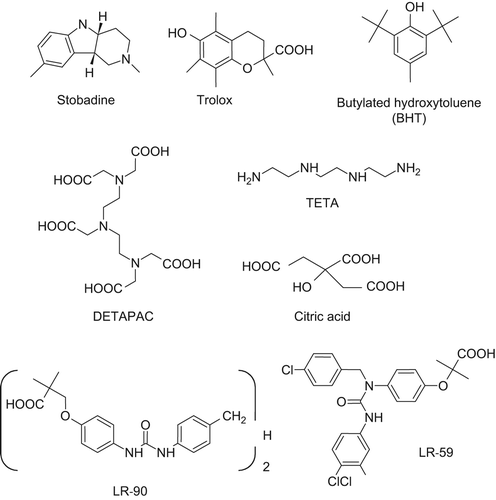
Using a model of streptozotocin (STZ)-diabetic rats in vivo, stobadine was found to attenuate pathological changes in the diabetic myocardium [Citation28,Citation29] and kidneys [Citation30–32], to decrease matrix collagen cross-linking [Citation30] and to reduce plasma cholesterol and triglyceride levels in diabetic animals [Citation28,Citation33]. Stobadine treatment normalized calcium homeostasis in diabetic heart and liver [Citation33] and produced beneficial effects on liver and leukocyte function in diabetic rats [Citation34,Citation35].
Long-term treatment of diabetic animals with stobadine led to a marked delay in the development of advanced stages of cataract [Citation36] and inhibited the development of retinal morphological abnormalities and lipid peroxidation, even under poor glycemic control [Citation37].
Treatment of diabetic animals with stobadine partially prevented the decrease in conduction velocity of the sciatic nerve measured in vitro. The protective effect was enhanced by co-therapy with vitamin E. On the other hand, resistance to ischemic conduction failure, elevated in diabetic animals, was not found to be affected by any of the drugs studied [Citation38,Citation39].
The antioxidants butylated hydroxytoluene (BHT, ) and trolox (6-hydroxy-2,5,7,8-tetramethyl chroman-2-carboxylic acid, ) were reported to protect sugar-induced cataractogenesis in cultured rat lens [Citation40], and BHT was found to prevent or delay also galactosemic cataractogenesis in rats [Citation41]. BHT ameliorated nerve dysfunction in STZ diabetic and galactosemic rats [Citation42].
Several observational studies on age-related and diabetic cataract have suggested that antioxidant micronutrients, such as α-tocopherol, retinol, and ascorbic acid, may help to protect against cataractogenesis [Citation43–45]. However, other studies reported non-significant effects or even negative results [Citation46,Citation47]; for review, see [Citation48].
Antioxidant action of natural flavonoids may be effective in reducing the oxidative pathways conducive to advanced glycation. It is beyond the scope of this review to give a thorough survey of the abundant literature covering numerous studies on the antioxidant activity of flavonoids. The key structural features required for the high antioxidant efficacy of flavonoids are summarized in Bors’ criteria [Citation49,Citation50]. The current data on natural polyphenols relevant to glycation-related pathologies demonstrate that flavonoids may play a role in the prevention of cataract [Citation51,Citation52] and some complications of diabetes [Citation53–55]. Antioxidant therapy may be of great interest in diabetic patients. Despite encouraging results obtained by in vitro and in vivo studies, a number of clinical intervention trials aimed at elucidating the effects of antioxidant micronutrients (vitamins E and C, β-carotene, and retinol) on the development of age-related and diabetic cataract, and other diabetic complications failed to show any beneficial effects. However, it is still too early to reach a definitive conclusion on the clinical benefits of antioxidants.
Metal chelators
ROS and free transition metal ions have been recognized as key players in advanced glycation. Redox metal chelators may therefore efficiently interfere with the process at the stage of both “autoxidative glycosylation” and “glycoxidation” [Citation1]. Chelation, which is sufficient to inhibit AGE formation independently of carbonyl trapping, is a common characteristic of many drugs with multiple functional groups [Citation56,Citation57]. The chelating activity of inhibitors of AGE formation varies widely, yet under in vivo conditions a complete chelation of all free redox metal ions may not be desirable.
The iron chelator diethylenetriaminepentaacetic acid (DETAPAC, ) efficiently inhibited autoxidation of glucose and glycoxidation changes of model proteins exposed to high glucose in experimental glycation models in vitro [Citation58–63]. The potent iron chelator, desferrioxamine, inhibited the formation of CML in experimental glycation models in vitro [Citation64] and accumulation of AGEs under diabetic conditions [Citation65,Citation66]. Desferrioxamine therapy ameliorated nerve dysfunction in STZ diabetic rats [Citation67].
Triethylenetetramine (TETA, ) has recently been identified as a highly selective divalent chelator of copper, and to a lesser extent of iron, preventing or reversing diabetic copper overload. Alterations in copper and iron homeostasis were reported in diabetic subjects [Citation68,Citation69]. In diabetes, AGE-modified proteins can act as endogenous chelators, thus increasing the copper content of organs such as the heart and kidneys. TETA tightly binds and extracts excess systemic Cu(II) into the urine whilst neutralizing its catalytic activity [Citation70–75]. Chelation of transition metals by TETA significantly attenuated structural and functional changes in the heart of the Zucker type 2 diabetic rat [Citation73] and ameliorated nerve dysfunction in STZ diabetic rats [Citation42,Citation67,Citation76]. In diabetic rat lenses, TETA decreased the levels of MG and 3-DG, the most potent precursors of AGEs, as well as of AGEs themselves [Citation77].
Dietary supplementation of citric acid (2-hydroxypropane-1,2,3-tricarboxylic acid, ), a relatively non-specific chelator, significantly prevented structural and functional changes in the heart of Zucker diabetic rats [Citation73] and inhibited development of cataract, proteinuria and ketosis in STZ diabetic rats [Citation78].
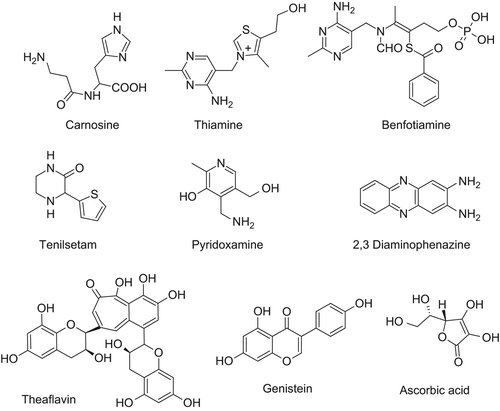
In their recent review, Nagai et al. proposed that the chelation activity may be responsible for AGE formation inhibitory activities of a variety of drugs commonly used for the treatment of diabetic complications [Citation57]. Indeed, strong metal-chelating action was reported for numerous inhibitors of AGE formation and AGE breakers, including pyridoxamine (PM) [Citation79], tenilsetam [Citation80–82], carnosine [Citation83,Citation84], metformin [Citation85–88], OPB-9195 [Citation84], angiotensin converting enzyme inhibitors, and angiotensin II receptor blockers [Citation89]. For most inhibitors of AGE formation, it is difficult to ascertain the contribution of alternative mechanisms of action, as trapping of carbonyl species, chelation of metal ions or scavenging of reactive oxygen intermediates.
To distinguish between the carbonyl trapping and chelation by AGE inhibitors, Price et al. measured the chelating activity of the inhibitors by determining the IC50 values for the rate of copper-catalyzed autoxidation of ascorbic acid in phosphate buffer. The apparent binding constants of copper ranged from approximately 2 mM for aminoguanidine and PM, to 10–100 μM for carnosine, phenazinediamine, OPB-9195, and tenilsetam. The AGE breakers, phenacylthiazolium and phenacyldimethylthiazolium bromide, and their hydrolysis products, were found to be the most potent inhibitors of ascorbate oxidation [Citation56]. Based on the above-mentioned results, it was concluded that, at millimolar concentrations of AGE inhibitors used in many in vitro studies, inhibition of AGE formation results primarily from the chelating or antioxidant activity of the AGE inhibitors, rather than from their carbonyl quenching activity. At therapeutic concentrations, the chelating activity of inhibitors of AGE formation and AGE-breakers may contribute to their inhibition of AGE formation.
Interestingly, compounds of the so-called LR series (phenyl ureido derivatives as seen in the LR-90 and LR-59 examples, ), the mechanism of which is almost exclusively ascribable to a very potent metal chelation, were found to prevent dyslipidemia, diabetic nephropathy, and retinopathy in several animal models with a protective effect comparable, if not better, with that exerted by PM or aminoguanidine [Citation90–92]. Notably, LR-90 also shown marked anti-inflammatory properties (as demonstrated in human monocytes) and might therefore have additional protective effects against diabetic vascular complications [Citation93]. Collectively, these results emphasize the prominent role that metal ions elicit in AGE/ALE formation thus indicating that their chelation can represent a very effective therapeutic strategy provided that the induced effect does not alter the fine homeostasis of the sequestered metal ions in human body.
Direct scavenging (trapping) of reactive carbonyls (GA)
Several reviews have explicitly discussed the biological activities as well as the potential therapeutic applications of RCS quenchers (see, e.g., [Citation94,Citation95]), and for this reason, these aspects will be not extensively covered but the literature referred to. In the present chapter, the RCS quenchers will be classified on the basis of the functional groups involved in the quenching reactions and the structure–activity relationship discussed, together with a detailed explanation of their mechanism in terms of reactivity and selectivity.
The analysis of carbonyl quenchers should start from the key concept that their biological activity is not based on fine recognition process between a ligand and a given biomacromolecular target but on a specific covalent reaction between a quencher and a given reactive carbonyl derivative. This implies a truly revolutionary change in the design approaches since all moieties of a ligand participate in the binding event, despite with a different relevance, while the quenching activity is heavily influenced by the presence of few reactive groups and the remaining part of the molecule acts rather as an inert framework which bears the key centers and at most modulates their reactivity and/or influences the overall pharmacokinetic profile. Therefore, the quencher design should shift attention from interacting groups which are chosen to stabilize short-range interactions with the crucial residues in the receptor binding site (e.g., H-bonds or hydrophobic contacts, [Citation96]) toward reactive centers which should be selected to match the specific electrophilicity of the RCS, thus promoting the formation of covalent adducts [Citation97].
In order to better understand the rational drug design of the compounds removing RCS so far reported, a brief description of the chemical reactivity of RCS acting as AGE and ALE precursors is reported here below. RCS can be firstly grouped into three main chemical classes: dialdehydes (α-dialdehydes such as GO and β dialdehydes such as MDA), keto-aldehydes (i.e., α oxoaldehydes such as 3-DG, MG) and α,β-unsaturated carbonyls. Generally speaking, the chemical reactivity of RCS is not ascribable to the sole carbonyl moiety but is essentially due to the combination of more chemical functions, which act synergistically making the compounds extremely reactive and hence damaging.
From a very general point of view, RCS can be seen as electrophilic compounds which require nucleophilic quenchers to yield condensation reactions which can be at least qualitatively predicted by the electronic properties of the reactants as computed by quantum mechanical parameters [Citation98]. It should be underlined that electrophiles do not react indiscriminately with nucleophiles, but, as discussed by LoPachin and coworkers [Citation99], the reactants can react provided that they have similar degree of polarizability, as predicted by Pearson's Hard and Soft, Acids and Bases (HSAB) theory [Citation100].
As described above, both AGE and ALE precursors contain at least two electrophilic moieties which can clearly explain their unique reactivity. Thus, the enhanced chemical reactivity of α-dicarbonyls is mainly associated with the powerful activation of one electron-withdrawing carbonyl group on the other [Citation101]. The enhanced reactivity of such compounds make them able to react at the same time with more nucleophilic centers such as two Lys residues to form protein cross-linking adducts such as imidazolium cross-links (i.e., the GO–Lys dimer, GOLD) and imidazole cross-links (i.e., the GO–Lys–Arg dimer, GODIC). Again, the specific reactivity of α,β-unsaturated carbonyls is mainly ascribable to their capacity to yield Michael-type adducts due to the marked reactivity of β carbon atom that is added to that of the carbonyl group, which however conserves the possibility to condense with suitable nucleophilic groups [Citation102]. In some cases, their reactivity is further modulated by the vicinal groups, such as the hydroxyl group in HNE or the keto function in 4-oxo-2-nonenal (ONE), that enhance the electrophilic property of α-carbon atom by electron-withdrawing effects.
On these grounds, it comes as no surprise that all reported quenchers are endowed with one or more nucleophilic centers, which differ in their physicochemical properties as well as in the capacity to trap different carbonyl species. As a trend, compounds possessing only one strong nucleophilic group act by a one-step mechanism, and as such they are rarely endowed by a suitable selectivity, trapping also physiological carbonyls. Differently, scavengers with two or more reactive groups generally show a softer reactivity, act by multistep mechanisms, and possess a satisfactory selectivity, thus avoiding the depletion of physiological compounds. By considering the number of their reactive centers, the reported carbonyl quenchers can be subdivided into monoreactive and polyreactive molecules. Based on the chemical nature of their reactive groups, the former can be further classified into thiol-containing compounds, guanidine and hydrazine derivatives, and β dicarbonyl analogs, while the latter can be grouped into heterocycle-based compounds, amino derivatives, and phenols and polyphenols.
Monoreactive molecules
Thiol-containing compounds. The prototypal example of thiol-containing carbonyl scavenger is offered by GSH, the endogenously reducing tripeptide (γ-L-glutamyl-L-cysteinylglycine) involved in several antioxidant processes given its capacity to prevent cellular damage caused by ROS such as free radicals and peroxides [Citation103]. Besides the known antioxidant properties, mainly based on its ability to oxidize into reversible disulfide species [Citation104], GSH, due to the marked sulfur nucleophilicity, can also yield covalent adducts with endogenous or xenobiotic electrophilic compounds [Citation105]. Once adducted, the peptide loses its terminal residues (L-glu and gly), thus yielding the corresponding mercapturic acid which detoxifies the quenched electrophile rendering it more hydrophilic and easier to be excreted [Citation106].
Although several electrophilic species can spontaneously react with GSH (as firstly demonstrated for α, β-unsaturated aldehydes by Esterbauer [Citation107]), the addition can also be catalyzed by cytosolic GSTs [Citation108], which represent a family of versatile enzymes able to catalyze the nucleophilic conjugation of GSH with a wide spectrum of electrophiles. Concerning the detoxification of ALE precursors [Citation109,Citation110], several studies revealed the involvement of GST enzymes as suggested by their upregulation in initial stages of oxidative stress. Among the involved GST isozymes, GSTA4-4 [Citation111] emerges for its high catalytic efficiency toward alkenal substrates, such as HNE, even though other subtypes can surely contribute to the RCS detoxification. Notably, this enzymatic HNE addition shows a certain degree of stereoselectivity, with a relative preference for (R)- versus (S)-HNE which is presumably due to the participation of the hydroxyl moiety to the substrate recognition [Citation112]. Such a selectivity tends to generate preferentially defined sets of diastereoisomeric adducts with potential biological implications for toxicity and excretion [Citation113].
Concerning the dicarbonyl detoxification, it should be reminded that the GSH serves as a cofactor for glyoxalase enzymes [Citation114], which catalyze the conversion of dicarbonyls into non-toxic products as a part of the cellular defense system against glycation [Citation115]. Given the remarkable nucleophilicity of the thiol function and, consequently, its facile reactivity with unsaturated carbonyls [Citation116], cysteine derivatives are more able to quench α,β-unsaturated aldehydes, through a Michael addition, compared to dicarbonyls even though also these latter can be trapped by formation of the corresponding thioacetals. Moreover, Nomi and coworkers have recently demonstrated that dicarbonyls can react with N-terminal group of the glutamate residue yielding stable dioxomorpholine adducts which appear to be favored by the α position of the primary amine (compared to the N-terminus of other peptides). This cyclic adduct should represent the most important products coming from quenching reactions between dicarbonyls and GSH [Citation117].
Free cysteine and N-acetyl cysteine (NAC) [Citation118] can be seen as both precursors of GSH [Citation119] and compounds able to directly quench RCS with a concentration- dependent activity comparable with that of the non- enzymatic GSH addition [Citation120]. Similar quenching activities are also shown by cysteamine (2-aminoethanethiol) and cysteine methyl ester, thus confirming that the carboxyl terminus is not involved in quenching mechanism [Citation121,Citation122]. Interestingly, also taurine (2- aminoethanesulfonic acid), that can be seen as an oxidized form of cysteamine, was reported to be able to directly quench MDA giving fluorescent 1,4-dihydropyridine and non-fluorescent enaminal derivatives, thus suggesting that the modulation of carbonyl stress may be included among the biological functions of taurine [Citation123].
Penicillamine (3-mercaptovaline) and its analogs [Citation124] show a significantly greater activity toward dicarbonyls which can be explained by considering their ability to yield stable thiazolidine adducts, which results from nucleophilic ring closure of the initial imine intermediate [Citation125]. Such a mechanism suggests that, although penicillamine is described here for structural analogy with the other sulfur-containing quenchers, it can be seen as an example for polyreactive quencher whose mechanism requires the concerted involvement of both reactive centers. The participation of both moieties can also explain the observed stereoselectivity since D-penicillamine is slightly more active than the L-enantiomer probably because it exposes the reactive groups in an arrangement more favorable to the final cyclization. Similarly, the 3,3 dialkyl substitution may favor the ring closure preventing the reverse hydrolysis reaction [Citation122]. The relevance of alkyl substitution is further confirmed by the notable activity of omo-derivative of penicillamine, namely 3-methyl, 3-ethyl cysteine (MEC) whose quenching ability induces apoptosis on malignant melanoma cell lines as reported by Wondrak and coworkers [Citation126].
Lastly, lipoic acid (6,8-dithiooctanoic acid), a naturally occurring nutraceutical [Citation127], is a well-known antioxidant able to directly quench free radicals as well as to regenerate other antioxidants due to its redox equilibrium between the reduced thiols and the oxidized disulfide. It is also able to trap both ALE and AGE precursors due to the presence of two nucleophilic thiols [Citation128,Citation129], thus resulting in protective effects against oxidative-based diseases as clearly demonstrated by animal models for diabetes, non-alcoholic steatohepatitis, and ischemia [Citation130]. As recently evidenced, the protective effect of lipoic acid can also be due to its restorative action on aldehyde dehydrogenase, ALDH2, whose activity is upregulated by about 60% after pretreatment with lipoic acid so inducing a significant decrease in all carbonyl species [Citation131].
However, it should be emphasized that, despite the strong sulfur nucleophilicity, thiol-containing drugs are potentially toxic compounds since they can react with cysteine thiol functions yielding protein-drug mixed disulfides. These adducts can impair the physiological functions of the modified proteins, even though cells and plasma possess protective dethiolation processes which are able to regenerate critical protein thiols [Citation132]. More importantly, thiol-containing drugs can behave as haptens when the mixed disulfides become antigenic determinants and initiate an immune response which can culminate in hypersensitivity and, consequently, idiosyncratic reactions (IDRs) depending on the body's capacity to detoxify these adducts [Citation133].
Guanidine and hydrazine derivatives. Due to the presence of more electron-rich nitrogen atoms, aminoguanidine (2-Aminoguanidine, aka Pimagedine [Citation134]; Figure 3) possesses the necessary nucleophilicity to rapidly react with both ALE and AGE precursors albeit through different reaction mechanisms [Citation135]. The α,β-unsaturated aldehydes can condense with aminoguanidine through Michael addition or imino formation [Citation136], while dicarbonyls cyclize with the formation of 1,2,4 triazine analogs [Citation137]. Aminoguanidine was found to reduce tissue levels of AGEs in experimental models of diabetes as well as to retard the development of several AGE-related pathologies, including diabetic vascular diseases, neuropathy, retinopathy, nephropathy, and cataract [Citation67,Citation135,Citation138–152]. However, due to its high reactivity, aminoguanidine does not possess a suitable selectivity trapping also physiological carbonyls [Citation136], and indeed, phase III clinical trials for aminoguanidine were discontinued because it induced vitamin B6 deficiency [Citation153]. Not to mention that aminoguanidine has other pharmacological activities since it inhibits both nitric oxide synthase [Citation154] and semicarbazide-sensitive amine oxidase (SSAO) [Citation155]). Despite the mentioned drawbacks, several aminoguanidine derivatives were proposed as carbonyl quenchers mainly with a view to removing the promiscuous biological activities as well as to improving the pharmacokinetic profile by enhancing the lipophilicity. Among them, β-resorcylidene aminoguanidine (RAG) [Citation156]; ) and ALT-946 [N-(2- acetamidoethyl) hydrazine-carboximidamide hydrochloride] () have attracted considerable interest for the efficient anti-glycation and anti-oxidant effects in both animal studies and in vitro experiments with minimal inhibitory effects on nitric oxide synthase [Citation157–159]. In addition to the indirect effect of all anti-diabetic agents on AGE formation by lowering blood glucose level, some anti-diabetic agents are also direct inhibitor of AGE formation and can be seen as aminoguanidine derivatives. Thus, the anti-diabetic agent metformin (1,1-dimethylbiguanide; ) was reported to decrease AGE formation in vitro, most likely by eliminating reactive carbonyl precursors [Citation88, Citation160–162]. In clinical studies, metformin reduced the levels of reactive dicarbonyls and AGEs [Citation86,Citation163]. These mechanisms contribute to medicinal effects of metformin beyond the benefits expected from its anti-hyperglycemic effect.
Figure 4. Relevant monoreactive carbonyl quenchers.
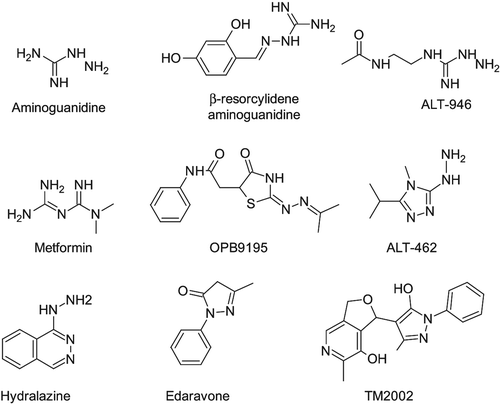
When serum albumin was incubated in vitro with glucose, ribose, or ascorbate, OPB-9195 (2-isopropylidenehydrazono-4-oxo-thiazolidin-5-yl acetanilide; ), a quite complex derivative in which the aminoguanidine moiety is embedded in a thiazolidin-5-acetanilide system, prevented AGE formation more efficiently than aminoguanidine, affecting especially the generation of pentosidine and carboxymethyl lysine [Citation164]. In animal studies using diabetic rats, OPB-9195 normalized AGE levels and afforded neuroprotective, renoprotective, and angioprotective effects [Citation157,Citation165–170]. Lastly, ALT-462 (4-amino-3-hydrazino-5-isopropyl-4H-1,2,4-triazole dihydrochloride; ) can be seen as a cyclic analogue of the aminoguanidine in which the reactive moiety is masked in a triazole ring and shows a dose-dependent scavenging activity at least 20 times more potent than aminoguanidine [Citation171].
Besides guanidine, a second nitrogen atom containing electron-rich function is represented by hydrazine (or hydrazide) group [Citation172]. Hydralazine (1-hydrazinylphthalazine, ) and dihydrohydralazine are the lead compounds of a series of hydrazine derivatives endowed by a marked scavenging ability toward both ALE and AGE precursors. Like aminoguanidine, hydrazine derivatives can react with α,β-unsaturated aldehydes through Michael addition or imine formation [Citation136,Citation173], while dicarbonyls cyclize yielding diazine analogs [Citation174]. Yet again, due to high hydrazine nucleophilicity, hydralazine lacks suitable selectivity trapping also physiological carbonyls through the formation of stable imine derivatives [Citation136]. Moreover, hydralazine and dihydrohydralazine also show a promiscuous activity since they are well-known vasodilator agents which have been used for the treatment of essential hypertension since the 1950s [Citation175]. Despite the mentioned pitfalls, such compounds have attracted considerable interest for their excellent scavenging activity toward reactive carbonyls, and indeed, they have been proven to be therapeutically efficient in preventing AGE and ALE formation in several oxidative-stress based disorders including diabetes, atherosclerosis, and Alzheimer disease [Citation122, Citation173,Citation174]. Hence, it is not surprising that numerous hydrazine-containing derivatives have been proposed with a view to separating the scavenging activity from the vasoactive effect, quite a feasible task considering that scavenging requires only the hydrazine group and, unlike vasoactivity, is rather independent on the remaining part of the molecule and (b) increasing hydrophobicity to enhance the pharmacokinetic profile and especially to increase blood–brain barrier permeability. Among the hydrazine derivatives, bisvanillyl-hydralazone shows notable antioxidant, carbonyl scavenger, and anti-apoptotic properties, thus resulting in a particularly efficient agent for therapeutic applications in atherosclerosis [Citation176]. Studies involving already-marketed drugs with a hydrazine (or hydrazide) function (e.g., isoniazid, iproniazid, and phenelzine) showed that all these drugs induce a significant carbonyl stress inhibition correlated with a reduction of atherosclerotic lesion development [Citation177]. By considering that oxidative damage is a major pathological feature of the Alzheimer disease, some authors developed new hydrazine derivatives endowed by extended aromatic systems able to interact with the amyloidogenic core of Aβ through p-p-stacking interactions so as to intercalate between Aβ fibrils. In this way, the quenchers so obtained (e.g., 2-hydrazino-4-phenylthiazole, indole-3-acetic hydrazide, and carbazochrome) induced a promising reduction of both Aβ misfolding and Aβ protein modification by RCS, thus suggesting their potential for further drug development [Citation178].
β dicarbonyl analogs. The nucleophilic reactivity of the β dicarbonyl analogs toward reactive carbonyls is essentially ascribable to the acidity of the α methylene due to the electron-drawing effect of the vicinal carbonyl groups. Although also β dicarbonyls (as in the well-known case of MDA) can derive from lipid peroxidation and promptly react with physiological nucleophiles due to the extreme reactivity of their β-hydroxyacrolein form [Citation179], β-dicarbonyl derivatives, in which the carbonyl reactivity is suitably modulated, may enhance their nucleophilicity thus resulting in very reactive carbonyl quenchers. A well-known example of this class of carbonyl quenchers is represented by edaravone (5-methyl-2-phenyl-2, 4-dihydro-3H-pyrazol-3-one; ) whose pyrazolone ring can be indeed seen as a cyclic (and masked) form of β dicarbonyl [Citation180]. Edaravone combines a potent free radical scavenging [Citation181] with a significant carbonyl quenching activity which is mostly due to the acidic character of the methylene in position 4 [Citation182]. Edaravone was approved in 2001 in Japan for the treatment of cerebral infarction, even though additional clinical studies are still necessary to verify the complete efficacy of edaravone and in particular to investigate which kind of ischemic patients is well suited for the edaravone treatment and to define the optimal dose and therapy duration in order to gain a significant efficacy within a reasonable therapeutic window [Citation183].
A recent study [Citation184] confirmed the excellent reactivity of edaravone toward GO, acrolein, and HNE while evidencing that it can react with α,β-unsaturated aldehydes acting as a polymerizing agent and forming adducts characterized by a very high molecular weight since all monitored adducts conserve a reactivity comparable with that of edaravone. The capacity to yield polymeric adducts can explain some side effects observed during prolonged edaravone treatments. Notwithstanding this, the notable quenching activity of edaravone prompted the development of derivatives mainly substituted in 4 with the aim to modulate the acidity of this carbon atom. Among them, TM2002 (), which derives from the 4-substitution of edaravone with a furo-pyridinol moiety, appears to be a non-toxic inhibitor of advanced glycation and oxidative stress devoid of effect on blood pressure with a neuroprotective efficacy comparable with that of the parent compound [Citation185].
By considering the known protective action of curcumin and other natural β-diketo derivatives [Citation186,Citation187], LoPachin and coworkers recently proposed a set of simplified diketo analogs (e.g., 2-acetylcyclopentanone), which showed a significant scavenging activity toward α,β-unsaturated aldehydes as well as metal ion chelation properties. The promising results obtained in cellular models of oxidative stress render these compounds potential candidates for the treatment of acute or chronic oxidative-stress based neurodegenerative conditions [Citation188].
Polyreactive compounds
Heterocycle-based compounds. The parent compound of all heterocycle-based scavengers is surely carnosine (β-Ala-His; ) [Citation189,Citation190], an endogenous peptide specifically found in millimolar concentration in some tissues as the brain, the heart, and the skeletal muscles and whose multistep mechanism, mainly based on the ability of the imidazole ring to yield Michael addition on the unsaturated imine intermediate, renders it markedly selective toward RCS [Citation191,Citation192]. The remarkable profile of this dipeptide has prompted the development of numerous derivatives which were extensively examined in recent reviews (see, e.g., [Citation193,Citation194]). While avoiding extensive analyses of its derivatives, it is interesting to remind here the two major objectives for which the carnosine analogues were designed. The first objective was to improve the quenching activity while maintaining the mentioned selectivity, and this aim was pursued essentially by replacing the primary amine with more reactive moieties such as hydrazines, hydrazides, and diammino groups (e.g., [Citation195]). The second objective involved the improvement of carnosine's pharmacokinetic profile since it is actively absorbed by intestinal transporter hPepT1 [Citation196] but rapidly hydrolyzed by serum carnosinase, a specific dipeptidase found in plasma and in the brain [Citation197]. To this end, carnosine derivatives were created by modification at the peptide bond or at the histidine configuration (i.e., D-carnosine, β-Ala-D-His) [Citation198]. These simple examples emphasized what was already discussed in the introductive part, namely that the structure of the scavengers can be clearly subdivided into few reactive moieties whose modification heavily influences the quenching activity and the remainder, which is virtually inert and can be vastly modified to improve the pharmacokinetic profile. Although no longer recognized by intestinal trasporter, D-carnosine and its lipophilic prodrugs showed a quenching activity almost superimposable on that of L-carnosine being totally stable in human plasma [Citation199]. Due to its promising bioactivity, D-carnosine underwent in vivo studies revealing that it is highly effective in attenuating experimental atherosclerosis and renal disease by reducing carbonyl stress and inflammation and that it may represent a promising therapeutic strategy in humans [Citation200].
Figure 5. Relevant polyreactive carbonyl quenchers.
The remarkable profile of both carnosine and other natural histidine containing peptides, as in the case of GHK (Gly-His-Lys), prompted also the development of novel short peptides more active and more stable than carnosine or GHK. In a recent patent, Lipotec (Barcelona, Spain) reported a set of tetrapeptides with a capacity to quench HNE and nonenal significantly greater than that of carnosine. All the reported tetrapeptides show the key histidine as a C-terminal residue and alanine in second position. Interestingly, the most active peptides have a 2,3-diaminopropionic acid residue as N-terminus, which might promote the formation of the initial imine intermediate [Citation201].
A second heterocycle-based natural compound is represented by thiamine (vitamin B1; ) whose antiglycation activity can have a dual mechanism. Thiamin is converted in the cell to thiamin pyrophosphate (TPP), the coenzyme for transketolase (TK), a rate-limiting step of the pentose phosphate pathway [Citation202]. TK activation could decrease the accumulation of glyceraldehyde-3-phosphate and fructose-6-phosphate during glycolysis, thus preventing AGE formation [Citation203]. Moreover, thiamine can directly quench reactive carbonyls [Citation204] due to the unique reactivity of its thiazolium nucleus [Citation205] with a mechanism similar to that exerted by thiamine in the enzymatic catalysis [Citation206]. A similar activity is offered by benfotiamine (), a soluble analogue of vitamin B1 [Citation207], which additionally prevents the lipopolysaccharide-induced activation of cPLA2 thus exerting a protective effect by modulating the arachidonic acid pathways [Citation208]. Both benfothiamine and thiamine were reported to be beneficial in experimental models of diabetic nephropathy [Citation209,Citation210]. Benfothiamine improved nerve conduction velocity in diabetic patients [Citation211,Citation212] and prevented vascular endothelial dysfunction after a high-AGE meal [Citation213]. Nevertheless, recent clinical studies on patients suffering from diabetes (types 1 and 2), diabetic nephropathy, and peripheral nerve inflammation showed that benfotiamine does not result in significant reductions in plasma or urinary AGEs or plasma markers of endothelial dysfunction and nerve inflammation compared to placebo [Citation214,Citation215].
Tenilsetam (3-thiophen-2-ylpiperazin-2-one; ), a derivative initially developed as cognition-enhancing drug for the treatment of patients suffering from Alzheimer's disease, shows beneficial effects which seem to be ascribable to its ability to reduce protein carbonylation by both directly quenching RCS and covalently attacking the already-glycated proteins, thus blocking the reactive sites for further polymerization reactions with favorable effects on the AGE-derived cross-linking of amyloid plaques [Citation82,Citation216]. Despite the fact that precise quenching mechanism is still unclear, the protective effect of tenilsetam indicates that carbonyl quenchers may represent a promising therapeutic strategy to reduce intracellular AGE-accumulation as well to decrease the RCS-induced impairment in aging and neurodegeneration [Citation217].
Amino derivatives. As a preamble, it should be emphasized that the main reactivity that a primary amine can exert toward RCS involves the formation of stable imine derivatives. Given the good correlation between basicity and nucleophilicity for amine derivatives [Citation218], a nucleophilicity increase is inevitably paralleled by an increase in the fraction of protonated forms which in fact cannot yield the imine formation since the protonated species cannot attack the carbonyl carbon atom to yield the corresponding carbinolamine [Citation219]. This means that the imine formation may be enhanced by lowering the basicity of the amino group so as to increase the fraction of neutral species even though such an approach should be cautiously pursued since the imine stability increases when nucleophilicity decreases due to the stabilizing effect of electron-drawing substituents on the C = N double bond [Citation220]. This implies that amino-based scavengers endowed with relatively basic amines lose their selectivity by trapping even physiological carbonyls. Differently stated, the reactivity of the amino groups should be carefully balanced to maximize the quenching activity without undermining the selectivity.
The amino reactivity is conveniently exploited by scavengers already examined in the previous sections. For example, the mechanism of carnosine reaction with RCS involves as a first step of the condensation between the carbonyl function and the carnosine primary amine, resulting in the formation of a reversible α,β-unsaturated imine intermediate which serves as an intramolecular catalyst to favor the key Michael addition involving the imidazole ring [Citation221]. Similarly, the cyclization process involving penicillamine with dicarbonyls starts with the formation of the initial imine intermediate. Nonetheless, the mentioned compounds were included in other sections since their quenching is heavily influenced by the nucleophilicity of the second reactive center (the imidazole for carnosine and the thiol for penicillamine). By contrast, this section will examine those derivatives whose quenching activity is almost exclusively determined by their amino groups.
PM (4-(aminomethyl)-5-(hydroxymethyl)-2-methylpyridin- 3-ol; ), a naturally occurring derivative of vitamin B6, has proved to be an effective inhibitor for protein glycation and lipoxidation both in vivo and in vitro [Citation222]. Its protective action involves different mechanism including chelation of metal ions with catalytic oxidation capacity [Citation56], neutralization of radical species due to the phenol acidity [Citation222], and scavenging of carbonyl species with a high glycation ability. Recent studies evidenced that PM can react with dicarbonyls by rapidly forming stable imine derivatives whose stability is enhanced by the low basicity of the benzilic amino group as well as by the capacity of second carbonyl group to interact with the phenol function yielding hemiacetalic cycles [Citation223,Citation224]. Moreover, some dicarbonyls can react with two PM molecules yielding a dimer fused through a central imidazolium or piperidinium ring depending on the structure of RCS and with a mechanism which reminds that of the MOLD adduct starting from two lysine residues [Citation225,Citation226]. Yet again, γ dicarbonyls, which can be derived from alkyl oxidation, rapidly cyclize with PM yielding pyrrole derivatives, according to Paal–Knorr reaction [Citation227]. Comparative analyses showed that PM is more reactive than the ε-amino group of lysine residues but remains still less reactive when compared to the strong reactivity of a cysteine thiol function [Citation228,Citation229]. This means that PM can successfully protect lysine residues by glycation processes but fails to protect also cysteines. Similarly, PM can quench α,β-unsaturated carbonyls by yielding both Michael adducts and imino derivatives which involve the PM amino group [Citation136]. Also here, the phenol group can promote these additions by yielding the already-mentioned hemiacetalic cycles for Michael adducts or condensed hemiaminal rings for adducted Schiff bases [Citation228,Citation229]. The protective effects of pyridoxime are also reinforced by its enzymatic role since the phosphorylation of the hydroxyl group in 5 induces an intracellular accumulation through the so-called metabolic trapping mechanism [Citation230]. Based on its excellent quenching activities, PM prevented retinal damage [Citation231] and improved renal function [Citation232,Citation233] in murine models of diabetes and, in contrast to other carbonyl quenchers, such as aminoguanidine, PM showed minimal toxicity [Citation234]. The promising results of the ongoing clinical studies involving PM in diabetes can be found in specialized reviews [Citation235].
As mentioned before, diaminopropionic acid (DAP) is a residue which possesses a significant reactivity toward RCS due to its capacity to give imino adducts. Sasaki and coworkers [Citation236] proposed a set of DAP-containing dipeptides endowed by a noteworthy capacity to quench AGE precursors. Among them, DAP-Val and DAP-Nle showed protective effects comparable with those of aminoguanidine. Interestingly, the prominent reactivity of DAP derivatives toward dicarbonyls was explained by their ability to form stable adducts containing piperazine ring systems. Recently, Audic and coworkers reported a set of 2,3- diaminopropionic acid-based molecules which proved to be excellent scavengers of both dicarbonyls and α,β-unsaturated aldehydes. In particular their remarkable quenching activity against MDA is ascribable to their capacity of yielding stable 2,3-dihydro-1H-1,4-diazepinium adducts, as indentified by LC-MS and NMR analyses [Citation237]. A similar reactivity is also shown by 2,3 diaminophenazine which reduces the formation of AGEs and attenuates the diabetes induced vascular hypertrophy with an overall effect at least comparable to that of aminoguanidine [Citation238]. Moreover, this quencher was also able to increase the collagen solubility, a clear effect of the inhibition of AGE cross-linking, whereas it was unable to ameliorate the diabetes-induced increase in the albumin clearance [Citation239].
Phenols and polyphenols. In the previous section, the ancillary reactivity of phenol group in determining the marked activity of PM was in-depth analyzed, even though the above described molecules cannot clarify whether the phenol derivatives possesses enough nucleophilicity to act as a carbonyl quenchers per se [Citation240]. Lo and coworkers in a recent systematic study analyzed the quenching activity of phenols, diphenols, and triphenols evidencing that the first two groups are substantially inactive toward MG, whereas some triphenols show relevant quenching activity not markedly influenced by the presence of other substituents [Citation241]. This finding clearly suggests that also natural polyphenols can possess efficient quenching activities and indeed in the last years several natural compounds were reported to be able to quench RCS, as recently reviewed by Wang [Citation242].
Concerning the capacity of dietary polyphenols to trap dicarbonyls, a systematic study [Citation243] with MG showed the significant inhibitory effects on MG mediated AGEs formation (greater than 60%) for luteolin, rutin, epigallocathin-3-gallate (EGCG), and quercetin, while catechin, epicatechin (EC), epicatechin gallate (ECG), epigallocatechin (EGC), kaempferol, and naringenin possess lower inhibitory effects. Among them, theaflavins (), the main components of black tea, were the most reactive polyphenols toward MG with an inhibition effect around 70% thus suggesting that theaflavins would be promising candidates for future in vivo studies [Citation244]. Collectively, these studies evidenced the presence of the flavan-3-ol (i.e., catechin) substructure as a key element for the quenching activity which is maximized at basic conditions and preferentially occurs at positions 6 and 8 of the A-ring of catechin while the gallate ring does not play an important role in the scavenging activity. Comparative analyses showed that the most efficient polyphenols are more reactive than lysine and arginine residues thus confirming their protective role against AGE-mediated protein carbonylation [Citation245]. The chemical relevance of the catechin's A-ring in determining carbonyl quenching was substantiated by quenching activity of genistein (4’,5,7-trihydroxyisoflavone; ) [Citation246], phloretin, and phloridzin [Citation247], thus confirming the major rules proposed by Matsuda and coworkers [Citation248], namely that (i) quenching activity increases with the number of hydroxyl groups; (ii) flavones are normally more active than the corresponding reduced analogs; (iii) modifications of hydroxyl groups decrease the activity apart from those on position 3’, and (iv) as a rule, there is a relationship between carbonyl quenching and free radical scavenging.
Even though most studies have been focused on the quenching of dicarbonyls by polyphenols, Beretta and coworkers demonstrated that green tea polyphenols can also trap α,β-unsaturated aldehydes always exploiting the specific reactivity of the A-ring despite with a different mechanism. Indeed, while polyphenols quench dicarbonyls through an electrophilic substitution in which the carbonyl carbon atoms attack the most electronegative phenyl carbon atoms, unsaturated carbonyls react through a Michael addition between a phenol oxygen atom and the acceptor β carbon atom followed by an intramolecular condensation to yield stable benzochromene adducts [Citation249].
Other natural phenolic compounds endowed with carbonyl quenching activities toward AGE precursors include the stilbene glucosides among which 2,3,5,4-O- tetrahydroxystilbene 2-O-β-D-glucoside (THSG), the major bioactive compound from Polygonum multiflorum, appeared much more active than resveratrol and its methylated derivative pterostilbene, the two major dietary stilbenes. Similar to flavones, the quenching occurs through an electrophilic substitution involving the electronegative positions 4 and 6 of the A ring [Citation250]. Besides the discussed polyphenols, also terpenoids [Citation251] and procyanidins [Citation252] displayed inhibitory effects on AGE formation, which are largely ascribable to both their antioxidant activities and carbonyl scavenging capacities. Overall, the reported studies indicate that many natural compounds exert their known protective and antioxidant effects also through a carbonyl quenching activity, and much remains to be done in order to elucidate the precise quenching mechanism for these molecules as well as their potential clinical applications.
Despite structurally different, also the quenching activity of ascorbic acid (aka Vitamin C, (5R)-[(1S)-1,2-dihydroxyethyl]-3,4-dihydroxyfuran-2(5H)-one; ) can be explained in terms of nucleophilicity of hydroxyl/enolic functions which act as Michael donor reacting with α,β-unsaturated aldehydes [Citation253]. In detail, ascorbic acid reacts with HNE yielding two major adducts, which share a common tetrahydrofuro[3,2-b]furan-5-one system and differ from the arrangement of the third tetrahydrofuran ring, which is linked in 6 position or fused in a spiro form similar to that observed with acrolein (see below [Citation254]). Similarly, ascorbic acid can react with acrolein giving a tricyclic intermediate which rearranges yielding 5,6,7, 8-tetrahydroxy-4-oxooctanal (THO) which in turn exists in solution in equilibrium with the corresponding 5- and 6-membered cyclic hemiketals and hemiacetals [Citation255]. Despite the capacity to generate covalent adducts, the ability of ascorbic acid to prevent protein carbonylation by reactive aldehydes is, however, mostly ascribable to its ability to increase the transport of GSH conjugates from the cells into the medium by enhancing the efflux of the multidrug-resistant protein (MRP) substrate as recently demonstrated in human monocytic THP-1 cells [Citation254].
Miscellaneous inhibition mechanisms
Anti-inflammatory drugs. Aspirin was one of the first molecules shown to prevent glycation. When present in incubations of lens proteins, aspirin inhibited glycation of the proteins treated by glucose, galactose, and other sugars in vitro [Citation256–258]. Aspirin was classified as a sugar competitor since it acts by acetylation of the reactive amino groups on proteins [Citation259–261]. Other anti-inflammatory drugs, ibuprofen and diclofenac, also inhibited glycation, yet the mechanism may be different since ibuprofen and diclofenac have no acetyl group. Aspirin and ibuprofen prevented cataract in diabetic rats [Citation262].
Pyruvic acid. The simplest α-keto acid pyruvate (2- oxopropanoic acid, an oxidized form of MG) was reported to protect rat eye lens proteins against sugar-induced glycoxidation [Citation263–265] and prevented cataract development in diabetic rats or mice. The effect was explained by the ability of pyruvate to bind competitively to protein primary amines, as well as by the antioxidant action of the compound [Citation264,Citation266].
Aldose reductase inhibitors. Under hyperglycemic conditions, fructose and 3-DG, compounds with high glycation potential, are produced by the polyol pathway [Citation267,Citation268]. Fructose formed via the polyol pathway can reach the concentrations same as glucose in certain tissues. In this way, polyol pathway may contribute to the process of advanced glycation. The aldose reductase inhibitor, epalrestat, was found to reduce plasma levels of markers of glycoxidation in diabetic patients [Citation269,Citation270]. Epalrestat suppressed the deterioration of diabetic peripheral neuropathy in association with a reduction of CML blood levels [Citation271]. The aldose reductase inhibitor, zopolrestat, significantly reduced MG levels in aged rat aorta and significantly improved endothelial-dependent relaxation in aged rats [Citation272].
ALEs/AGEs elimination
Physiological pathways of AGE and ALE removal(NC)
Enzymatic defense against AGEs and ALEs
Enzymatic deglycation at the Amadori level. Fructosamine-3-kinase (FN3K) is a key player in the repair of Amadori products. It was discovered almost at the same time by Delpierre et al. [Citation273] and Szwergold et al. [Citation274]. The enzyme was purified from human erythrocytes and expressed in Escherichia coli. FN3K is a 35-kDa monomeric protein whose tertiary structure is still not resolved, even though it should show a certain degree of similarity with the recently resolved structure of the putative FN3K from Thermobifida fusca YX-ER1 (YP_290396.1, Pdb Id: 3f7w). FN3K phosphorylates low-molecular mass and protein-bound fructosamines on the third carbon of the hexose. The enzyme phosphorylates 1-deoxy-1- morpholinofructose, fructoselysine, fructosevaline, and fructoseglycine in order of decreasing affinity. Histone, bovine serum albumin, lysozyme, or hemoglobin-bound fructoselysine was also found to be phosphorylated by FN3K. The affinity of protein-bound fructoselysines is about 75-fold higher as compared with free fructoselysines. The reaction of fructoseamine phosphorylation is ATP dependent. The phosphorylation product fructosamine-3-phosphate is unstable and spontaneously decomposes by β-elimination into 3-DG, inorganic phosphate, and non-glycated amine (). As an example, a half-live of fructoselysine-3-phosphate is about 5–6 h at 37°C [Citation274].
Figure 6. Reaction of fructosamine-3-kinase.
The discovery of FN3K in human erythrocytes revealed the enzymatic defense mechanism against glycation at its early stage. It became obvious that mammalian cells are able to prevent further conversion of Amadori products into AGEs and to clear fructosamines formed on proteins in the cell [Citation275–277]. Physiological importance of FN3K as a protein repair enzyme was confirmed in the experiment with FN3K-deficient mice. Their level of hemoglobin-bound fructosamines was approximately 2.5-fold higher than that of control mice. Moreover, other intracellular proteins were 1.8- to 2.2-fold more glycated in erythrocytes and 1.2- to 1.8-fold more glycated in brain, kidney, liver, and skeletal muscle [Citation278]. On the other hand, the physiological role of FN3K is not fully clear even despite the fact that the enzyme is able to reduce the glycation of intracellular proteins in vivo. Recently, it has been shown that the survival, functions, and glucotoxic alterations of pancreatic β-cells from both FN3K-knockout and wild-type mice are almost identical in the presence of high glucose concentration [Citation279].
The enzyme is present in mammals and birds, and was not found in fishes, plants, and bacteria [Citation280], even though homologous proteins (FN3K-related proteins) are present in all taxa, where they retain intracellular repair functions. FN3K is particularly active in brain, heart, kidney, and skeletal muscle. The activity of FN3K in erythrocytes from different species correlates well with the concentration of glucose. Indeed, FN3K is active in erythrocytes from rat, mouse and man where the intracellular concentration of glucose is close to that of the plasma, and the activity of FN3K is very low in erythrocytes from pig and chicken in which the concentration of glucose is low or near zero [Citation281,Citation282].
Besides FN3K, other deglycating enzymes of Amadori products are known. In fungi and bacteria, fructosyl amino acid oxidases called amadoriases catalyze the oxidative degradation of Amadori products leading to the formation of unmodified amine and 2-ketoaldose as well as H2O2. In bacteria, fructosamine 6-phosphate deglycases and fructoseamine 6-kinases decompose Amadori products, although these enzyme use different catalytic mechanisms and have different physiological role as compared with FN3K [Citation283].
Enzymatic detoxification of reactive α-oxoaldehydes. GO, MG, and 3-DG are important glycating agents besides glucose and fructose (). They are up to 20,000-fold more reactive than glucose and form AGEs bypassing the stage of early glycation [Citation284,Citation285]. There are two important pathways involved in the enzymatic defense against glycation and the prevention of accumulation of α-oxoaldehydes as AGE precursors in vivo, that is, aldose reductase and glyoxalase pathways.
Human aldose reductase (AKR1B1; EC 1.1.1.21) belongs to the AKR superfamily. AKR are oxidoreductases found in organisms from prokaryotes to eukaryotes including yeast, plants, amphibia, and mammals. A systematic nomenclature for AKR was adopted in 1996. More than 110 enzymes of AKR superfamily are grouped into 14 families (AKR1–AKR14) having less than 40% amino acid identity with any other family. Families are divided into subfamilies (> 60% amino acid identity among members) described by a letter, and an Arabic number signs the unique protein sequence [Citation286,Citation287].
Human aldose reductase is expressed in almost all tissues [Citation288–294]. AKR1B1 is a cytosolic monomeric protein of 315 amino acids with a molecular mass of about 36-kDa and known three-dimensional structure. The enzyme shows an eight-stranded β/α -barrel folding and catalyzes the NADPH-dependent reduction of a wide variety of carbonyl-containing compounds to their corresponding alcohols with a broad range of catalytic efficiencies [Citation295–299]. AKR1B1 is the first enzyme in the polyol pathway of glucose metabolism. It catalyzes the reduction of glucose to sorbitol that is subsequently converted to fructose by sorbitol dehydrogenase. Under normal physiological conditions, this pathway plays a minor role, and cellular glucose enters the glycolytic pathway. For aldose reductase, glucose is not a preferred substrate: KM = 100 mM and kcat/KM = 2.8 × 102 M− 1min− 1 [Citation295]. It seems that the primary role of AKR1B1 is to remove reactive aldehydes. Kinetics constants kcat/KM for MG, 3-DG, and GO are equal to 1.8 × 107, 2.5 × 106, and 3.0 × 105 M− 1min− 1, respectively. Thus, α-oxoaldehydes are better substrates than glucose as glycating agents leading to AGEs formation [Citation300,Citation301]. Moreover, the cytoprotective role of AKR1B1 against MG was revealed using cultured vascular smooth muscle cells. An MG-induced dose- and time-dependent increase in aldose reductase mRNA accompanied by higher enzyme activity and protein level was found. However, the cytotoxic effect of MG was enhanced when the activity of aldose reductase was suppressed by ponalrestat, an inhibitor of the enzyme [Citation302]. Recently, the in vivo role of aldose reductase in mammalian metabolism of AGE precursors has been studied. The results show that the reduction of α-oxoaldehydes catalyzed by aldose reductase is a significant pathway in AGE precursor removal in human endothelial cells. Mice aldose reductase AKR1B3 is also an efficient catalyst for the reduction of AGEs precursors. The process is diminished in hearts of aldose reductase-null mice. Moreover, diabetic aldose reductase –null mice accumulate more AGEs in the plasma and the heart than wild-type mice [Citation303,Citation304].
Cellular metabolism of 3-DG was studied using human umbilical vein endothelial cells. The results indicated that 3-DG was internalized by the cells and reduced to 3- deoxyfructose by AKRs. The reaction was inhibited by an AKR inhibitor [Citation305].
The glyoxalase system consists of two enzymes, glyoxalase 1 (EC 4.4.1.5) and glyoxalase 2 (EC 3.2.1.6); GSH is used as a cofactor. The glyoxalase system catalyzes the conversion of α-oxoaldehydes into the corresponding α- hydroxyacids. Glutathione-methylglyoxal hemithioacetal formed non-enzymatically from MG and GSH is isomerized by glyoxalase 1 to S-D-lactoylglutathione. The latter serves as a substrate for glyoxalase 2 and is converted to D-lactate and GSH [Citation114,Citation306]. Human glyoxalase 1 is found in all tissues. This is a cytosolic dimeric Zn2+ metalloenzyme of molecular mass 42 kDa. For glutathione-methylglyoxal hemithioacetal KM is 71–130 μM and kcat is 7–11 × 104 min− 1. Thus, kcat/KM for glyoxalase 1 is approximately 100-fold higher than for aldose reductase, thus suggesting that glyoxalase system should be more effective in MG detoxification than aldose reductase. Besides MG, physiological substrates of glyoxalase are also GO and 4,5-dioxovalerate [Citation307,Citation308]. Glyoxalase 2 is also a metalloenzyme and contains a Fe2+-Zn2+ center. Zinc plays an essential role in its catalytic mechanism. The enzyme has two isoforms and can be expressed in cytosol (cytosolic form of molecular mass 29 kDa) or in mitochondria (the mitochondrial form has molecular mass of 34 kDa). Glyoxalase 2 has a broad substrate specificity for GSH thiol esters. KM and kcat values for S-D-lactoylglutathione are equal to 146 μM and 727 s− 1, respectively [Citation308]. The physiological role of the mitochondrial isoform is not clear [Citation309].
Some studies show the biological significance of glyoxalase system in the enzymatic defense against glycation reactions mediated by α-oxoaldehydes. Shinohara et al. demonstrated that overexpression of glyoxalase 1 in bovine endothelial cells reduced intracellular AGEs when the cells were exposed to high glucose concentration in vitro [Citation310]. Moreover, the ability of glyoxalase system to reduce the level of α-oxoaldehydes and AGEs was demonstrated in an in vivo model of diabetes. Diabetic transgenic rats with a glyoxalase 1 overexpression had less AGEs as compared with wild-type diabetic rats [Citation311]. Glyoxalase 1 is also critical for human retinal capillary pericyte survival under hyperglycemic conditions. Human retinal capillary pericyte incubation with a combination of high glucose and bromobenzyl-S-p-glutathione cyclopentyl diester, a competitive inhibitor of glyoxalase 1, results in cells apoptosis along with an increase in MG concentration. However, overexpression of glyoxalase 1 in the cells protects against apoptosis induced by glyoxalase 1 inhibitor under high glucose [Citation312]. Investigations of pathophysiological role of glyoxalase 1 in rat renal ischemia-reperfusion injury revealed that ischemia-reperfusion induced tubulointerstitial injury and that the histological changes are associated with a significant decrease in renal glyoxalase activity and an increase in MG level. However, in rats overexpressing human glyoxalase 1, renoprotective effects of that enzyme were observed and the reduction of MG accumulation in tubular cells was noticed [Citation313,Citation314].
Glyoxalase 1 expression declines on aging, and oxidative stress results in increased glycation and tissue damage [Citation308]. The decrease in the enzyme activity was observed in aging human lenses [Citation315], skeletal muscles [Citation316], and human brain [Citation317]. In AD, glyoxalase 1 is upregulated in a compensatory manner to maintain physiological level of α-oxoaldehydes [Citation318]. However, with advancing stage of AD, the decrease in the level of glyoxalase I was observed. In both age- and AD-affected brains the level of glyoxalase 1 correlated with AGEs deposits [Citation319].
Intracellular degradation of AGEs by lysosomal and proteasomal system
Introduction to proteolytic systems. AGEs are the final products of the non-enzymatic reaction of reducing sugars and reactive aldehydes with amino-groups on macromolecules such as proteins, lipids, or nucleic acids [Citation320,Citation321]. These structures are rather stable and capable of forming cross-links between proteins. As said above, AGEs have been reported to be accumulated during several pathological conditions and have been detected in various foods where they are mainly produced by heating [Citation322]. Hence, they can be ingested thus promoting clear physiological consequences [Citation323]. Among the mentioned strategies to reduce their amount in vivo, the proteolytic catabolism requires the precise identification of the enzymatic pathways that are involved in the AGE degradation in order to modulate them exogenously, thus to increase the cell clearance of AGEs.
There are two main cellular proteolytic systems that are responsible for the degradation of the various macromolecules: the lysosomes and the proteasomes. With regard to lysosomal degradation, macromolecules are internalized from the extracellular space by endocytosis and they are then transferred by fusion with phagosomes ending up in complete degradation by the endosomal–lysosomal system. Additionally, cytoplasmic molecules that need to be degraded are also processed through autophagy [Citation324]. Autophagy can be functionally divided into three known pathways including the chaperone-mediated autophagy, the macrophagy, and the microphagy. The chaperone-mediated autophagy is a highly specific process where substrates to be degraded are unfolded in a chaperon- assisted process and transported directly into the lysosomes via the LAMP-2A membrane channel [Citation325]. Also microphagy is a direct uptake into lysosomes, via a not very well-understood process of invagination of the lysosomal membrane and enclosure of small parts of the surrounding cytosol. In contrast, macrophagy is the new formation of a membrane from a starting point. This membrane encircles larger parts of the cytosol, including organelles as mitochondria and protein aggregates. Macrophagy is terminated by the complete engulfment of the material to be degraded in a structure called autophagosome, which is later on fusing with lysosomes. It is widely accepted that damaged proteins, if not degraded by the proteasome, are taken up by macrophagy into the lysosomal autosomal compartment. This refers especially to protein aggregates, as lipofuscin, and results in the fact that only a low percentage of lipofuscin is actually located in the cytosol [Citation326]. Interestingly, there seems to be a different approach of cells to sequester protein aggregates: either in lysosomes or in aggresomes. While terminally oxidized, non-degradable protein aggregates are more located in lysosomes [Citation326], while under certain unknown circumstances oxidized protein aggregates and AGE-modified proteins might be located in aggresomes [Citation327,Citation328].
The proteasome is an important intracellular protease complex responsible for the regulated degradation of both normal and abnormal proteins. The ubiquitin-proteasome system (UPS) and autophagy have long been regarded as two separate cellular pathways with distinct functions. It was previously thought that autophagy predominantly degraded long half-life proteins whereas the clearance of the short half-life proteins that were typical UPS substrates, would not be degraded by autophagy. However, studies in the last decade suggest that the two systems “communicate,” at least under certain circumstances whereas compensatory functions have also been revealed in some cases. More specifically, concerning modified proteins, it has been shown that moderately modified proteins are degradable by the proteasome whereas heavily modified proteins are more likely to be processed by the lysosomal proteases [Citation329].
Lysosomes. Lysosomes were discovered by Christian de Duve and were then recognized as the terminal compartment where the degradation procedure of the endocytic pathway occurs. These organelles are highly heterogeneous in size and shape, and they are often found to contain electron-dense material and membranous aggregates [Citation330]. These single or double membrane organelles possess various acidic hydrolases, such as peptidases, lipases, and nucleases for the degradation of the corresponding biological polymers. A proton pump in their membrane actively transports protons (H+ ions) into the lumen, thus maintaining the internal acidic pH that is a prerequisite for the optimal activity of hydrolases. These acid hydrolases are responsible for degradation of the delivered cargo, whereas the derived essential nutrients (amino acids, fatty acids, sugars, etc) are then returned to the cytosol through the action of permeases that reside at the lysosomal membrane [Citation331].
Briefly, lysosomal formation is functionally connected with endocytic and secretory pathways. Clathrin-coated endocytic vesicles are extorted from the plasma membrane engulfing extracellular material. They are then fused with early endosomes, and a gradual maturation into late endosomes occurs [Citation332]. These late endosomes fuse with transport vesicles, originated from the trans-Golgi network, to form the lysosome. The trans-Golgi vesicles have mannose-6-phosphate receptors (MPRs) that are necessary to attract acid hydrolases. Gradually the internal pH decreases to approximately 5 that is characteristic for endosome maturation [Citation333]. This pH decrease permits the release of acid hydrolases from the MPRs into the lysosomal lumen to produce the active lysosomes. At the same time, the receptors are recycled back to the Golgi network.
Previous work has confirmed that AGEs undergo receptor-mediated endocytosis by various scavenger receptors [Citation334]. More specifically, several cell surface proteins have been identified to recognize and to bind AGEs, like the RAGE [Citation335,Citation336], CD36 receptor (belonging to scavenger receptor family type B) [Citation337], macrophage scavenger receptor A I and II (a member of the scavenger receptor family type A) [Citation334] and the receptor complex p60, p90, and galectin-3 [Citation334,Citation338]. For few of them like CD36 [Citation339] and MSR [Citation340], a direct role in endocytosis has been shown. Nevertheless, although endocytosis of AGEs through specific receptors has been demonstrated, the proteases that take over to degrade the glycated proteins are not fully elucidated. However, more and more evidence accumulates that the RAGE receptor is just a signaling receptor recognizing the presence of AGEs in the extracellular environment, but not responsible for the uptake of the material [Citation341]. This is clearly the function of the scavenger receptors. Miyata et al. have shown that AGEs-modified proteins (in specific, pyrraline-modified albumin) accumulate in phagocytes under conditions of reduced lysosomal- mediated degradation, thus implying that the lysosomal enzymes are pivotal for the AGE proteolysis [Citation342]. Saito et al. have also confirmed the endocytic/lysosomal fate of 125I-AGE-BSA [Citation343]. More specifically, they have shown that 125I-AGE-BSA is less degraded following chloroquine and leupeptin addition; both agents are known to inhibit the lysosomal enzymes function. A similar partial effect of lysosomal degradation has also been suggested to occur in microglial cells [Citation344]. Grune and colleagues have identified cathepsin D as one of the major enzymes involved in the degradation of AGE-modified proteins both in vitro and in vivo [Citation345]. More specifically, they showed that AGE- modified albumin is degraded by cathepsin D, while cathepsin B is less effective in the degradation of aldehyde-modified albumin. Additionally, the authors suggested that the 20S proteasome was completely unable to degrade these modified proteins. With regard to in vivo data, extensive intracellular AGE accumulation was revealed in mouse primary embryonic fibroblasts derived from cathepsin D knockout animals; these aggregates were mainly located in lysosomes. Moreover, in these cells, reduced degradation rate of AGE-modified proteins was recorded as compared to the relative levels in cells isolated from wild-type animals [Citation345]. More recently, along with cathepsin D, the same group has also identified cathepsin L to possess a pivotal role in the cellular response against AGEs [Citation346].
Grimm et al. also tested the degradation of AGEs by proteases from the gastrointestinal tract. Interestingly, pepsin was able to degrade AGE-modified proteins to a good extent; however, trypsin and chymotrypsin had a lower efficiency [Citation345]. It seems that due to the established pH optimum of the used cathepsins and the other proteases, in the case of a low pH (perhaps due to the unfolding of the substrate), proteases are able to degrade AGE-modified proteins. However, to our knowledge, there are no conditions found where AGE-modified proteins are degraded with higher efficiency compared to the normal substrates. In other words, these AGE substrates are not degraded preferentially as oxidized proteins in the case of the proteasome [Citation347,Citation348]. So it might well be that due to the unfolding in the case of a low pH, the protein is just degraded, not because it was AGE-modified but regardless that it is modified.
Aggrephagy. Today, all statements about the fate of AGE-modified protein aggregates are highly speculative. However, a few wage hints exist, that AGE-modified proteins, if aggregated, might be further processed. First, it was shown that AGE-modified proteins, in particular AGE-modified vimentin are located in aggresomes [Citation328]. Whether this is a special property of vimentin or important for more proteins remains obscure. Since non-modified vimentin is a part of the aggresome caging structure [Citation349], it might well be that vimentin is – modified or not – targeted to aggresomes, once they are formed. However, since aggresomes in general sequester proteins which are not degraded efficiently by the UPS, also AGE-modified proteins, might be targeted toward aggresomes. In general, aggresomes are known to be ubiquitinated in vitro and in vivo [Citation350]. Interestingly, it was more recently detected that ubiquitination of aggresomes happens as well in the K48, K63, and K11 positions of the ubiquitin [Citation351]. In particular, the K63-lysine binding of ubiquitin draws the attention of researchers, since this kind of ubiquitination is targeting for autophagy-mediated lysosomal degradation [Citation352]. This gave hints that aggregated proteins at least if packed in aggresomes might be targeted toward autophagy. This process is referred to as “aggrephagy”. Of special interest is, of course, the recognition of the aggresome during the initial steps of autophagy. It is generally acknowledged that aggrephagy is a special form of macrophagy. The process of aggrephagy is until now not well understood, but it is known already that a wealth of regulatory proteins are playing a role, including HDAC6, p66, NBR1, mTOR, and Alfy, just to name a few. For an extensive review of aggrephagy, see [Citation353].
To conclude, it should be mentioned that the targeting of AGE-modified protein aggregates toward the lysosomes does not mean that lysosomal proteases are able to degrade the uptaken material completely. Most studies reporting the degradation of aggregates/aggresomes targeted to lysosomes refer to just unfolded proteins, but not to modified proteins or AGE-modified proteins [Citation353].
Proteasomes. The proteolytic core, the 20S proteasome, is a 700-kDa multisubunit enzyme complex composed by 7 different α- and 7 different β-subunits. The subunits of the same type form heptameric rings; thus, two α- and two β-type rings give rise to the barrel-shaped proteasome. The final structure of the proteasome is the α1–7β1–7β1–7α1–7 structure [Citation354,Citation355]. Three of the β-subunits, β1, β2, and β5, are responsible for the proteasome-hydrolyzing activities that cleave peptide bonds on the carboxyl site of acidic (peptidylglutamylpeptide-hydrolyzing or caspase-like activity, PGPH or C-L), basic (trypsin-like activity, T-L) and hydrophobic (chymotrypsin-like activity, CT-L) amino acids, respectively [Citation356]. The N-terminal threonine residues of these active β-type subunits act as nucleophiles during hydrolysis, thus forming the catalytic centers of the complex. In mammals, upon specific conditions, the constitutively expressed β1, β2, and β5 subunits can be substituted during de novo proteasome biosynthesis by the immunosubunits, namely β1i, β2i, and β5i, thus forming the immunoproteasome [Citation357]. Immune and non-immune functions have been attributed to this special proteasome type [Citation358], but most importantly, the immunoproteasome has been demonstrated to conduct the efficient turnover of oxidatively damaged proteins and thus to reduce the formation of harmful protein aggregates [Citation359,Citation360]. Additional regulatory complexes can be attached at the ends of the 20S core, thus giving rise to various proteasome supra-complexes with the 26S proteasome being the most known (two 19S regulatory complexes that bind either to one or both ends of the 20S core) [Citation356]. Controlled protein degradation is predominately catalyzed by the proteasome. Therefore, this complex is responsible for cell clearance of abnormal, denatured or, in general, damaged proteins as well as for the regulated degradation of short-lived proteins [Citation356,Citation361].
Regarding the proteolysis of AGEs by the proteasome, Grimm et al. have demonstrated that although the 20S proteasome is able to degrade oxidized BSA, it is completely inert to degrade AGE-modified albumin [Citation345]. More specifically, they clearly showed that although AGEs could be degraded by cathepsin D, they could not be affected by the proteasome. This study is in accordance with the study of Bulteau et al. who have demonstrated that CML, a known AGE structure, is resistant to the 20S proteasome degradation [Citation362]. In contrast, activation in terms of both content and activities of nuclear proteasomes and degradation of carboxymethylated histones was reported in human keratinocytes following GO treatment [Citation363]. Stolzing et al. have suggested that microglial cells are able to degrade BSA-AGEs both through lysosomal and also through proteasomal pathways [Citation344]. A functional interaction between both lysosomal and UPS-mediated degradation was also shown in both live cell and cell-free systems by Taylor and colleagues [Citation364]. Finally, a recent study has shown that AGEs induce the expression of immunoproteasomes [Citation346]. More specifically, AGEs inhibited the activities of the constitutive proteasomes and promoted the decrease in the expression of the constitutive β-type subunits, while they increased both expression and activities of immunoproteasomes. This induction involved in the activation of RAGE and the downstream modulation of Jak2/STAT1 [Citation341]. Although the authors have not checked whether the increased immunoproteasome function leads to increased clearance of AGEs, one cannot rule out the possibility that immunoproteasomes may degrade AGEs instead of constitutive proteasomes that have been tested up to now. However, it seems that the AGE degradation by the immunoproteasome if happens at all is a slow, non-efficient process. Finally, it is noteworthy that regardless of the negative or the positive effect of the proteasome-mediated degradation on the AGEs-modified proteins, the majority of these studies report that these proteins additionally reveal a deleterious effect on the proteasome function mainly through its inhibition [Citation344, Citation363,Citation365].
Strategies to activate the proteasome (NC)
20S complex modulation
Given that it is still debatable whether the proteasome (constitutive or immunoproteasome) is involved in the degradation of AGEs, a potential induction of the proteasome would be interesting in case the AGEs are finally shown to be degraded by the proteasome. Nevertheless, although the proteasome biology is highly investigated, the existing knowledge with regard to activation/upregulation of the proteasome is still limited [Citation354,Citation366]. There are few studies in the literature dealing with proteasome activation through genetic manipulation of the 20S proteasome core subunits or through the use of natural or synthetic compounds [Citation367].
Genetic manipulation of the proteasome
With regard to genetic manipulation of the 20S proteasome, it has been shown that overexpression of β5i subunit in lymphoblasts and HeLa cells promotes enhancement of cleavage after hydrophobic amino acids (CT-L activity) and basic amino acids (T-L activity) while enhancement of the basic activity (T-L) was also recorded upon transfection of the β1i subunit [Citation368]. With regard to overexpression of a constitutive catalytic subunit, overexpression of β1 subunit in HeLa cells was shown to stimulate the acidic activity (PGPH or C-L) [Citation369]. We have also shown that upon overexpression of the constitutive β5 catalytic subunit either in WI-38/T and HL60 cells or in human primary fibroblasts (IMR90), new and functional proteasomes are produced. In the presence of these enhanced levels of proteasomes, the cells exhibit higher rates of proteolysis following treatment with various oxidants, they possess an enhanced capacity to cope with stress and to survive, and they have decreased levels of oxidized proteins. Moreover, upon β5 transfection in primary human embryonic fibroblasts (IMR90 cells), an ˜ 15–20% life span extension is recorded [Citation370]. Similar proteasome modulation has been also produced through β5 transfection in lens epithelial cells [Citation371] and murine neuroblastoma cells [Citation372]. On top of that, upon upregulation of the levels of proteasome subunits through genetic manipulation in aged human fibroblasts, the levels of various aging biomarkers were markedly reduced [Citation373]. Accordingly, proteasome activation has been succeeded genetically through overexpression of: a) hUMP1/POMP in human fibroblasts [Citation374], and b) UMP1 in yeast [Citation375], the necessary chaperones for β-ring formation during proteasome assembly [Citation376,Citation377]. Rpn4 transcription factor in yeast has been shown to be responsible for the transcription of most proteasome genes [Citation378], thus regulating the homeostasis of the yeast proteasome [Citation379] and its response under stress conditions [Citation380]. It was recently shown that Rpn4 manipulation can also lead to proteasome activation and thus to extension of the replicative life span of S. cerevisiae and to improved clearance of toxic huntingtin fragments in the relative yeast model for neurodegenerative diseases [Citation381]. In total, these studies support the pivotal role of the proteasome activation in the retardation of senescence. Given that AGEs have been also shown to increase with aging and senescence, proteasome activation in aged cells could accordingly has increased significance in AGEs elimination through degradation.
Compound-mediated proteasome activation
Given that there are ethical restrains of any manipulation in humans, the interest of scientists and companies has been turned recently into the identification of natural or synthetic compounds with proteasome-activating properties. It was initially shown that this activation is feasible through the “opening” of the closed/latent structure of the proteasome and thus through conformational changes. Proteasome activities were shown to be stimulated in the test tube upon the addition of SDS or fatty acids such as olein, linoleic, and linolenic acids [Citation382,Citation383], in contrast to the inhibition of these activities in the presence of potassium chloride that seemed to promote the latent form of the complex [Citation384]. Similar activating properties have also been exhibited by hydrophobic peptides that were bound as modifiers at non-catalytic sites and opened the α-gated pore of the 20S complex, thus mimicking the action of the 11S complex [Citation385]. Synthetic peptidyl alcohols, p-nitroanilides, esters, and nitriles along with cellular lipid components such as lysophosphatidylinositol, ceramides, and cardiolipin were also reported to activate the proteasome through binding to the 11S-complex- binding site [Citation386–389]. More recently, S-glutathionylation was also reported to constitute a post-translational modification that triggers gate opening and, thus, proteasome activation, whereas following treatment with high concentrations of dithiothreitol, the gate was closed [Citation390]. We have also identified oleuropein, a phenolic compound in Olea europaea leaf extract, olive oil, and olives, as a potent stimulator of proteasome activities in vitro. Human primary fibroblasts treated with oleuropein were also more resistant to oxidative stress and exhibited extended cellular life span [Citation391]. We have suggested that oleuropein most likely alters the conformation of 20S α-gated channels reminiscent to the SDS action. Similar stimulatory effects on 20S proteasome activities and thus protective outcomes were also described for a lipid algae extract (Phaeodactylum tricornutum) in human keratinocytes following UVA and UVB irradiation [Citation392]. Finally, betulinic acid, a pentacyclic triterpene, has also been shown to act as a proteasome activator only enhancing the CT-L proteasome activity [Citation393], while recently lithocholic acid derivatives 3α-O-pimeloyl-lithocholic acid methyl ester and its isosteric isomer were also identified as activators of the chymotrypsin-like activity [Citation394].
The above-mentioned studies investigated compounds that are mainly interfering with the structure of the 20S complex with a consequent induction of its activities. Nevertheless, several exogenous stimuli have been shown to modulate the expression of proteasome subunits and the relative activities in mice through the regulation by nuclear factor (erythroid-derived 2)-like 2 (Nrf2). This transcription factor has been shown to be induced in response to antioxidants such as 3H-1,2-dithiole-3-thione (D3T) [Citation395] or sulforafane [Citation372], thus promoting the expression of cytoprotective genes and the downstream elevated protection against various oxidants. More specifically, Nrf2 has been suggested as a guardian of healthspan and gatekeeper of species longevity [Citation396]. Nrf2 belongs to the family of Cap'n'collar (Cnc) transcription factors, and it is the central mediator of a prominent antioxidant response system by orchestrating the transcriptional response of cells to oxidative stressors and electrophilic xenobiotics. The genes that are included in the list of Nrf2 target genes encompass multiple phase 2 genes, such as GST, γ-glutamylcysteine ligases, heme-oxygenese 1 (HO-1) and NADPH quinone oxidoreductase [Citation397], molecular chaperones and proteasome subunits [Citation395]. The regulation occurs through a common DNA regulatory cis-acting element, called antioxidant response element (ARE) or electrophile responsive element (EpRE; 5’-TGA[C/T]NNNGC-3’) [Citation398]. The main regulator of Nrf2 is the Kelch-like ECH-associated protein 1 (Keap1) that carries multiple cysteine residues that are oxidatively modified according to the changes in the intracellular redox state [Citation399]. Under non-stimulated conditions, Nrf2 is bound by Keap1 and thus sequestered in the cytoplasm while it is actively targeted for ubiquitination and proteasomal degradation [Citation400]. Moreover, Nrf2 may became phosphorylated and/or Keap1 may be modified/phosphorylated [Citation401], resulting in the disruption of the Keap1–Nrf2 complex and the nuclear translocation of Nrf2 [Citation402]. Once transferred into the nucleus, Nrf2 heterodimerizes with a small musculo-aponeurotic fibrosarcoma (MafF, G, or K) protein, thus forming a heterodimer that binds to AREs of the target genes to stimulate their expression [Citation403,Citation404]. Several chemopreventive agents and natural compounds have been reported as potent Nrf2 inducers at low doses. D3T, sulforaphane, curcumin, lycopene, carnosol, quercetin, epigallocatechin-3-gallate, resveratrol, and wasabi are included in this list [Citation405,Citation406]. Nevertheless, the interaction with the proteasome has only been investigated for few of them [Citation372,Citation395]. Two triterpenoids namely hederagenin and 18α-glycyrrhetinic acid have been identified by our group as potential Nrf2 inducers. Treatment of human primary fibroblasts with 18α-glycyrrhetinic acid resulted in Nrf2 activation and the downstream proteasome induction in terms of RNA and protein expression levels as well as in terms of proteasome activities. These increased levels of proteasome function coincided with increased stress resistance and cellular life span extension. These positive outcomes are dependent on the Nrf2-mediated proteasome activation since inhibition of the proteasome induction abrogated the enhanced cell survival despite the presence of the compound [Citation407]. We have also shown elevation of CT-L proteasome activity and induction of the proteasome content upon treatment of HFL-1 human primary cells with quercetin [Citation408], a known Nrf2 enhancer [Citation409]. Given that genes of the 26S proteasome complex have been shown to be regulated by Nrf2 [Citation395], the observed quercetin-mediated proteasome activation could be Nrf2 mediated. Constant treatment of cells with quercetin leaded to enhanced cellular life span [Citation408] while similar life span-extending results were also observed upon feeding of C. elegans with quercetin [Citation410,Citation411] without, however, investigating a possible link with proteasome induction. On top of the above studies regarding the cross talk between the proteasome and Nrf2, it has also been shown that Nrf2 is activated by lipid peroxidation end products (e.g., 4-hydroxynonenal (HNE)) [Citation412], thus further linking the Nrf2 signaling pathway with the response to different types of modified proteins.
Cross-link breakers (GA)
Collagens and elastin are cross-linked in a tightly regulated manner by the copper-containing quinoprotein lysyl oxidase (enzymatic cross-linking), and these bonds provide mechanical strength and confer resistance against non-specific proteases [Citation413]. In contrast to this specific enzymatic oxidation, non-enzymatic cross-linking is not selective and may affect the structure and function of fibrillar collagen and extracellular matrix, causing vascular and myocardial stiffness: this leads to elevated systolic and pulse pressures, impaired ventricular relaxation, and diastolic dysfunction [Citation414]. Once formed, non-enzymatic cross-links are highly resistant to proteolysis degradation and they apparently inhibit activation of matrix metalloproteinase (MMP) thereby promoting collagen accumulation and fibrosis [Citation415]. Non-enzymatic cross-linking of collagen is mainly due to glycation and to other covalent modifications induced by RCS, forming irreversible AGE and ALE adducts, including glyoxal–lysine dimer (GOLD), methylglyoxal–lysine dimer (MOLD), glyoxal-derived imidazolium cross-link (GODIC), lysine-arginine cross-links derived from glucose (GLUCOSEPANE), and MG-derived imdazolium cross-link (MODIC)(). Of these, GLUCOSEPANE, GODIC, and MODIC have been recognized as being the most important in vitro and in vivo, although the molecular steps by which they arise are uncertain [Citation416–418]. Recently, the cross-linking mechanisms of GO and MG with lysine and arginine have been studied using a molecular modeling approach. One proposed route involves the formation of an aldimine (Schiff base) by reaction of GO or MGO with lysine, followed by reaction with the guanidine group on the side chain of an arginine residue. The reaction finishes by some rearrangement and proton transfer reactions that lead to the cross-linked product [Citation419].
Figure 7. Structures of Lys–Arg (panel A) and Lys–Lys cross-links (panel B).
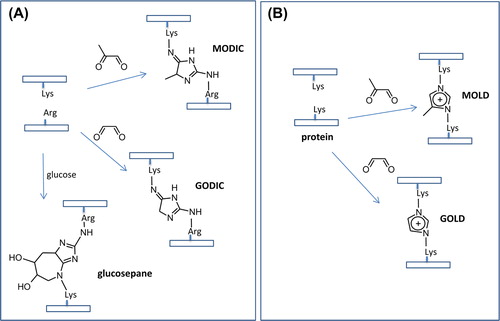
It is quite clear that inhibitors of AGEs/ALEs as reported in 2.1 cannot affect preexisting cross-linking. The first study on a rational design of AGEs breakers was published in 1996 by Vasan et al. [Citation420]. The strategy was based on the design of compounds able to covalently react and then cleave the α-dicarbonyl derivatives which were proposed as intermediates of AGEs formation. The prototype was identified as N-phenacylthiazolium (PTB), and the proposed reaction mechanism is reported in , consisting of the covalent adduction of the thiazolium ring to the dicarbonyl intermediate to form a condensed thiazolium intermediate which rearranges to regenerate the AGE breaker and leaving the CML moiety on a peptide fragment and the aldehydic function on the other peptide involved in the cross-link. Further studies on PTB were discontinued due to the chemical instability of the compound, undergoing a rapid hydrolysis at pH 7.4 (kHydrolysis was 2.6 ± 0.1 × 10− 4 s; half-life ca. 44 min), forming two isomeric 2,3-dihydro-4-formyl-2-hydroxy-2-phenyl-1,4-thiazines [Citation421]. Stabilization of the thiazolium ring was obtained by inserting two methyl groups, leading to the stable 4,5-dimethylthiazolium derivative and named ALT-711 or Alagebrium (N- phenacyl-4,5-dimethyl-1,3 thiazolium bromide) [Citation422]. ALT-711 represents the lead compound of this class and several studies have been thus far reported, showing the effects of ALT-711 in different animal models as well as in clinical trials in reversing the cardiovascular complications of aging and diabetes. ALT-711 was firstly reported by academic researchers and was then developed by Alteon Corporation which changed its name to Synvista Therapeutics, Inc. The company seems to have discontinued operations, and the clinical trials were terminated early in 2009 as reported in the clinicaltrial.gov web site due to financial constraints. The web site of the company is no longer available.
Figure 8. Structure of AGEs breakers (lower panel) and proposed reaction mechanism of thiazolium-derived AGEs breakers (upper panel). The simplified reaction mechanism is referred to the dinucleophilic attack of the thiazolium ring toward the dicarbonyl AGE cross-link followed by internal rearrangement and hydrolysis.
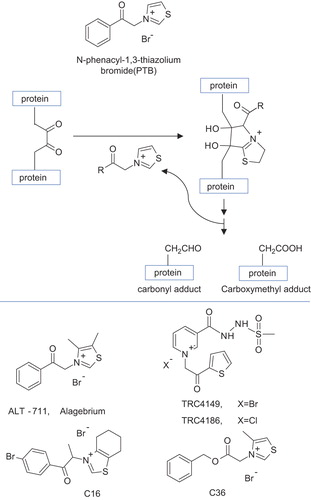
Belonging to the class of thiazolium AGE breakers, two derivatives coded C16 [Citation423] and C36 [Citation424], which were found to be as effective as ALT-711 in animal models, but exhibiting lower LD50 in mice and neither mutagenesis nor teratogenesis in ex vivo studies. More recently, a novel class of AGE breakers has been reported by Torrent Research Centre and characterized by a pyridinium (PMT) ring. TRC4186 and TRC4149 are the chloride and bromide salts, respectively, of the same novel active base, PMT, 3-[[2-(methylsulfonyl) hydrazino] carbonyl]-1-[2-oxo-2-2-thienyl) ethyl] (PMT) [Citation425]. The compounds have demonstrated AGE-breaking activities in in vitro experiments improving the endothelial and myocardial functions in animal models of diabetes mellitus with reduction of AGEs accumulation in tissues over time. A phase I clinical study of TRC4186 was carried out to evaluate the safety, tolerability, and pharmacokinetics of TRC4186 in healthy human subjects after single and multiple ascending doses, fixed doses in elderly male and female subjects, and with food and different formulations of the compound [Citation426].
While the AGE breakers have been found to exert beneficial effects and, in particular, to increase vascular elasticity, and to improve cardiovascular functions in aging humans, in animal models of diabetes, and in other diseases, several questions remain unanswered regarding the real mechanism of action, as recently pointed out by Nagai et al. [Citation427]. The AGE breakers have been designed to react with the dicarbonyl intermediates of AGEs but, as noted by Nagai et al. [Citation427], not a single cross-link structure identified in tissue proteins to date contains a dicarbonyl structure susceptible to the proposed mechanism of action. Indeed, the proposed target of AGE breakers, the dicarbonyl compounds, would be too reactive to accumulate.
In model systems, the AGE breakers were tested with phenylpropanedione (PPD), a model for putative dicarbonyl cross-links in tissue proteins, and the cleavage product, benzoic acid, was readily measured by reversed-phase HPLC. However, when the AGE breakers were applied to cross-linked proteins, isolated from diabetic animals, the AGE breakers were found to be ineffective and this is easily explained by considering that the well-recognized AGE cross-links detected in tissue proteins including pentosidine, crosslines, vesperlysines, fluorolink, GOLD, MOLD, and GLUCOSEPANE, which, based on their structures, would not be susceptible to cleavage by AGE breakers [Citation428]. Similar results were obtained by [Citation429], reporting that treatment with PMT did not reverse the cross-linking of rat skin and tail collagen, cross-linked by incubation with glucose or ribose in vitro.
The beneficial effect of the AGE beakers so far reported, which is not questionable and confirmed by several research groups, could be ascribed to alternative mechanisms as pointed out by Nagai et al. [Citation427]. For instance, PTB and ALT-711 have been reported to act as metal ion-chelating agents and their hydrolysis products have even stronger chelating properties than the intact compounds [Citation430]. Moreover, it should be noted that the AGE breakers designed to react with the dicarbonyl protein AGEs intermediates are also effective quenchers of MG [Citation431,Citation432] and possibly other dicarbonyl compounds which are well-recognized AGE precursors. In conclusion, although the AGE breaking mechanism is very promising since it would permit the recovery of oxidized proteins, the beneficial effects of the so-called AGE breakers are unlikely to be the result of cleavage or reversal of preexisting AGE cross-links. It is more likely that AGE breakers may have more direct effects on the formation of AGEs through their antioxidant and chelating effects and their reaction mechanism with MG and other dicarbonyl intermediates in the Maillard reaction.
AGE and ALE clearance (MS)
Lysozyme, an antimicrobial enzyme with the ability to mediate degradation of the peptidoglycan component of bacterial wall, was shown to bind AGEs with high selectivity and affinity [Citation433]. Lysozyme administration decreased circulating AGE levels and enhanced renal AGE excretion [Citation434]. The AGE-binding properties of lysozyme have been used to reduce AGE levels in the dialysate from diabetic patients with kidney disease [Citation435]. Oral administration of microencapsulated lysozyme decreased serum levels of AGEs and attenuated nephropathy in STZ diabetic rats [Citation436]. Overexpression of the native lysozyme in transgenic mice was found to protect against acute oxidative stress and to confer resistance to chronic oxidative stress against milder oxidants, such as AGEs, with a subsequent reduction in vascular cell inflammation and proliferation [Citation437,Citation438]. Under in vitro conditions, lysozyme was found to bind more efficiently to AGE-modified low-density lipoprotein (LDL), increased in type 2 diabetic patients, than to unmodified LDL [Citation439].
Inhibition of absorption of dietary AGEs may be a novel approach to reduce the deleterious effects of AGEs. AST-120 (Kremezin) is an oral adsorbent that attenuated the progression of chronic renal failure by removing uremic toxins, including AGEs [Citation440–442]. As reported by Hayashino et al., treating type 2 diabetic patients with AST-120 slowed the progression of the end-stage renal disease [Citation443]. Long-term treatment with AST-120 was found beneficial for chronic renal failure patients in the predialysis stage [Citation444].
Reducing exogenous exposure (MS)
AGEs are not only generated endogenously under hyperglycemic conditions but also derived exogenously from AGE rich meal. Diet is a major environmental source of AGEs with the highest yield in foods rich in carbohydrates and fats [Citation445]. Formation of AGE is enhanced by exposure to heat. According to Sgarbieri et al. and Koschinsky et al., an estimated 10% of AGEs ingested are absorbed into the body's circulation, and two-thirds of those absorbed are retained [Citation446,Citation447]. Numerous studies in animals and humans have confirmed the substantial toxic role of exogenous AGE derivatives in multiple target systems, as reviewed by Huebschman et al. [Citation448]. For example, mice on high AGE diet exhibited significant albuminuria and glomerular sclerosis, effects not seen in mice fed with low AGE diet [Citation449]. A clinical study by Uribarri et al. showed that restriction of dietary glycotoxins reduced excessive AGEs in renal failure patients [Citation445]. These observations suggest that restriction of food-derived AGEs or inhibition of absorption of dietary AGEs may also be a target for therapeutic anti-AGE strategy [Citation445,Citation448,Citation450–454].
Blockade of the ligand–RAGE axis (GB)
Studies of several last decades demonstrated that a significant part of oxidative stress in cells exposed to products of glycoxidation is due to the stimulation of special receptors present on cell surface, namely RAGE.
RAGE
This protein is a multiligand transmembrane receptor, which was originally identified as a receptor for the heterogeneous AGE [Citation455]. RAGE is expressed in several cells including endothelial, smooth muscle cells, and neurons, at a very low level in physiological conditions but at higher levels during embryonic development. This suggests that RAGE signaling may play an important role during development, although deletion of RAGE does not result in fertility disturbances or overt phenotypes excepting hyperactivity and increased sensitivity to auditory stimuli in mice [Citation455–457]. RAGE being also involved in tissue homeostasis and regeneration/repair upon acute injury, and in resolution of inflammation [Citation458,Citation459]. Conditions such as diabetes and atherosclerosis lead to overexpression of RAGE [Citation460]. Final effects of RAGE axis activation include endothelial dysfunction, activation of coagulation, and increase in the activity of the renin– angiotensin system [Citation461]. RAGE-overexpressing diabetic mice show progressive glomerulosclerosis with renal dysfunction, compared with diabetic littermates lacking the RAGE transgene. Diabetic homozygous RAGE null mice fail to develop significantly increased mesangial matrix expansion or thickening of the glomerular basement membrane. Taken together, these findings suggest that the activation of AGE–RAGE axis contributes to the expression of VEGF and enhanced attraction/activation of inflammatory cells in the diabetic glomerulus, thereby setting the stage for mesangial activation and TGF-beta production [Citation462]. RAGE has also a pro-adipogenic function in senescent preadipocytes by suppressing p53 function, which may contribute to aging-dependent adiposity by glycated proteins derived from the diet and/or chronically dysregulated metabolic conditions such as hyperglycemia [Citation463].
RAGE ligands fall into several distinct families. They include the high-mobility group family proteins including the prototypic HMGB1/amphoterin, members of the S100/calgranulin protein family, matrix proteins such as collagens I and IV, amyloid β peptide, some AGEs such as CML, and phosphatidylserine. The S100/calgranulin family consists of closely related calcium-binding polypeptides, which act as proinflammatory extracellular cytokines. Not all members of these families have been identified as RAGE ligands, and many RAGE ligands have a variety of RAGE-independent effects. AGEs, which represent the first RAGE ligand discovered in the 90s, are prevalent in pathological conditions marked by oxidative stress, generation of methoxyl species, and contribute to the increase in blood sugar level, as found in type 2 diabetes mellitus [Citation456,Citation458,Citation460].
RAGE has been described as a pattern recognition receptor, able to recognize a common pattern within its different ligands. RAGE ligands do not seem to share much similarity. Nevertheless, all have a net negative charge at neutral pH: S100 proteins are very acidic, AGE-modified proteins accumulate negative surface charge during their transformation by glycation and oxidation, amyloid β-peptide has a net negative charge, and amyloid fibrils expose a regular pattern of negative charges on their surface, while HMGB1 contains a highly acidic domain. Phosphatidylserine is also negatively charged at physiological pH. The second common feature is the tendency of most RAGE ligands to oligomerize [Citation464]. The interactions contributing to this effect include also hydrophobic regions of the not charged and lipophilic AGEs. AGEs are negatively charged as the result of the Maillard formation reaction, by which reducing sugars or aldehydes modify arginines and lysines to produce glycated proteins, adding a net negative charge on the proteins at physiological pH. AGE binding to the V region of RAGE is initially facilitated by the ionic attraction between the positive charges of RAGE and the negative charges of AGE. The binding complex is then stabilized with hydrophobic interaction after conformational changes. It is not still well understood, which among the different AGEs products so far identified act as a binder of RAGE, which is the order or affinity, nor whether ALEs, such as HNE or MDA protein adducts also bind RAGEs. In fact, with the exception of CML and CEL generated in a peptide or protein environment, most of the studies so far reported on RAGE activation by AGEs are based on not structurally resolved AGEs which are usually generated by incubating proteins (very often albumin) with glucose or fructose or with GO/MG resulting in the formation of a different set of glycoxidation products, spanning from either early glycation to cross-linking products, depending on the incubation times [Citation465].
RAGE consists of an extracellular region, which is composed of one ligand-binding V-type Ig domain and two Ig-like C-domains (C1 and C2), a short hydrophobic transmembrane-spanning region, and a signal-transducing cytoplasmic domain. The V and C1 domains form an integrated structural unit (VC1), which is involved in the ligand recognition. The V domain is characterized by at least two distinct regions involved in the ligand engagement. In particular, X-ray and NMR studies have revealed the presence of a large hydrophobic region together with a positively charged region on the surface of the V- domain. The former is involved in the hydrophobic interactions, and the latter in the electrostatic bindings, as those involved with the negative charged sites of the ligands. As a matter of fact, the exact molecular interactions of RAGE with its ligands are still far from being elucidated, and this is particularly true in the case of AGEs. The lack of information on AGE–RAGE interaction is in part due to the fact that AGEs are an heterogeneous class of oxidized products where the chemical variability is not only referred to the oxidized moiety such as CML, CEL, and methylglyoxal-hydroimidazolone (MGH-1), but also to the surrounding chemical environment, which is also required for the RAGE interaction. In fact, it has been found that CML and CEL do not bind RAGE as such, but only when they are embedded in a peptide or protein, suggesting that both the oxidized moiety and the surrounding backbone are required for RAGE interaction. Hence, the oxidized moiety of AGEs represents a necessary but not sufficient condition for the RAGE recognition [Citation459,Citation464].
The ligand–RAGE engagement induces the activation of multiple signaling pathways that may vary depending on the nature of the ligand and on the cell and tissue microenvironment, thus mediating different cellular responses. For example, the RAGE activation by AGEs induces NADPH oxidase activation leading to ROS formation which on the one hand further propagates the oxidative damage and hence the AGEs production. Another effect is the NFκB activation which promotes an increased gene transcription of pro-inflammatory and pro-fibrotic cytokines and chemokines leading to an inflammatory condition [Citation455]. The diversity of signaling cascades identified in RAGE-mediated cellular signaling implies that different RAGE ligands might induce different pathways, especially in different cell types [Citation466]. It has been hypothesized that the specific signaling pathways activated by RAGE ligands are greatly dependent on the duration of ligand stimulation and the specific functions of each cell type [Citation455]. For example, RAGE ligands result in increased serine 9 phosphorylation of GSK-3β in smooth muscle cells, a process which causes increased smooth muscle cell migration [Citation467]. On the other hand, in primary murine adult cardiomyocytes, RAGE ligands mediate decreased phosphorylation of serine 9 GSK-3β, which is linked to increased activation of cell death pathways in these cells [Citation468]. These data suggest that the actions of RAGE ligands are highly dependent on the type, properties, stress responses, and fate of the impacted cells.
RAGE activation is known to be involved in the pathogenetic mechanism of some oxidative-based diseases including diabetes, atherosclerosis, AD, and chronic airway diseases. Based on these premises, the AGE–RAGE damaging axis is now considered as a promising drug target and hence the compounds acting by inhibiting the RAGE-mediated signal have been found able to exert a beneficial effect in various pathologies. The main molecular approaches used to inhibit the RAGE activation can be grouped into the following classes: (i) inactivating ligands or using RAGE-truncated and soluble forms able to bind the circulating RAGE agonists; (ii) inactivating RAGE, for example, through antibodies or by use of RAGE antagonists, blocking the AGE/RAGE damaging axis; (iii) downregulating RAGE expression, and (iv) inhibiting the signal transduction pathway that follows ligand–RAGE interaction.
Inactivating ligand
Apart from full-length RAGE present in the plasma membrane (fRAGE), there exist a population of shortened RAGE (sRAGE) circulating in blood and body fluids, which lack the transmembrane domain. sRAGE comprises a heterogeneous population that includes an endogenous secretory isoform generated via alternative splicing (endogenous secretory receptor for AGEs, esRAGE) and a form generated through proteolytic cleavage of the full-length cell surface receptor by the action of membrane-associated matrix MMPs such as a disintegrin and MMP10 and MMP9 (). The sRAGE may be subject to further proteolytic cleavage, generating miniRAGE comprising only the V and C1 domains; miniRAGE is not subjected to further degradation and is still able to bind ligands due to the presence of the V domain. It has an even higher sensitivity to AGEs compared with that of the full-length sRAGE [Citation469]. Circulating sRAGEs are detectable in human plasma and are able to bind ligands, competing with membrane-bound RAGE. However, sRAGEs lack the intracellular signaling domain, and therefore, its engagement with ligands does not trigger signal transduction so, competing for the ligands of RAGE, it can have cytoprotective effects [Citation470,Citation471]. The balance between the levels of RAGE ligands, cell surface RAGE, and sRAGE/esRAGE represents, thus represent a complex dynamic system, which state is influenced by diseases and drugs, and which may be of diagnostic importance.
Figure 9. The structure of full-length RAGE and its variants. The V-type domain is critical for binding of RAGE–ligand axis. Deletion of this domain results in an N-truncated form that does not bind ligands. The C-truncated, circulating soluble RAGE contains only the extracellular domain of the receptor. It may be a result of alternate endogenous splicing (esRAGE) or proteolytic cleavage.
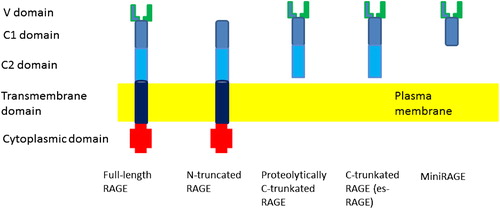
In liver pathology, the expression of RAGE and its ligands (AGEs, CML, and HMGB1) increases, whereas the expression of sRAGE decreases. A negative feedback has been shown to exist for sRAGE when RAGE interacts with its ligands [Citation470]. Higher circulating levels of sRAGE are associated with reduced risks of coronary artery disease, hypertension, metabolic syndrome, and other chronic diseases. In a colonoscopy-based case–control study, the associations between plasma levels of sRAGE, sTNF-αRI, sTNF-αRII, sIL-6R, EGF, IFNα2, G-CSF, MCP1, TNFβ, and VEGF with the risk of colorectal adenoma were estimated. An inverse association between sRAGE and colorectal adenoma was found among those without hypertension [Citation471]. In Italian male subjects without diabetes, lower levels of plasma esRAGE were associated with enhanced risk of angiographically detected coronary artery disease [Citation472]. In type 1 diabetic patients, circulating esRAGE levels were significantly lower than in non- diabetic subjects and were inversely associated with the severity of some diabetic vascular complications [Citation473]. Low circulating esRAGE levels were found to be predictive for cardiovascular mortality in both non-diabetic and diabetic patients with end-stage renal diseases [Citation473,Citation474]. It should be noted, however, that another authors found higher serum sRAGE levels in type 2 diabetic patients than in non-diabetic subjects, positively associated with the presence of coronary artery disease [Citation475].
18F-Fluorodeoxyglucose positron emission tomography (FDG-PET) is a novel imaging technique for detecting vascular inflammation. Vascular 18F-FDG uptake was measured as the blood-normalized standardized uptake value (SUV), known as the target-to-background ratio (TBR). Circulating sRAGE showed a significant association with TBR values measured using FDG-PET, which reflect vascular inflammation [Citation476]. A recent study suggested that sRAGE may be one of the biomarkers diagnosing post-stroke cognitive impairment [Citation477]. Decreased levels of sRAGE in patients with acute Kawasaki disease were found, suggesting the potential anti-inflammatory effect of sRAGE in inflammatory vascular disorders [Citation478]. The level of circulating sRAGE decreases in rats after middle cerebral artery occlusion and may be a peripheral biomarker of focal ischemia [Citation479]. sRAGE may reflect some peripheral plasma features of the pathophysiological process of AD. Decreased plasma levels of sRAGE in patients with AD have been documented. Comparison of the plasma levels of sRAGE in patients with AD, vascular dementia (VaD), non-AD neurodegenerative dementias (NND), and cognitively normal controls (NC) showed a significantly lower concentration of plasma sRAGE in the group with AD compared with those in the VaD or NC group [Citation480]. These examples demonstrate that the level of sRAGE may be a useful biomarker of diseases.
sRAGE has indeed potent anti-inflammatory properties by acting as a decoy for RAGE ligands. sRAGE binds with high affinity to atherogenic low-density lipoprotein (LDL) modified by hypochlorous acid (HOCl), the major oxidant generated by the myeloperoxidase-H2O2-chloride system of phagocytes activated during inflammation, acting as a sink for HOCl-LDL, which is abundantly present in human atherosclerotic lesions. sRAGE can be coprecipitated with HOCl-LDL from spiked serum. It has been proposed that sRAGE represents a physiological antagonist that interferes with scavenger receptor-mediated cholesterol accumulation and foam cell formation of macrophages [Citation481]. A significant inverse correlation between circulating levels of sRAGE and oxLDL has, indeed, been found, which suggests that part of the anti-atherosclerotic effects of sRAGE may be related to oxLDL quenching [Citation482].
The endogenous phospholipid lysophosphatidic acid (LPA) regulates fundamental cellular processes such as proliferation, survival, motility, and invasion implicated in homeostatic and pathological conditions. In vitro, RAGE was found to be required for LPA-mediated signal transduction in vascular smooth muscle cells and C6 glioma cells, proliferation, and migration. In vivo, the administration of soluble RAGE mitigated LPA-stimulated vascular Akt signaling, autotaxin/LPA-driven phosphorylation of Akt and cyclin D1 in the mammary tissue of transgenic mice vulnerable to carcinogenesis, and ovarian tumor implantation and development. A novel role for RAGE as a conduit for LPA signaling has been identified and targeting LPA-RAGE interaction has been suggested as a therapeutic strategy to modify the pathological actions of LPA [Citation483].
The protective role of sRAGE prompted interest in studies on how various drugs affect the level of circulating sRAGE. Interestingly, traditional cardiovascular drugs (statins, thiazolidinediones, angiotensin-converting enzyme (ACE) inhibitors, and angiotension AT-1 receptor antagonists) as well as nutraceuticals (grape seed proanthocyanidin extract) can modulate RAGE expression and circulating sRAGE levels in cardiovascular disease states characterized by enhanced RAGE activation, which may represent a novel mechanism of their action [Citation484].
Introduction of recombinant sRAGE may be expected to become a new way of therapy to prevent the activation of RAGE by proinflammatory ligands. In experiments on acetaminophen-induced liver injury, animals treated with sRAGE displayed increased survival compared with vehicle treatment, and markedly decreased hepatic necrosis. Consistent with an important role for RAGE-triggered oxidant stress in hepatic injury, a significant reduction of protein nitrotyrosine was observed in hepatic tissue in sRAGE-treated versus vehicle-treated mice receiving acetaminophen, in parallel with significantly increased level of GSH. In addition, pro-regenerative cytokines tumor necrosis factor-alpha and interleukin-6 were increased in sRAGE-treated versus vehicle-treated mice [Citation485]. RAGE blockade by genetically engineered sRAGE was found to reduce vascular complications in a diabetic animal model [Citation486,Citation487].
Inactivating RAGE
Another therapeutical approach consists in inactivating RAGE. The receptor can be inactivated by high- molecular-weight substrate analogs, low-molecular-weight inhibitors, or anti-RAGE antibodies, neutralizing the receptor. A set of compounds have been proposed as RAGE inhibitors and tested in animal models.
Low-molecular-weight antagonists of RAGE
Probably due to the complexity of the molecular interactions between AGEs-RAGE, only few studies on AGE antagonists have so far been reported. Most of the studies on the rational design of RAGE antagonists have been carried out by considering amyloid β-peptide (Aβ) as a ligand [Citation488]. Also in this case, the molecular interaction between Aβ and RAGE is not completely elucidated: by using fluorescence and MS, it has been found that the major V-RAGE binding region of Aβ involves a highly hydrophobic stretch, 17LVFFAED21 and a flanked negatively charged residue D22E23 at the C terminus. The interest of Aβ as RAGE ligand is due to the fact that the Aβ-RAGE axis is involved in the AD and represents an emerging drug target. In particular, in brain endothelium, RAGE mediates the influx of circulating Aβ into the brain while in neurons, it mediates Aβ-induced oxidant stress and Aβ intraneuronal transport, causing mitochondrial dysfunction. Aβ–RAGE interaction also activates nuclear factor-κB (NF-κB), which plays a crucial role in various inflammatory responses.
Zlokovic et al. reported the structure of RAGE antagonist with a Kd in a nanomolar range. The lead compounds from the first screening shared common structural moieties such as a tertiary amide moiety substituted with a hydrophobic moiety and a monosubstituted aromatic group. A second-generation library of compounds was then synthesized on the basis of the chemical features retrieved from the first library. From the second library, a compound coded as FPS-ZM1 was identified and characterized by a favorable inhibitory and dissociation constants (Ki/Kd = 0.66) as well as suitable pK and bioavailability properties. Structure–activity relationship studies revealed that the tertiary amide and aromatic rings are essential for activity and that the chlorine atom on the electron poor aromatic group may enhance its binding to the receptor [Citation489]. Besides in vitro studies, FPS-ZM1 was also found effective in a mouse model of AD inhibiting the RAGE-mediated influx of circulating Aβ40 and Aβ42 into the brain, β-secretase activity, Aβ production and microglia activation and neuroinflammation, and normalizing cognitive performance and cerebral blood flow responses [Citation488].
PF-04494700 (previously known as TTP488), an orally bioavailable low-molecular-weight inhibitor of RAGE, which inhibits sRAGE from binding to RAGE ligands, S100B, amphoterin, and CML has been developed as a potential treatment for AD (as well as diabetic nephropathy) and has reached a phase II clinical trial [Citation490] but was discontinued in 2011 due to a poor efficacy. Feng et al. demonstrated that CM-1, a cell protecting agent able to prevent AGE-induced endothelial dysfunction, blocks the interaction of MGH-1 with RAGE by acting as a competitive high-affinity antagonist, with IC50 values in the nanomolar range [Citation491]. A screen of 50 compounds for their ability to prevent binding to RAGE of a fluorescent AGE-BSA derivative in RAGE-overexpressing cells demonstrated that genistein inhibits this binding in a dose-dependent manner at nM concentrations [Citation492]. Thiazolidinediones, calcium channel blockers, angiotensin-converting enzyme inhibitors, angiotensin II receptor blockers, and statins are reported to suppress RAGE expression [Citation493].
There is no essential difference in the binding with RAGE between the macromolecular and monomeric AGE. Monomeric AGEs, which generally consist of an N-heterocyclic moiety and an amino acid moiety as a part of the AGE− protein linkage are thus attractive as a template for the design of novel small-molecule RAGE antagonists. In particular, argpyrimidine 1, a monomeric AGE, was selected for design of the incipient RAGE antagonists, because the key structure of argpyrimidine seems to provide more synthetic accessibility and more drug-like features compared with that of other monomeric AGEs. Using the approach of ligand-based drug design, the group of Suh discovered a novel series of 4,6-disubstituted 2-aminopyrimidines as RAGE antagonists. In transgenic mouse models of AD, one of the 4,6-bis(4-chlorophenyl)pyrimidine analogs (59) significantly lowered the concentration of toxic soluble Aβ in the brain and improved cognitive function. 59 binds directly to RAGE and thus inhibits the RAGE–Aβ interaction. A docking study predicted also the binding mode of the 4,6-bis(4-chlorophenyl)pyrimidine analogs to the RAGE V-domain [Citation459].
Peptides and polysaccharides
A study of interaction of a series of truncated versions of Aβ with RAGE demonstrated that such peptides may also be RAGE antagonists, preventing binding of true Aβ [Citation494]. It has not yet been demonstrated whether a RAGE antagonist designed and tested by considering Aβ as ligand is also effective as inhibitor of the AGE–RAGE interaction; however, literature data suggest that it may be the case.
Hearst et al. reported that the newly designed S100B inhibitory peptide, Synb1-ELP-TRTK, has a capacity to block the RAGE-mediated uptake of S100B in neuroblastoma cells, what makes it a promising agent against cerebellar neurodegenerative disorders [Citation495]. Systemic administration of another RAGE antagonist, a S100P- derived peptide, reduced the growth and the metastasis of pancreatic tumors and also inhibited glioma tumor growth in animal models [Citation496].
Heparin and its derivatives, 2-O,3-O-desulfated heparin (ODSH), also block ligation of RAGE with a number of its important ligands, including the AGE-product CML-bovine serum albumin, HMGB-1, and S100 calgranulins. Heparin inhibits many proteins through electrostatic charge interactions with cationic amino acids. Electrostatic charge interactions may also play an important role in ligand–RAGE binding. Heparin and non-anticoagulant heparins such as ODSH might be expected to disrupt these interactions by competing with AGEs for binding to the cationic V region. Because heparin and ODSH are large, long polymers, their electrostatic attachment to the V domain of RAGE might also cause steric hindrance for S100/calgranulin binding to adjacent C1 and C2 domains. Low-molecular-weight heparin (LMWH) also can bind to RAGE and act as an antagonist to RAGE. LMWH treatment of mice showed preventive and therapeutic effects on albuminuria and increased glomerular cell number, mesangial expansion, and advanced glomerulosclerosis in a dose-dependent manner [Citation497].
Antibodies
The most common approach to the prevention of deleterious outcomes of RAGE activation in in vitro system and animal models is the application of inactivating monoclonal or polyclonal antibodies against RAGE. Such antibodies were found to prevent the NADPH oxidase activation induced by RAGE ligand binding, subsequent oxidative stress in the cells (generation of reactive oxygen and nitrogen species, increase in MDA and protein carbonyl content) [Citation498] and various indirect sequelae of this activation. For example, application of anti-RAGE antibodies attenuated AGE-induced increased expression of KCa3.1 channels in cardiac fibroblasts [Citation499], activation of the renin- angiotensin system through the RAGE/phosphoinositide 3-kinase signaling pathway [Citation500], pulmonary metastasis of tumor cells [Citation501], delayed-type hypersensitivity reactions, collagen-induced arthritis, experimental autoimmune encephalomyelitis, and alloimmunity [Citation502].
Pretreatment with endogenous anti-RAGE antibodies isolated from transgenic APPSWE-PS1 mice expressing human presenilin 1 (A246E variant) and a chimeric amyloid precursor protein prevented Aβ1-42-induced neurotoxicity in cultured primary rat cortical neurons [Citation503]. HMGB1, like AGE, reduces collagen synthesis by fibroblasts; this effect was abrogated by co-treatment with an anti-RAGE antibody [Citation504].
One of the antibodies against RAGE, designated XT-M4, a rat-derived anti-mouse RAGE monoclonal antibody, binds the extracellular region of RAGE and inhibits the interaction of RAGE with multiple ligands. XT-M4 has a broad species cross-reactivity, a binding affinity of 0.3 nM for murine dimeric RAGE and was protective in the mouse cecal ligation and puncture-induced (CLP) model of sepsis. The humanized XT-M4 antibody maintains all the inhibitory and binding properties of the parental rat XT-M4 antibody and also showed efficacy in the mouse model of pneumococcal pneumonia [Citation505]. Administration of humanized anti-RAGE monoclonal antibody after intratracheal infection with Streptococcus pneumoniae decreased mortality in mice and modulated expression of genes, especially those involved in the inflammatory response [Citation506]. In vitro, anti-RAGE antibody protected podocytes against AGE-induced dysfunction [Citation507].
Downregulation of RAGE expression
There is a considerable interest in the possibility of downregulation of RAGE expression by drugs and natural substances. For some drugs, this mechanism, interestingly, may represent a secondary mechanism of their action.
Peroxisome proliferator-activated receptor gamma (PPAR-γ), a nuclear receptor, is highly expressed in all cells involved in vascular pathologies, including macrophages, endothelial cells, and smooth muscle cells [Citation508]. PPAR-γ synthetic ligands such as pioglitazone and rosiglitazone improve insulin resistance in diabetic patients, and now become one of the most popular anti-diabetic drugs in the developed countries [Citation509]. Accumulating data indicate that PPAR-γ agonists can downregulate the expression of RAGE both in vitro and in vivo [Citation508]. This effect has been demonstrated for several of the thiazolidinedione class of insulin-sensitizing PPAR-γ agonists including pioglitazone [Citation510,Citation511] and rosiglitazone [Citation512]. RAGE activation-induced inflammation promotes aortic valve calcification in hypercholesterolemic rabbits, which can be attenuated by pioglitazone treatment [Citation508]. Nifedipine, a calcium-channel blocker, also suppresses RAGE expression through PPAR-γ activation [Citation513].
An anti-diabetic drug commonly used in the treatment of Type II diabetes, metformin, downregulates RAGE expression, which may be another facet of its action [Citation514]. Metformin inhibits the AGEs-induced growth and VEGF expression in MCF-7 breast cancer cells by suppressing RAGE gene expression via AMP-activated protein kinase pathway. Due to this effect, metformin may protect against breast cancer expansion in diabetic patients by blocking the AGE–RAGE axis [Citation514]. Irbesartan, an angiotensin II type 1 receptor antagonist used mainly for the treatment of hypertension, was also found to downregulate RAGE expression [Citation515]. The combination therapy with metformin and irbesartan may have therapeutic potential in diabetic nephropathy. It can play a protective role against tubular injury in diabetes not only by inhibiting AGEs formation, but also by attenuating the deleterious effects of AGEs via downregulating RAGE expression [Citation516]. Downregulation of RAGE expression was observed for other angiotensin II type 1 receptor blockers, telmisartan [Citation517], and olmesartan [Citation518], too, and ascribed mainly to the activation of PPAR-γ [Citation517].
Vardenafil, an inhibitor of phosphodiesterase 5, can block the AGE-induced upregulation of monocyte chemoattractant protein-1 (MCP-1) mRNA levels in human umbilical vein endothelial cells (HUVECs) by suppressing RAGE expression via the elevation of cGMP [Citation519]. RAGE downregulation was also noted for glucagon-like peptide-1 [Citation520] and pigment epithelium-derived factor [Citation521]. Another class of drugs used to lower cholesterol levels, statins (e.g., pravastatin), known to ameliorate renal function and reduce proteinuria in patients with chronic kidney disease, decreases RAGE expression [Citation522]. It was also demonstrated that pentoxifylline (PTX), a non-selective phosphodiesterase inhibitor, may show protective effects on hepatic and arterial functions, partially through the inhibition of RAGE expression [Citation523].
As mentioned above, many natural compounds can also downregulate RAGE expression. RAGE downregulation was reported, that is, for resveratrol [Citation524] and for various plant extracts, of complex composition. Curcumin downregulated RAGE expression in rats with experimental diabetes [Citation525]. Paeoniflorin, isolated from the root of Paeonia, reported to exert an anti-inflammatory effect, downregulates RAGE expression in HUVECs [Citation526]. Puerarin, a major isoflavonoid derived from the Chinese medical herb Radix puerariae (kudzu root), decreases or inhibits RAGE expression in retinas of STZ-induced early diabetic rats [Citation527]. Pinocembrin, a flavonoid abundant in propolis, inhibits upregulation of RAGE in an animal model of AD [Citation528]. Ursolic acid, a pentacyclic triterpene acid present in many plants, including apples (especially apple peels), basil, bilberries, cranberries, elder flower, peppermint, rosemary, lavender, oregano, thyme, hawthorn, and prunes, also downregulates RAGE expression [Citation529]. Administration of extract of Salvia miltiorrhiza to early diabetic rats decreased the expression of RAGE in the kidneys [Citation530]. In a similar model, increased RAGE expression in the aorta was attenuated by a Hibiscus sabdariffa polyphenolic extract [Citation531]. Xu et al. [Citation532] evaluated the potential pathological role of RAGE and nuclear factor-kappa BP65 in diabetic encephalopathy. The results obtained by these authors indicated that grape seed proanthocyanidin extracts decrease the expression of RAGE and suggest that grape seed proanthocyanidin extracts might be a useful remedy in the treatment of diabetic encephalopathy [Citation532]. RAGE downregulation was also demonstrated for extract from the Japanese apricot, MK615 [Citation533], methanol extract of Lycium barbarum (Goji Berry) as well as its main component, taurine [Citation534]. Extract of Ginkgo biloba, a traditional Chinese medicine (EGb761), found to protect brain from hypoxic damage and ROS generation and considered as a therapeutic agent against AD, was demonstrated to downregulate RAGE expression in immortalized mouse endothelial cells [Citation525,Citation535]. Aqueous extract of Rehmannia glutinosa, a plant traditionally prescribed in Korean medicine to reduce fever and regulate immunity, suppresses inflammation and RAGE expression [Citation536]. Recently, retinol was documented to decrease RAGE levels, suggesting that RAGE downregulation may exert a role in the deleterious effects observed in some retinol supplementation therapies, especially in the lungs [Citation537]. Selenium supplementation downregulated the expressions of both NFkB and RAGE is rats with STS-induced diabetes [Citation538]. Oral administration of fish oil with high levels of (n-3 PUFAs) following small bowel transplantation significantly reduced the HMGB1 and RAGE expression, which coincided with the amelioration of chronic allograft vasculopathy. In contrast, feeding of corn oil that contained low levels of n-3 PUFAs had no favorable effects on chronic allograft vasculopathy development and failed to decrease the HMGB1 and RAGE expression [Citation539].
The development of nanomedicine brings new possibilities of modulation of RAGE expression. It was reported that combination of 3- to 5-nm gold nanoparticles, epigallocatechin gallate, and α-lipoic acid prevents upregulation of RAGE expression in diabetic wound [Citation540].
Inhibiting the signal transduction pathway that follows ligand-RAGE interaction
Reports from multiple laboratories indicate that ligand–RAGE interaction stimulates signal transduction and that such signaling is essential for RAGE-dependent modulation of gene expression and fundamental cellular properties [Citation455]. RAGE activation results in the activation of Ras-extracellular signal-regulated kinase 1/2 (ERK1/2), p21ras, Cdc42/Rac, stress-activated protein kinase/c-Jun- NH2-terminal kinase (SAPK/JNK) and p38 MAP kinases (p38 MAPK), phosphoinositol-3 kinase and rhoGTPases, eventually impinging on canonical NF-κB, AP-1, CREB, STAT3, NFAT signaling pathways, and myogenin with consequent changes in the transcription of an array of genes coding for cytokines, inflammatory enzymes, chemokines, chemokine receptors, adhesion molecules, matrix MMPs, and/or cytoskeletal constituents which affect cell proliferation, survival, differentiation and migration, phagocytosis and authophagy [Citation458,Citation541] ().
Figure 10. Intracellular effects of AGEs mediated by RAGE.
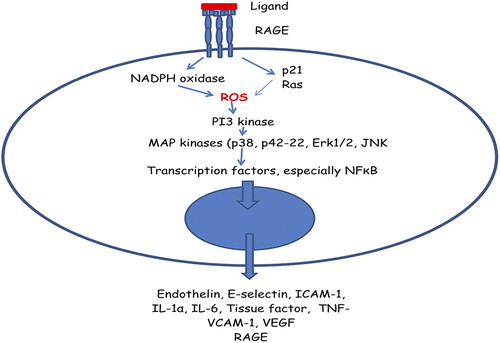
NF-κB-DNA binding is associated with the expression of an altered cellular phenotype including the expression of adhesion molecules for circulating inflammatory cells such as VCAM-1 and ICAM-1, monocyte chemoattractant protein-1 (MCP-1), plasminogen activator inhibitor-1 (PAI-1), tissue factor, vascular endothelial growth factor, endothelin-1, E-selectin, thrombomodulin, and proinflammatory cytokines, including interleukin (IL)-1α, IL-6, tumor necrosis factor-α and RAGE itself [Citation541–543]. These cytokines and adhesion molecules promote an inflammatory response with associated cellular migration and proliferation and play have central roles in both inflammation and atherosclerosis [Citation544]. What is important, ligation of RAGE causes a positive feed-forward loop, in which inflammatory stimuli activate NF-κB, which in turn induces RAGE expression, followed again by NF-κB activation [Citation493,Citation545].
The cytoplasmic domain of RAGE is phosphorylated at Ser391 by PKCζ upon binding of ligands. TIRAP and MyD88, which are known to be adaptor proteins for Toll-like receptor-2 and Toll-like receptor-4 (TLR2/4), bound to the phosphorylated RAGE and transduced a signal to downstream molecules. Blocking of the function of TIRAP and MyD88 largely abrogated intracellular signaling from ligand-activated RAGE. These findings indicate that functional interaction between RAGE and TLRs coordinately regulates inflammation, immune response, and other cellular functions [Citation546].
AGE–RAGE-mediated ROS generation activates TGF-beta–Smad signaling and subsequently induces mesangial cell hypertrophy and fibronectin synthesis by autocrine production of angiotensin II. This pathway may provide an important link between the AGE–RAGE axis and the RAS in promoting the development and progression of CKD [Citation547].
Recently, mDia1 (a mammalian homologue of Drosophila gene Diaphanous 1) has been identified as a direct binding partner of an intracellular domain of RAGE and as a part of the machinery of RAGE intracellular signaling. RAGE binds the FH1 domain of mDia1 at least in part through the interaction of R5/Q6 amino acids of the human RAGE cytoplasmic tail (ctRAGE) [Citation455,Citation483]. ctRAGE contains an unusual α-turn that mediates the mDia1–ctRAGE interaction and is required for RAGE-dependent signaling. mDia1 is required for AGE-mediated upregulation of Egr-1 in macrophages exposed to hypoxia [Citation548]; it is required for RAGE ligand (AGE and S100B)-stimulated migration; activation of Rac-1 and Cdc42 in transformed cells [Citation549]; S100B-mediated activation of Rac1, Akt and GSK-3β; generation of oxidative stress; and cellular migration in primary murine aortic smooth muscle cells [Citation467]. Experiments are underway to probe the impact of mDia1 in murine models of diabetic complications. If these studies identify that mDia1 is essential for macro- and microvascular complications, such data may highlight novel areas for therapeutic intervention in diabetes. In addition, diaphanous 1 recruits Rac1 and Cdc42 in glioma cells, thereby driving RAGE-dependent cell migration. Also, following RAGE ligation by S100B in microglia, mDia1 recruits Rac1/Cdc42 with ensuing activation of a RhoA/ROCK and a Src kinase/Ras/PI3K/RhoA/ROCK pathway governing microglial motility and a JNK/AP-1 pathway responsible for chemokine expression [Citation458].
Extensive literature analysis identified 95 molecules involved in the AGE/RAGE axis signaling. The map of these interactions is available freely in NetSlim at http://www.netpath.org/netslim/age_signaling_pathways.html [Citation550].
One of the best documented consequences of RAGE activation is the generation of ROS. ROS are produced by upregulation of subunits of NADPH oxidase, including p47phox (11), nox-4 (12), and p22phox, and activation of this superoxide-producing enzyme [Citation551,Citation552]. AGEs increase the expression of both the mRNA and protein levels of NADPH oxidase p47phox that are in turn blocked by anti-RAGE antibodies, confirming that NADPH oxidase is a downstream target of RAGE in AGE-induced ROS pathway in SH-SY5Y cells and rat cortical neurons. AGE-induced oxidative stress contributes to ER stress, and the cell death induced by ER stress is involved by C/EBP homologous protein (CHOP) and caspase-12. Inhibiting both AGE–RAGE–NADPH-ROS scavenging pathways and ER stress can lead to the prevention of cell death. The signaling pathway involved during AGE-induced apoptosis identified in the current study suggested that targeting RAGE, NADPH oxidase, ROS scavenging, or ER stress may also be a potential novel approach for the prevention of RAGE-induced neurodegeneration [Citation553].
The primary ROS generated by NADPH oxidase is superoxide but it dismutates to hydrogen peroxide in a reaction catalyzed by SOD or occurring spontaneously (albeit at a much lower rate). Superoxide may react with nitric oxide to form peroxynitrite while hydrogen peroxide may produce hypochloric acid/hypochlorite in a reaction catalyzed by myeloperoxidase. Thus, RAGE activation produces a set of ROS damaging constituents of cells and of extracellular matrix. Although produced mainly for the sake of signaling, these ROS contribute significantly to oxidative stress occurring in diabetes and other diseases. Sustained NADPH oxidase activation compromises several anti-oxidant systems by depleting NADPH [Citation554].
Blockade of the RAGE downstream signaling may be a promising target for therapeutic intervention in diabetic vascular complication, apart from downregulation of RAGE expression. Various attempts in this direction have been made. In many cases, it is hard to decide whether the observed effect is due to downregulation of RAGE expression, inhibition of downstream signaling, or both. One obvious target intervention is the ROS production which may be affected by NADPH oxidase inhibitors; otherwise, antioxidants may attenuate the RAGE-initiated signaling by reacting with ROS. However, ROS generation occurs also at the earlier stage of glycoxidation so the interpretation of the effect of antioxidants is not simple. RAGE overexpression was found to inhibit osteoblast proliferation in vitro via suppression of Wnt, PI3K, and ERK signaling. Prevention of Wnt signaling using Sfrp1 or DKK1 rescues RAGE-decreased PI3K and ERK signaling and cell proliferation [Citation508].
AGEs induce proliferation of vascular smooth muscle cells, which involves autophagy. Treatment with AGEs activates ERK, JNK, and p38/MAPK, but inhibits Akt. Pretreatment with an ERK inhibitor and an Akt activator inhibits AGE-induced autophagy [Citation555]. Similarly, excessive production of matrix MMP IX by HaCaT keratinocytes dependent on RAGE activation is attenuated by inhibitors of downstream elements of the RAGE signaling pathway: ERK1/2 (U0126), p38 mitogen-activated protein kinase (SB203580), and NF-κB [Citation556].
Treatment of neutrophils with AGEs increases generation of superoxide and nitric oxide by NADPH oxidase and iNOS, respectively, which is inhibited by diphenyleneiodonium, an inhibitor of both enzymes, hexamethylphosphoric triamide (HMPA), inhibitor of NADPH oxidase, or anti-RAGE antibodies [Citation542,Citation557], although other classes of NOS, especially eNOS, are rather attenuated than activated by RAGE activation. The inhibitor of NADPH oxidase, apocynin, improves renal function in STZ-induced diabetic rats, albeit over a relatively short period [Citation558]. SOD and antioxidants such as probucol, α-tocopherol, α-lipoic acid, and N-acetyl-cysteine (NAC) provide cytoprotection against AGEs [Citation559,Citation560]; the effects of the burst of ROS production are enhanced by GSH depletion [Citation545]. Activation of RAGE by AGE induces oxidative stress in endothelial and other cells, including the generation of thiobarbituric acid-reactive substances (TBARS), activation of NF-kappa B, and induction of heme oxygenase I; these effects are blocked by antibodies to RAGE and by antioxidants [Citation559]. Nifedipine, a calcium-channel blocker, inhibits the AGE-induced RAGE upregulation [Citation513]; this effect was ascribed to inhibition of ROS generation [Citation561]. Irbesartan, an angiotensin II type 1 receptor blocker, also downregulates RAGE expression and attenuates ROS formation in kidney tubular cells [Citation515]. Similarly, olmesartan was found to decrease AGE-evoked ROS generation in endothelial cells [Citation562]. Pioglitazone inhibits NF-κB activation as well [Citation508,Citation563]. Statins have a similar effect as demonstrated for pravastatin in proximal tubular epithelial cells from human kidney [Citation522]. From among natural compounds, quercetin was found to downregulate RAGE expression and inhibit the RAGE-initiated ERK/CREB/BDNF signaling pathway [Citation564]. Rosiglitazone and pioglitazone diminish the RAGE protein expression in human endothelial cells, decreasing AGE-BSA and beta-amyloid-induced MCP-1 production. A recent study highlights the detrimental role of RAGE activation and RAGE signaling in the development and progression of aortic valve (AV) calcification [Citation508]. Pretreatment with Bay 11–7082, an NF-κB inhibitor, inhibited RAGE-mediated upregulation of Runx2 and osteopontin. These results indicate that NF-κB activation mediates AGE-BSA-induced proinflammatory effects and osteoblastic differentiation of cultured porcine aortic valve interstitial cells (VICs).
Curcumin alleviates diabetic cardiomyopathy in a rat model decreasing RAGE expression markers of oxidative stress, Rac1 activity, NADPH oxidase subunits expression of gp91(phox) and p47(phox), inflammatory factors (TNF-α and IL-1β) and inhibiting Akt and GSK-3β phosphorylation [Citation525]. Similar effects were noted for soybean isoflavones [Citation565]. Pinocembrin significantly inhibits the upregulation of RAGE transcripts and protein expression both in vivo and in vitro, and also markedly depresses the activation of p38 mitogen-activated protein kinase (MAPK)-MAPKAP kinase-2 (MK2)-heat shock protein 27 (HSP27) and stress-activated protein kinase (SAPK)/c-Jun N-terminal kinase (JNK)-c-Jun pathways and the downstream NFκB inflammatory response subsequent to Aβ-RAGE interaction. Pinocembrin was shown to infer cognitive improvement and neuronal protection in Alzheimer disease models [Citation528]. Kaempferol abrogates age-related upregulation of the RAGE axis by inhibition of NADPH oxidase and preventing activation of NFkappaB [Citation554]. Taurine blocks the AGE-induced Raf-1/extracellular signal-regulated kinase (ERK) activation in epithelial cells and the subsequent cell hypertrophy [Citation566]. Quercetin treatment for Aβ(25–35)-induced amnesic mice improved the learning and memory capabilities and conferred robust neurovascular coupling protection, involving maintenance of the NVU integrity, reduction of neurovascular oxidation, modulation of microvascular function, improvement of cholinergic system, and downregulation of neurovascular RAGE signaling pathway and ERK/CREB/BDNF pathway [Citation567]. Ursolic acid attenuates D-gal-induced inflammatory response in the prefrontal cortex of D-gal-treated mice through decreasing AGEs, ROS, and protein carbonyl levels and inhibits AGE/RAGE/NF-κB-mediated inflammatory response [Citation568].
Final remarks
Formation of AGEs and ALEs is a feature of parametabolism, that is, metabolic reactions which are not programmed and teleonomic but occur due to the chemical properties of metabolites [Citation569]. Although they may be involved in cellular signaling [Citation570,Citation571], their excess contributes to the development of diabetic complications and other pathologies. The complexity of formation and interactions of AGEs and ALEs makes it possible to interact with these processes at various levels. The compounds discussed in this review and new, which are to be discovered, can be invaluable in combating diseases affecting millions of persons in the world.
Acknowledgements
This article was based on works performed and interactions within the COST CM1001 action.
Declaration of interest
The authors report no declarations of interest. The authors alone are responsible for the content and writing of the paper.
References
- Baynes JW. Role of oxidative stress in development of complication in diabetes. Diabetes 1991;40:405–412.
- Baynes JW, Thorpe SR. Perspectives in diabetes: role of oxidative stress in diabetic complication-a new perspective on an old paradigm. Diabetes 1999;48:1–9.
- Wohaieb SA, Godin DV. Alterations in free radical tissue-defense mechanisms in streptozotocin-induced diabetes. Diabetes 1987;36:1014–1018.
- Lyons TJ, Silvestri G, Dunn JA, Dyer DG, Baynes JW. Role of glycation in modification of lens crystallins in diabetic andnondiabetic senile cataracts. Diabetes 1991;40: 1010–1015.
- Brownlee M. Nonenzymatic glycosylation of macromolecules. Prospects of pharmacologic modulation. Diabetes 1992;41:57–60.
- Monnier VM, Sell DR, Nagaraj RH, Miyata S, Grandhee S, Odetti P, Ibrahim SA. Maillard reaction-mediated molecular damage to extracellular matrix and other tissue proteins in diabetes, aging, and uremia. Diabetes 1992;41:36–41.
- Monnier VM, Glomb M, Elgawish A, Sell DR. The mechanism of collagen cross-linking in diabetes: a puzzle nearing resolution. Diabetes 1996;45:S67–S72.
- Monnier VM, Mustata GT, Biemel KL, Reihl O, Lederer MO, Zhenyu D, Sell DR. Cross-linking of the extracellular matrix by the maillard reaction in aging and diabetes: an update on “a puzzle nearing resolution”. Ann N Y Acad Sci 2005;1043:533–544.
- Ghanem AA, Elewa A, Arafa LF. Pentosidine and N-carboxymethyl-lysine: biomarkers for type 2 diabetic retinopathy. Eur J Ophthalmol 2011;21:48–54.
- Hirata K, Kubo K. Relationship between blood levels of N-carboxymethyl-lysine and pentosidine and the severity of microangiopathy in type 2 diabetes. Endocr J 2004;51: 537–544.
- Dworacka M, Winiarska H, Szymańska M, Szczawińska K, Wierusz-Wysocka B. Serum N-epsilon-(carboxymethyl)lysine is elevated in nondiabetic coronary heart disease patients. J Basic Clin Physiol Pharmacol 2002;13:201–213.
- Kaufmann E, Boehm BO, Süssmuth SD, Kientsch-Engel R, Sperfeld A, Ludolph AC, Tumani H. The advanced glycation end-product N epsilon-(carboxymethyl)lysine level is elevated in cerebrospinal fluid of patients with amyotrophic lateral sclerosis. Neurosci Lett 2004;371:226–229.
- Southern L, Williams J, Esiri MM. Immunohistochemical study of N-epsilon-carboxymethyl lysine (CML) in human brain: relation to vascular dementia. BMC Neurol 2007;7:35.
- Fukuda M, Kanou F, Shimada N, Sawabe M, Saito Y, Murayama S, et al.. Elevated levels of 4-hydroxynonenal-histidine Michael adduct in the hippocampi of patients with Alzheimer's disease. Biomed Res 2009;30:227–233.
- Podborska M, Sevcikova A, Trna J, Dite P, Lojek A, Kubala L. Increased markers of oxidative stress in plasma of patients with chronic pancreatitis. Neuro Endocrinol Lett 2009;30:116–120.
- Grimsrud PA, Picklo MJS, Griffin TJ, Bernlohr DA. Carbonylation of adipose proteins in obesity and insulin resistance: identification of adipocyte fatty acid-binding protein as a cellular target of 4-hydroxynonenal. Mol Cell Proteomics 2007;6:624–637.
- Wang G, Pierangeli SS, Papalardo E, Ansari GA, Khan MF. Markers of oxidative and nitrosative stress in systemic lupus erythematosus: correlation with disease activity. Arthritis Rheum 2010;62:2064–2072.
- Monnier VM. Intervention against the Maillard reaction in vivo. Arch Biochem Biophys 2003;419:1–15.
- Monnier VM, Sell DR. Prevention and repair of protein d amage by the Maillard reaction in vivo. Rejuvenation Res 2006;9:264–273.
- Ellis EM. Reactive carbonyls and oxidative stress: potential for therapeutic intervention. Pharmacol Ther 2007;115:13–24.
- Iacobini C, Menini S, Ricci C, Scipioni A, Sansoni V, Mazzitelli G, et al.. Advanced lipoxidation end-products mediate lipid-induced glomerular injury: role of receptor-mediated mechanisms. J Pathol 2009;218:360–369.
- Fu MX, Wells-Knecht KJ, Blackledge JA, Lyons TJ, Thorpe SR, Baynes JW. Glycation, glycosylation, and cross-linking of collagen by glucose. Kinetics, mechanims, and inhibition of the late stages of the Maillard reaction. Diabetes 1994;43:676–683.
- Di Mario U, Pugliese G. 15th Golgi lecture: from hyperglycaemia to the dysregulation of vascular remodelling in diabetes. Diabetologia 2001;44:674–692.
- Nishikawa T, Edelstein D, Brownlee M. The missing link: a single unifying mechanism for diabetic complications. Kidney Int 2000;58:S26–S30.
- Augustyniak A, Bartosz G, Cipak A, Duburs G, Horáková L, Luczaj W, et al.. Natural and synthetic antioxidants: an updated overview. Free Radic Res 2010;44:1216–1262.
- Stefek M, Drozdikova I, Vajdova K. The pyridoindole antioxidant stobadine inhibited glycation-induced absorbance and fluorescence changes in albumin. Acta Diabetol 1996; 33:35–40.
- Stefek M, Krizanova L, Trnkova Z. Oxidative modification of serum albumin in an experimental glycation model of diabetes mellitus in vitro: Effect of the pyridoindole antioxidant stobadine. Life Sci 1999;65:1995–1997.
- Stefek M, Sotnikova R, Okruhlicova L, Volkovova K, Kucharska J, Gajdosik A, et al.. Effect of dietary supplementation with the pyridoindole antioxidant stobadine on antioxidant state and ultrastructure of diabetic rat myocardium. Acta Diabetol 2000;37:111–117.
- Karasu C. Glycoxidative stress and cardiovascular complications in experimentally-induced diabetes: effects of antioxidant treatment. Open Cardiovasc Med J 2010;4:240–256.
- Stefek M, Gajdosik A, Tribulova N, Navarova J, Volkovova K, Weismann P, et al.. The pyridoindole antioxidant stobadine attenuates albuminuria, enzymuria, kidney lipid peroxidation and matrix collagen cross-linking in streptozotocin- induced diabetic rats. Meth Find Exp Clin Pharm 2002a;24: 565–571.
- Stefek M, Tribulova N, Gajdosik A, Gajdosikova A. The pyridoindole antioxidant stobadine attenuates histochemical changes in kidney of streptozotocin-induced diabetic rats. Acta Histochem 2002b;104:413–417.
- Cumaoglu A, Stefek M, Bauer V, Ari N, Aricioglu A, Karasu C. Glycoxidative and nitrosative stress in kidney of experimental diabetic rats: effects of the prydoindole antioxidant stobadine. Neuro Endocrinol Lett 2010;31:313–318.
- Pekiner B, Ulusu NN, Das-Evcimen N, Sahilli M, Aktan F, Stefek M, et al.. In vivo treatment with stobadine prevents lipid peroxidation, protein glycation and calcium overload but does not ameliorate Ca2+-ATPase activity in heart and liver of streptozotocin-diabetic rats: comparison with vitamin E. Biochim Biophys Acta 2002;1588:71–78.
- Cumaoglu A, Cevik C, Rackova L, Ari N, Karasu C. Effects of antioxidant stobadine on protein carbonylation, advanced oxidation protein products and reductive capacity of liver in streptozotocin-diabetic rats: role of oxidative/nitrosative stress. Biofactors 2007;30:171–178.
- Demiryurek AT, Karasu C, Stefek M, Stolc S. Effect of stobadine on leukocyte free radical generation in streptozotocin-diabetic rats: comparison with vitamin E. Pharmacology 2004;70:1–4.
- Kyselova Z, Gajdosik A, Gajdosikova A, Ulicna O, Mihalova D, Karasu C, Stefek M. Effect of the pyridoindole antioxidant stobadine on development of experimental diabetic cataract and on lens protein oxidation in rats: comparison with vitamin E and BHT. Mol Vis 2005;11:56–65.
- Yulek F, Or M, Ozogul C, Isik AC, Ari N, Stefek M, et al.. Effects of stobadine and vitamin E in diabetes-induced retinal abnormalities: Involvement of oxidative stress. Arch Med Res 2007;38:503–511.
- Skalska S, Kyselova Z, Gajdosikova A, Karasu C, Stefek M, Stolc S. Protective effect of stobadine on NCV in streptozotocindiabetic rats: augmentation by vitamin E. Gen Physiol Biophys 2008;27:106–114.
- Skalska S, Kucera P, Goldenberg Z, Stefek M, Kyselova Z, Jariabka P, et al.. Neuropathy in a rat model of mild diabetes induced by multiple low doses of streptozotocin: effects of the antioxidant stobadine in comparison with a high-dose alpha-lipoic acid treatment. Gen Physiol Biophys 2010;29: 50–58.
- Ansari NH, Bhatnagar A, Fulep E, Khanna P, Srivastava SK. Trolox protects hyperglycemia-induced cataractogenesis in cultured rat lens. Res Commun Chem Pathol Pharmacol 1994;84:93–104.
- Srivastava SK, Ansari NH. Prevention of sugar-induced cataractogenesis in rats by butylated hydroxytoluene. Diabetes 1988;37:1505–1508.
- Love A, Cotter MA, Cameron NE. Nerve function and regeneration in diabetic and galactosaemic rats: antioxidant and metal chelator effects. Eur J Pharmacol 1996;314:33–39.
- Nourmohammadi I, Modarress M, Khanaki K, Shaabani M. Association of serum alpha-tocopherol, retinol and ascorbic acid with the risk of cataract development. Ann Nutr Metab 2008;52:296–298.
- Jacques PF, Chylack LTJ, Hankinson SE, Khu PM, Rogers G, Friend J, et al.. Long-term nutrient intake and early age-related nuclear lens opacities. Arch Ophthalmol 2001; 119:1009–1019.
- Valero MP, Fletcher AE, De Stavola BL, Vioque J, Alepuz VC. Vitamin C is associated with reduced risk of cataract in a Mediterranean population. J Nutr 2002;132: 1299–1306.
- Cumming RG, Mitchell P, Smith W. Diet and cataract: the Blue Mountains Eye Study. Ophthalmology 2000;107: 450–456.
- Hankinson SE, Stampfer MJ, Seddon JM, Colditz GA, Rosner B, Speizer FE, Willett WC. Nutrient intake and cataract extraction in women: a prospective study. Br Med J 1992;305:335–339.
- Ravindran RD, Vashist P, Gupta SK, Young IS, Maraini G, Camparini M, et al.. Inverse association of vitamin C with cataract in older people in India. Ophthalmology 2011; 18:1958–1965.
- Bors W, Michel C. Chemistry of the antioxidant. Effect of polyphenols. Ann NY Acad Sci 2002;957:57–69.
- Amic D, Davidovic-Amic D, Beslo D, Rastija V, Lucic B, Trinajstic N. SAR and QSAR of the antioxidant activity of flavonoids. Curr Med Chem 2007;14:827–845.
- Stefek M. Natural flavonoids as potential multifunctional agents in prevention of diabetic cataract. Interdiscip. Toxicol 2011;4:69–77.
- Stefek M, Karasu C. Eye lens in aging and diabetes: effect of quercetin. Rejuvenation Res 2011;14:525–534.
- Sternberg MC, Borsos AM, Roux A, Adam C, Urios P. Compared inhibition of pentosidine formation in type-I collagen incubated with glucose by catechin, myricetin, kaempferol and quercetin: role of flavonoid structure. Diabetologia 2002;45:A393–A394.
- Odetti PR, Borgolio A, De Pascale A, Rolandi R, Adezati L. Prevention of diabetes-increased aging effect on rat collagen-linked fluorescence by aminoguanidine and rutin. Diabetes 1990;39:796–801.
- Urios P, Kassab I, Borsos AM, Guillot R, Peyroux J, Sternberg M. Long-term treatment with a purified micronized flavonoid fraction reduces urinary albumin clearance and restores albuminemia in normotensiveand hypertensive diabetic rats. Diabetes Res Clin Pract 2000;50:S362.
- Price DL, Rhett PM, Thorpe SR, Baynes JW. Chelating activity of advanced glycation end-product (AGE) inhibitors. J Biol Chem 2001;276:48967–48972.
- Nagai R, Murray DB, Metz TO, Baynes JW. Chelation: a fundamental mechanism of action of AGE inhibitors, AGE breakers, and other inhibitors of diabetes complications. Diabetes 2012;61:549–559.
- Hunt JV, Dean RT, Wolff SP. Hydroxyl radical production and autoxidative glycosylation. Glucose autoxidation as the cause of protein damage in the experimental glycation model of diabetes mellitus and ageing. Biochem J 1988; 256:205–212.
- Hunt JV, Wolff SP. Oxidative glycation and free radical production: a causal mechanism of diabetic complications. Free Radic Res Commun 1991;1:115–123.
- Wolff SP, Bascal ZA, Hunt JV. Autoxidative glycosylation: free radicals and glycation theory. Prog Clin Biol Res 1989;304:259–275.
- Wolff SP, Jiang ZY, Hunt JV. Protein glycation and oxidative stress in diabetes mellitus and ageing. Free Radic Biol Med 1991;10:339–352.
- Dunn JA, Ahmed MU, Murtiashaw MH, Richardson JM, Walla MD, Thorpe SR, Baynes JW. Reaction of ascorbate with lysine and protein under autoxidizing conditions: formation of N epsilon-(carboxymethyl)lysine by reaction between lysine and products of autoxidation of ascorbate. Biochemistry 1990;29:10964–10970.
- Wells-Knecht MC, Thorpe SR, Baynes JW. Pathways of formation of glycoxidation products during glycation of collagen. Biochemistry 1995;34:15134–15141.
- Smith PR, Thornalley PJ. Mechanism of the degradation of non-enzymatically glycated proteins under physiological conditions. Studies with the model fructosamine, N epsilon-(1-deoxy-D-fructos-1-yl)hippuryl-lysine. Eur J Biochem 1992;210:729–739.
- Young IS, Tate S, Lightbody JH, McMaster D, Trimble ER. The effects of desferrioxamine and ascorbate on oxidative stress in the streptozotocin diabetic rat. Free Radic Biol Med 1995;18:833–840.
- Schleicher ED, Wagner E, Nerlich AG. Increased accumulation of the glycoxidation product N(epsilon)-(carboxymethyl)lysine in human tissues in diabetes and aging. J Clin Invest 1997;99:457–468.
- Cameron NE, Cotter MA. Rapid reversal by aminoguanidine of the neurovascular effects of diabetes in rats: modulation by nitric oxide synthase inhibition. Metabolism 1996;45: 1147–1152.
- Zheng Y, Li XK, Wang Y, Cai L. The role of zinc, copper and iron in the pathogenesis of diabetes and diabetic complications: therapeutic effects by chelators. Hemoglobin 2008;32:135–145.
- Urui-Adams JY, Keen CL. Copper, oxidative stress, and human health. Mol Asp Med 2005;26:268–298.
- Lu J, Gong D, Choong SY, Xu H, Chan YK, Chen X, et al.. Copper(II)-selective chelation improves function and antioxidant defences in cardiovascular tissues of rats as a model of diabetes: comparisons between triethylenetetramine and three less copper-selective transition-metal-targeted treatments. Diabetologia 2010a;53:1217–1226.
- Lu J. Triethylenetetramine pharmacology and its clinical applications. Mol Cancer Ther 2010b;9:2458–2467.
- Cooper GJ. Therapeutic potential of copper chelation with triethylenetetramine in managing diabetes mellitus and Alzheimer's disease. Drugs 2011;71:1281–1320.
- Baynes JW, Murray DB. The metal chelators, trientine and citrate, inhibit the development of cardiac pathology in the Zucker diabetic rat. Exp Diabetes Res 2009;2009:696378.
- Gong D, Lu J, Chen X, Choong SY, Zhang S, Chan YK, et al.. Molecular changes evoked by triethylenetetramine treatment in the extracellular matrix of the heart and aorta in diabetic rats. Mol Pharmacol 2006;70:2045–2051.
- Cooper GJ, Phillips AR, Choong SY, Leonard BL, Crossman DJ, Brunton DH, et al.. Regeneration of the heart in diabetes by selective copper chelation. Diabetes 2004; 53:2501–2508.
- Nakamura J, Hamada Y, Chaya S, Nakashima E, Naruse K, Kato K, et al.. Transition metals and polyol pathway in the development of diabetic neuropathy in rats. Diabetes Metab Res Rev 2002;18:395–402.
- Hamada Y, Nakashima E, Naruse K, Nakae M, Naiki M, et al.. A copper chelating agent suppresses carbonyl stress in diabetic rat lenses. J Diabetes Complications 2005;19: 328–334.
- Nagai R, Nagai M, Shimasaki S, Baynes JW, Fujiwara Y. Citric acid inhibits development of cataracts, proteinuria and ketosis in streptozotocin (type 1) diabetic rats. Biochem Biophys Res Commun 2010;393:118–122.
- Adrover M, Vilanova B, Frau J, Munoz F, Donoso J. The pyridoxamine action on Amadori compounds: A reexamination of its scavenging capacity and chelating effect. Bioorg Med Chem 2008;16:5557–5569.
- Dukic-Stefanovic S, Schinzel R, Riederer P, Munch G. AGES in brain ageing: AGE-inhibitors as neuroprotective and antidementia drugs?Biogerontology 2001;2:19–34.
- Munch G, Taneli Y, Schraven E, Schindler U, Schinzel R, Palm D, Riederer P. The cognition-enhancing drug tenilsetam is an inhibitor of protein crosslinking by advanced glycosylation. J Neural Transm Park Dis Dement Sect 1994;8:193–208.
- Shoda H, Miyata S, Liu BF, Yamada H, Ohara T, Suzuki K, et al.. Inhibitory effects of tenilsetam on the Maillard reaction. Endocrinology 1997;138:1886–1892.
- Hipkiss AR, Preston JE, Himsworth DT, Worthington VC, Keown M, Michaelis J, et al.. Pluripotent protective effects of carnosine, a naturally occurring dipeptide. Ann NY Acad Sci 1998;854:37–53.
- Reddy VP, Beyaz A. Inhibitors of the Maillard reaction and AGE breakers as therapeutics for multiple diseases. Drug Discov Today 2006;11:646–654.
- Ruggiero-Lopez D, Lecomte M, Moinet G, Patereau G, Lagarde M, Wiernsperger N. Reaction of metformin with dicarbonyl compounds. Possible implication in the inhibition of advanced glycation end produc formation. Biochem Pharmacol 1999;58:1765–1773.
- Beisswenger P, Ruggiero-Lopez D. Metformin inhibition of glycation processes. Diabetes Metab 2003;29:6S95–103.
- Ouslimani N, Mahrouf M, Peynet J, Bonnefont-Rousselot D, Cosson C, Legrand A, Beaudeux JL. Metformin reduces endothelial cell expression of both the receptor for advanced glycation end products and lectin-like oxidized receptor 1. Metabolism 2007;56:308–313.
- Rahbar S, Natarajan N, Yerneni K, Scott S, Gonzales N, Nadler J. Evidence that pioglitazone, metformin and pentoxifylline are inhibitors of glycation. Clin Chim Acta 2000;301:65–77.
- Miyata T, van Ypersele de Strihou C. Angiotensin II receptor blockers and angiotensin converting enzyme inhibitors: implication of radical scavenging and transition metal chelation in inhibition of advanced glycation endproduct formation. Arch Biochem Biophys 2003;419:50–54.
- Figarola JL, Scott S, Loera S, Tessler C, Chu P, Weiss L, et al.. LR-90 a new advanced glycation endproduct inhibitor prevents progression of diabetic nephropathy in streptozotocin-diabetic rats. Diabetologia 2003;46:1140–1152.
- Figarola JL, Loera S, Weng Y, Shanmugam N, Natarajan R, Rahbar S. LR-90 prevents dyslipidaemia and diabetic nephropathy in the Zucker diabetic fatty rat. Diabetologia 2008;51:882–891.
- Bhatwadekar A, Glenn JV, Figarola JL, Scott S, Gardiner TA, Rahbar S, Stitt AW. A new advanced glycation inhibitor, LR-90, prevents experimental diabetic retinopathy in rats. Br J Ophthalmol 2008;92:545–547.
- Figarola JL, Scott S, Loera S, Xi B, Synold T, Weiss L, Rahbar S. Prevention of early renal disease, dyslipidemia and lipid peroxidation in STZdiabetic rats by LR-9 and LR-74 novel AGE inhibitors. Diabetes Metab Res Rev 2005;21: 533–544.
- Krautwald M, Münch G. Advanced glycation end products as biomarkers and gerontotoxins – a basis to explore methylglyoxal-lowering agents for Alzheimer's disease?Exp Gerontol 2010;45:744–751.
- Burcham PC, Kaminskas LM, Tan D, Pyke SM. Carbonyl-scavenging drugs & protection against carbonyl stress- associated cell injury. Mini Rev Med Chem 2008;8:319–330.
- Zhou P, Huang J, Tian F. Specific noncovalent interactions at protein-ligand interface: implications for rational drug design. Curr Med Chem 2012;19:226–238.
- Mayr H, Bug T, Gotta MF, Hering N, Irrgang B, Janker B, et al.. Reference scales for the characterization of cationic electrophiles and neutral nucleophiles. J Am Chem.Soc 2001;123:9500–9512.
- Cárdenas C, Rabi N, Ayers PW, Morell C, Jaramillo P, Fuentealba P. Chemical reactivity descriptors for ambiphilic reagents: dual descriptor, local hypersoftness, and electrostatic potential. J Phys Chem A 2009;113:8660–8667.
- LoPachin RM, Gavin T, Decaprio A, Barber DS. Application of the Hard and Soft, Acids and Bases (HSAB) theory to toxicant–target interactions. Chem Res Toxicol 2012;25: 239–251.
- Pearson RG, Songstad J. Application of the principle of hard and soft acids and bases to organic chemistry. J Am Chem Soc 1967;89:1827–1836.
- Contreras R, Andrés J, Domingo LR, Castillo R, Pérez P. Effect of electron-withdrawing substituents on the electrophilicity of carbonyl carbons. Tetrahedron 2005;61:417–422.
- LoPachin RM, Barber DS, Gavin T. Molecular mechanisms of the conjugated alpha,beta-unsaturated carbonyl derivatives: relevance to neurotoxicity and neurodegenerative diseases. Toxicol Sci 2008;104:235–249.
- Halliwell B. Antioxidants in human health and disease. Annu Rev Nutr 1996;16:33–50.
- Schafer FQ, Buettner GR. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic Biol Med 2001;30: 1191–1212.
- Blair IA. Endogenous glutathione adducts. Curr Drug Metab 2006;7:853–872.
- Testa B, Krämer SD. The biochemistry of drug metabolism–an introduction: part 4. reactions of conjugation and their enzymes. Chem Biodivers 2008;5:2171–2336.
- Esterbauer H, Zollner H, Scholz N. Reaction of glutathione with conjugated carbonyls. Z Naturforsch C 1975;30: 466–473.
- Oakley A. Glutathione transferases: a structural perspective. Drug Metab Rev 2011;43:138–151.
- Awasthi YC, Yang Y, Tiwari NK, Patrick B, Sharma A, Li J, Awasthi S. Regulation of 4-hydroxynonenal-mediated signaling by glutathione S-transferases. Free Radic Biol Med 2004;37:607–619.
- Balogh LM, Atkins WM. Interactions of glutathione transferases with 4-hydroxynonenal. Drug Metab Rev 2011;43: 165–178.
- Raza H. Dual localization of glutathione S-transferase in the cytosol and mitochondria: implications in oxidative stress, toxicity and disease. FEBS J 2011;278:4243–4251.
- Balogh LM, Roberts AG, Shireman LM, Greene RJ, Atkins WM. The stereochemical course of 4-hydroxy-2-nonenal metabolism by glutathione S-transferases. J Biol Chem 2008;283:16702–16710.
- Balogh LM, Le Trong I, Kripps KA, Shireman LM, Stenkamp RE, Zhang W, et al. Substrate specificity combined with stereopromiscuity in glutathione transferase A4-4- dependent metabolism of 4-hydroxynonenal. Biochemistry 2010;49:1541–1548.
- Mannervik B. Molecular enzymology of the glyoxalase system. Drug Metabol Drug Interact 2008;23:13–27.
- Thornalley PJ. Glutathione-dependent detoxification of alpha-oxoaldehydes by the glyoxalase system: involvement in disease mechanisms and antiproliferative activity of glyoxalase I inhibitors. Chem Biol Interact 1998;111–112: 137–151.
- Wlodek L. The reaction of sulfhydryl groups with carbonyl compounds. Acta Biochim Pol 1988;35:307–317.
- Nomi Y, Aizawa H, Kurata T, Shindo K, Nguyen CV. Glutathione reacts with glyoxal at the N-terminal. Biosci Biotechnol Biochem 2009;73:2408–2411.
- Dodd S, Dean O, Copolov DL, Malhi GS, Berk M. N-acetylcysteine for antioxidant therapy: pharmacology and clinical utility. Expert Opin Biol Ther 2008;8:1955–1962.
- Atkuri KR, Mantovani JJ, Herzenberg LA, Herzenberg LA. N-Acetylcysteine–a safe antidote for cysteine/glutathione deficiency. Curr Opin Pharmacol 2007;7:355–359.
- Esterbauer H, Ertl A, Scholz N. Reaction of cysteine with alpha,beta-unsaturated aldehydes. Tetrahedron 1976;32: 285–289.
- Wondrak GT, Cervantes-Laurean D, Roberts MJ, Qasem JG, Kim M, Jacobson EL, Jacobson MK. Identification of alpha-dicarbonyl scavengers for cellular protection against carbonyl stress. Biochem Pharmacol 2002;63:361–373.
- Mehta R, Wong L, O’Brien PJ. Cytoprotective mechanisms of carbonyl scavenging drugs in isolated rat hepatocytes. Chem Biol Interact 2009;178:317–323.
- Li G, Tang T, Peng M, He H, Yin D. Direct reaction of taurine with malondialdehyde: evidence for taurine as a scavenger of reactive carbonyl species. Redox Rep 2010;15: 268–274.
- Perrett D. The metabolism and pharmacology of D-penicillamine in man. J Rheumatol Suppl 1981;7:41–50.
- Szwergold BS. Alpha-thiolamines such as cysteine and cysteamine act as effective transglycating agents due to formation of irreversible thiazolidine derivatives. Med Hypotheses 2006;66:698–707.
- Wondrak GT, Jacobson MK, Jacobson EL. Antimelanoma activity of apoptogenic carbonyl scavengers. J Pharmacol Exp Ther 2006;316:805–814.
- Shay KP, Moreau RF, Smith EJ, Smith AR, Hagen TM. Alpha-lipoic acid as a dietary supplement: molecular mechanisms and therapeutic potential. Biochim Biophys Acta 2009;1790:1149–1160.
- Korotchkina LG, Yang H, Tirosh O, Packer L, Patel MS. Protection by thiols of the mitochondrial complexes from 4-hydroxy-2-nonenal. Free Radic Biol Med 2001;30: 992–999.
- Kim HY, Oi Y, Kim M, Yokozawa T. Protective effect of lipoic acid against methylglyoxal-induced oxidative stress in LLC-PK(1) cells. J Nutr Sci Vitaminol (Tokyo) 2008;54: 99–104.
- Gorąca A, Huk-Kolega H, Piechota A, Kleniewska P, Ciejka E, Skibska B. Lipoic acid - biological activity and therapeutic potential. Pharmacol Rep 2011;63:849–858.
- He L, Liu B, Dai Z, Zhang HF, Zhang YS, Luo XJ, et al.. Alpha lipoic acid protects heart against myocardial ischemia-reperfusion injury through a mechanism involving aldehyde dehydrogenase 2 activation. Eur J Pharmacol 2012;678: 32–38.
- Di Simplicio P, Frosali S, Priora R, Summa D, Cherubini Di Simplicio F, Di Giuseppe D, Di Stefano A. Biochemical and biological aspects of protein thiolation in cells and plasma. Antioxid Redox Signal 2005;7:951–963.
- Elsheikh A, Lavergne SN, Castrejon JL, Farrell J, Wang H, Sathish J, et al.. Drug antigenicity, immunogenicity, and costimulatory signaling: evidence for formation of a functional antigen through immune cell metabolism. J Immunol 2010;185:6448–6460.
- Bolton WK, Abdel-Rahman E. Pimagedine: a novel therapy for diabetic nephropathy. Expert Opin Investig Drugs 2002;11:565–574.
- Brownlee M, Vlassara H, Kooney A, Ulrich P, Cerami A. Aminoguanidine prevents diabetes-induced arterial wall protein crosslinking. Science 1986;232:1629–1632.
- Vistoli G, Orioli M, Pedretti A, Regazzoni L, Canevotti R, Negrisoli G, et al.. Design, synthesis, and evaluation of carnosine derivatives as selective and efficient sequestering agents of cytotoxic reactive carbonyl species. Chem MedChem 2009;4:967–975.
- Thornalley PJ. Use of aminoguanidine (Pimagedine) to prevent the formation of advanced glycation endproducts. Arch Biochem Biophys 2003;419:31–40.
- Soulis-Liparota T, Cooper M, Papazoglou D, Clarke B, Jerums G. Retardation by aminoguanidine of development of albuminuria, mesangial expansion, and tissue fluorescence in streptozocin-induced diabetic rat. Diabetes 1991;40: 1328–1334.
- Huijberts MS, Wolffenbuttel BH, Boudier HA, Crijns FR, Kruseman AC, Poitevin P, Levy BI. Aminoguanidine treatment increases elasticity and decreases fluid filtration of large arteries from diabetic rats. J Clin Invest 1993;92: 1407–1411.
- Hammes HP, Ali SS, Uhlmann M, Weiss A, Federlin K, Geisen K, Brownlee M. Aminoguanidine does not inhibit the initial phase of experimental diabetic retinopathy in rats. Diabetologia 1995;38:269–273.
- Hammes HP, Brownlee M, Edelstein D, Saleck M, Martin S, Federlin K. Aminoguanidine inhibits the development of accelerated diabetic retinopathy in the spontaneous hypertensive rat. Diabetologia 1994;37:32–35.
- Swamy-Mruthinti S, Green K, Abraham EC. Inhibition of cataracts in moderately diabetic rats by aminoguanidine. Exp Eye Res 1996;62:505–512.
- Soulis T, Cooper ME, Vranes D, Bucala R, Jerums G. Effects of aminoguanidine in preventing experimental diabetic nephropathy are related to the duration of treatment. Kidney Int 1996;50:627–634.
- Kelly DJ, Gilbert RE, Cox AJ, Soulis T, Jerums G, Cooper ME. Aminoguanidine ameliorates overexpression of prosclerotic growth factors and collagen deposition in experimental diabetic nephropathy. J Am Soc Nephrol 2001; 12:2098–2107.
- Thornalley PJ. Glycation in diabetic neuropathy: characteristics, consequences, causes and therapeutic options. Int. Rev. Neurobiol. 2002;50:37–57.
- Moreau R, Nguyen BT, Doneaunu CE, Hagen TM. Reversal by aminoguanidine of the age-related increase in glycoxidation and lipoxidation in the cardiovascular system of Fischer 344 rats. Biochem Pharmacol 2005;69:29–40.
- Chang KC, Tseng CD, Wu MS, Liang JT, Tsai MS, Cho YL, Tseng YZ. Aminoguanidine prevents arterial stiffening in a new rat model type 2 diabetes. Eur J Clin Invest 2006;36: 528–535.
- Kern TS, Engerman RL. Pharmacological inhibition of diabetic retinopathy. Aminoguanidine and aspirin. Diabetes 2001;50:1636–1642.
- Yagihashi S, Kamijo M, Baba M, Yagihashi N, Nagai K. Effects of aminoguanidine on functional and structural abnormalities in peripheral nerve of STZ-induced diabetic rats. Diabetes 1992;41:47–52.
- Miyauchi Y, Shikama H, Takasu T, Okamiya H, Umeda M, Hirasaki E, et al.. Slowing of peripheral motor nerve conduction was ameliorated by aminoguanidine in streptozotocin-induced diabetic rats. Eur J Endocrinol 1996;134: 467–473.
- Li YM, Steffes M, Donnely T, Liu C, Fuh H, Basgen J, et al.. Prevention of cardiovascular and renal pathology of aging by the advanced glycation inhibitor aminoguanidine. Proc Natl Acad Sci USA 1996;93:3902–3907.
- Corman B, Duriez M, Poitevin P, Heudes D, Bruneval P, Tedgui A, Levy B. Aminoguanidine prevents age-related stiffening and cardiac hypertrophy. Proc Natl Acad Sci USA 1998;95:1301–1306.
- Freedman BI, Wuerth JP, Cartwright K, Bain RP, Dippe S, Hershon K, et al.. Design and baseline characteristics for the aminoguanidine Clinical Trial in Overt Type 2 Diabetic Nephropathy (ACTIONII). Control Clin Trials 1999;20: 493–510.
- Corbett JA, Tilton RG, Chang K, Hasan KS, Ido Y, Wang JL, et al.. Aminoguanidine, a novel inhibitor of nitric oxide formation, prevents diabetic vascular dysfunction. Diabetes 1992;41:552–556.
- Yu PH, Zuo DM. Aminoguanidine inhibits semicarbazide-sensitive amine oxidase activity: implications for advanced glycation and diabetic complications. Diabetologia 1997; 40:1243–1250.
- Čàsrki J, Lazarova M, Beño A. Study of β-resorcylidene aminoguanidine. I. Spectral and acid-basic properties of the onium compounds. Acta FRN Univ Comen Chimia 1978; 26:89–101.
- Forbes JM, Soulis T, Thallas T, Panagiotopoulos S, Long DM, Vasan S, et al.. Renoprotective effects of a novel inhibitor of advanced glycation. Diabetologia 2001;44: 108–114.
- Wilkinson-Berka JL, Kelly DJ, Koerner SM, Jaworski K, Davis B, Thallas V, Cooper ME. ALT-946 and aminoguanidine, inhibitors of advanced glycation, improve severe nephropathy in the diabetic transgenic (mREN-2)27 rat. Diabetes 2002;51:3283–3289.
- Labieniec-Watala M, Siewiera K, Jozwiak Z. Resorcylidene aminoguanidine (RAG) improves cardiac mitochondrial bioenergetics impaired by hyperglycaemia in a model of experimental diabetes. Int J Mol Sci 2011;12:8013–8026.
- Ruggiero-Lopez D, Lecomte M, Moinet G, Patereau G, Lagarde M, Wiernsperger N. Reaction of metformin with dicarbonyl compounds. Possible implication in the inhibition of advanced glycation end product formation. Biochem Pharmacol 1999;58:1765–1773.
- Battah S, Ahmed N, Thornalley PJ. Kinetics and mechanism of the reaction of metformin with methylglyoxal. Int Congr Ser 2002;1245:355–356.
- Kiho T, Kato T, Usui S, Hirano K. Effect of buformin and metformin on formation of advanced glycation end products by methylglyoxal. Clin Chim Acta 2005;358:139–145.
- Beisswenger PJ, Howell SK, Touchette AD, Lal S, Szwergold BS. Metformin reduces systemic methylglyoxal levels in type 2 diabetes. Diabetes 1999;48:198–202.
- Miyata T, Ueda Y, Asahi K, Izuhara Y, Inagi R, Saito A, et al.. Mechanism of the inhibitory effect of OPB-9195 [(+/2)-2-isopropylidenehydrazono-4-oxo-thiazolidin-5-yla cetanilide] on advanced glycation end product and advanced lipoxidation end product formation. J Am Soc Nephrol 2000;11:1719–1725.
- Nakamura S, Makita Z, Ishikawa S, Yasumura K, Fujii W, Yanagisawa K, et al.. Progression of nephropathy in spontaneous diabetic rat is prevented by OPB-9195, a novel inhibitor of advanced glycation. Diabetes 1997;46:895–899.
- Tsuchida K, Makita Z, Yamagishi S, Atsumi T, Miyoshi H, Obara S, et al.. Suppression of transforming growth factor beta and vascular endothelial growth factor in diabetic nephropathy in rats by a novel advanced glycation end product inhibitor, OPB-9195. Diabetologia 1999;42:579–588.
- Miyata T, Ishikawa S, Asahi K, Inagi R, Suzuki D, Horie K, et al.. 2-Isopropylidenehydrazono-4-oxo-thiazolidin-5-ylacetanilide (OPB-9195) treatment inhibits the development of intimal thickening after balloon injury of rat carotid artery: Role of glycoxidation and lipoxidation reactions in vascular tissue damage. FEBS Lett 1999;445:202–206.
- Wada R, Nishizawa Y, Yagihashi N, Takeuchi M, Ishikawa Y, Yasumura K, et al.. Effects of OPB-9195, anti-glycation agent, on experimental diabetic neuropathy. Eur J Clin Invest 2001;31:513–520.
- Kass DA. Getting better without AGE: new insights into the diabetic heart. Circ Res 2003;92:704–706.
- Mizutani K, Ikeda K, Tsuda K, Yamori Y. Inhibitor for advanced glycation end products formation attenuates hypertension and oxidative damage in genetic hypertensive rats. J Hypertens 2002;20:1607–1614.
- Kochakian M, Manjula BN, Egan JJ. Chronic dosing with aminoguanidine and novel advanced glycosylation end product-formation inhibitors ameliorates cross-linking of tail tendon collagen in STZ-induced diabetic rats. Diabetes 1996;45:1694–1700.
- O’Donnell JP. The reaction of amines with carbonyls: its significance in the nonenzymatic metabolism of xenobiotics. Drug Metab Rev 1982;13:123–159.
- Burcham PC, Pyke SM. Hydralazine inhibits rapid acrolein-induced protein oligomerization: role of aldehyde scavenging and adduct trapping in cross-link blocking and cytoprotection. Mol Pharmacol 2006;69:1056–1065.
- Nangaku M, Miyata T, Sada T, Mizuno M, Inagi R, Ueda Y, et al.. Anti-hypertensive agents inhibit in vivo the formation of advanced glycation end products and improve renal damage in a type 2 diabetic nephropathy rat model. J Am Soc Nephrol 2003;14:1212–1222.
- Kandler MR, Mah GT, Tejani AM, Stabler SN, Salzwedel DM. Hydralazine for essential hypertension. Cochrane Database Syst Rev 2011;9:CD004934.
- Bouguerne B, Belkheiri N, Bedos-Belval F, Vindis C, Uchida K, Duran H, et al.. Antiatherogenic effect of bisvanillyl-hydralazone, a new hydralazine derivative with antioxidant, carbonyl scavenger, and antiapoptotic properties. Antioxid Redox Signal 2011;14:2093–2106.
- Galvani S, Coatrieux C, Elbaz M, Grazide MH, Thiers JC, Parini A, et al.. Carbonyl scavenger and antiatherogenic effects of hydrazine derivatives. Free Radic Biol Med 2008; 45:1457–1467.
- Maheshwari M, Roberts JK, Desutter B, Duong KT, Tingling J, Fawver JN, et al.. Hydralazine modifies Aβ fibril formation and prevents modification by lipids in vitro. Biochemistry 2010;49:10371–11080.
- Del Rio D, Stewart AJ, Pellegrini N. A review of recent studies on malondialdehyde as toxic molecule and biological marker of oxidative stress. Nutr Metab Cardiovasc Dis 2005; 15:316–328.
- Tabrizchi R. Edaravone Mitsubishi-Tokyo. Curr Opin Investig Drugs 2000;1:347–354.
- Pérez-González A, Galano A. On the outstanding antioxidant capacity of edaravone derivatives through single electron transfer reactions. J Phys Chem B 2012;116:1180–1188.
- Izuhara Y, Nangaku M, Takizawa S, Takahashi S, Shao J, Oishi H, et al.. A novel class of advanced glycation inhibitors ameliorates renal and cardiovascular damage in experimental rat models. Nephrol Dial Transplant 2008; 23:497–509.
- Lapchak PA. A critical assessment of edaravone acute ischemic stroke efficacy trials: is edaravone an effective neuroprotective therapy?Expert Opin Pharmacother 2010; 11:1753–1763.
- Aldini G, Vistoli G, Regazzoni L, Benfatto MC, Bettinelli I, Carini M. Edaravone inhibits protein carbonylation by a direct carbonyl-scavenging mechanism: focus on reactivity, selectivity, and reaction mechanisms. Antioxid Redox Signal 2010;12:381–392.
- Takizawa S, Izuhara Y, Kitao Y, Hori O, Ogawa S, Morita Y, et al.. A novel inhibitor of advanced glycation and endoplasmic reticulum stress reduces infarct volume in rat focal cerebral ischemia. Brain Res 2007;1183:124–137.
- Sajithlal GB, Chithra P, Chandrakasan G. Effect of curcumin on the advanced glycation and cross-linking of collagen in diabetic rats. Biochem. Pharmacol. 1998;56:1607–1614.
- Hu TY, Liu CL, Chyau CC, Hu ML. Trapping of methylglyoxal by curcumin in cell-free systems and in human umbilical vein endothelial cells. J Agric Food Chem 2012; 60:8190–8196.
- LoPachin RM, Gavin T, Geohagen BC, Zhang L, Casper D, Lekhraj R, Barber DS. β-Dicarbonyl enolates: a new class of neuroprotectants. J Neurochem 2011;116:132–143.
- Hipkiss AR. Carnosine and its possible roles in nutrition and health. Adv Food Nutr Res 2009;57:87–154.
- Boldyrev AA. Carnosine: new concept for the function of an old molecule. Biochemistry (Mosc). 2012;77:313–326.
- Aldini G, Carini M, Beretta G, Bradamante S, Facino RM. Carnosine is a quencher of 4-hydroxy-nonenal: through what mechanism of reaction?Biochem Biophys Res Commun 2002;298:699–706.
- Liu Y, Xu G, Sayre LM. Carnosine inhibits (E)-4-hydroxy-2-nonenal-induced protein cross-linking: structural characterization of carnosine-HNE adducts. Chem Res Toxicol 2003;16:1589–1597.
- Vistoli G, Carini M, Aldini G. Transforming dietary peptides in promising lead compounds: the case of bioavailable carnosine analogs. Amino Acids 2012;43:111–126.
- Bellia F, Vecchio G, Rizzarelli E. Carnosine derivatives: new multifunctional drug-like molecules. Amino Acids 2012; 43:153–163.
- Guiotto A, Calderan A, Ruzza P, Osler A, Rubini C, Jo DG, et al.. Synthesis and evaluation of neuroprotective alpha, beta-unsaturated aldehyde scavenger histidyl-containing analogues of carnosine. J Med Chem 2005;48:6156–6161.
- Meredith D. The mammalian proton-coupled peptide cotransporter PepT1: sitting on the transporter-channel fence?Philos Trans R Soc Lond B Biol Sci 2009;364: 203–207.
- Pegova A, Abe H, Boldyrev A. Hydrolysis of carnosine and related compounds by mammalian carnosinases. Comp Biochem Physiol B Biochem Mol Biol 2000;127:443–446.
- Aldini G, Orioli M, Rossoni G, Savi F, Braidotti P, Vistoli G, et al.. The carbonyl scavenger carnosine ameliorates dyslipidaemia and renal function in Zucker obese rats. J Cell Mol Med 2011;15:1339–1354.
- Orioli M, Vistoli G, Regazzoni L, Pedretti A, Lapolla A, Rossoni G, et al.. Design, synthesis, ADME properties, and pharmacological activities of β-alanyl-D-histidine (D-carnosine) prodrugs with improved bioavailability. ChemMedChem 2011;6:1269–1282.
- Menini S, Iacobini C, Ricci C, Scipioni A, Blasetti Fantauzzi C, Giaccari A, et al.. D-carnosine octylester attenuates atherosclerosis and renal disease in ApoE null mice fed a Western diet through reduction of carbonyl stress and inflammation. Br J Pharmacol 2012;166:1344–1356.
- Van Den Nest W, Domenech NA, Puche JC, Carreno Serraima C. Peptides used in the treatment and/or care of the skin, mucous membranes and/or scalp and their use in cosmetic or pharmaceutical compositions. US: Lipotec; 2011.
- Zhao J, Zhong CJ. A review on research progress of transketolase. Neurosci Bull 2009;25:94–99.
- Shangari N, Bruce WR, Poon R, O’Brien PJ. Toxicity of glyoxals–role of oxidative stress, metabolic detoxification and thiamine deficiency. Biochem Soc Trans 2003;31: 1390–1393.
- Tarwadi KV, Agte VV. Effect of micronutrients on methylglyoxal-mediated in vitro glycation of albumin. Biol Trace Elem Res 2011;143:717–725.
- Breslow R. The mechanism of thiamine action: predictions from model experiments. Ann N Y Acad Sci 1962;98: 445–452.
- Pohl M, Sprenger GA, Müller M. A new perspective on thiamine catalysis. Curr Opin Biotechnol 2004;15: 335–342.
- Balakumar P, Rohilla A, Krishan P, Solairaj P, Thangathirupathi A. The multifaceted therapeutic potential of benfotiamine. Pharmacol Res 2010;61:482–488.
- Shoeb M, Ramana KV. Anti-inflammatory effects of benfotiamine are mediated through the regulation of the arachidonic acid pathway in macrophages. Free Radic Biol Med 2012;52:182–190.
- Karachalias N, Babaei-Jadidi R, Ahmed N, Thornalley P. Accumulation of fructosyllysine and advanced glycation end products in the kidney, retina and peripheral nerve of streptozotocin-induced diabetic rats. Biochem Soc Trans 2003; 31:1423–1425.
- Babei-Jadidi R, Karachalian N, Ahmed N, Battab S, Thornalley PJ. Prevention of incipient diabetic nephropathy by high-dose thiamine and benfotiamine. Diabetes 2003;52:2110–2120.
- Stracke H, Lindemann A, Federlin K. A benfotiamine- vitamin B combination in treatment of diabetic polyneuropathy. Exp Clin Endocrinol Diabetes 1996;104:311–316.
- Haupt E, Ledermann H, Kopcke W. Benfotiamine in the treatment of diabetic polyneuropathy-a three-week randomized, controlled pilot study (BEDIP study). Int J Clin Pharmacol Ther 2005;43:71–77.
- Stirban A, Negrean M, Stratmann B, Gawlowski T, Horstmann T, Gotting C, et al.. Benfotiamine prevents macro- and microvascular endothelial dysfunction and oxidative stress following a meal rich in advanced glycation end products in individuals with type 2 diabetes. Diabetes Care 2006;29:2064–2071.
- Alkhalaf A, Klooster A, van Oeveren W, Achenbach U, Kleefstra N, Slingerland RJ, et al.. A double-blind, randomized, placebo-controlled clinical trial on benfotiamine treatment in patients with diabetic nephropathy. Diabetes Care 2010;33:1598–1601.
- Fraser DA, Diep LM, Hovden IA, Nilsen KB, Sveen KA, Seljeflot I, Hanssen KF. The effects of long-term oral benfotiamine supplementation on peripheral nerve function and inflammatory markers in patients with type 1 diabetes: a 24-month, double-blind, randomized, placebo-controlled trial. Diabetes Care 2012;35:1095–1097.
- Münch G, Taneli Y, Schraven E, Schindler U, Schinzel R, Palm D, Riederer P. The cognition-enhancing drug tenilsetam is an inhibitor of protein crosslinking by advanced glycosylation. J Neural Transm Park Dis Dement Sect 1994;8:193–208.
- Webster J, Urban C, Berbaum K, Loske C, Alpar A, Gärtner U, et al.. The carbonyl scavengers aminoguanidine and tenilsetam protect against the neurotoxic effects of methylglyoxal. Neurotox Res 2005;7:95–101.
- Jaramillo P, Perez P, Fuentealba P. Relationship between basicity and nucleophilicity. J Phys Org Chem 2007;20: 1050–1057.
- Hall NE, Smith BJ. High-Level ab Initio molecular orbital calculations of imine formation. J Phys Chem A 1998;102:4930–4938.
- Tahmassebi DC. Substituent effects on the stability of carbodiimides. J Chem Soc Perkin Trans 2 2001;4:613–617.
- Shimizu M, Hachiya I, Mizota I. Conjugated imines and iminium salts as versatile acceptors of nucleophiles. Chem Comm 2009;8:874–889.
- Voziyan PA, Hudson BG. Pyridoxamine as a multifunctional pharmaceutical: targeting pathogenic glycation and oxidative damage. Cell Mol Life Sci 2005;62:1671–1681.
- Caldés C, Vilanova B, Adrover M, Muñoz F, Donoso J. Phenol group in pyridoxamine acts as a stabilizing element for its carbinolamines and Schiff bases. Chem Biodivers 2011;8:1318–1332.
- Adrover M, Vilanova B, Frau J, Muñoz F, Donoso J. The pyridoxamine action on Amadori compounds: A reexamination of its scavenging capacity and chelating effect. Bioorg Med Chem 2008;16:5557–5569.
- Nagaraj RH, Sarkar P, Mally A, Biemel KM, Lederer MO, Padayatti PS. Effect of pyridoxamine on chemical modification of proteins by carbonyls in diabetic rats: characterization of a major product from the reaction of pyridoxamine and methylglyoxal. Arch Biochem Biophys 2002;402:110–119.
- Kang Z, Li H, Li G, Yin D. Reaction of pyridoxamine with malondialdehyde: mechanism of inhibition of formation of advanced lipoxidation end-products. Amino Acids 2006; 30:55–61.
- Amarnath V, Amarnath K, Amarnath K, Davies S, Roberts LJ II. Pyridoxamine: an extremely potent scavenger of 1,4-dicarbonyls. Chem Res Toxicol 2004;17:410–415.
- Adrover M, Vilanova B, Muñoz F, Donoso J. Unexpected isomeric equilibrium in pyridoxamine Schiff bases. Bioorg Chem 2009;37:26–32.
- Adrover M, Vilanova B, Frau J, Muñoz F, Donoso J. A comparative study of the chemical reactivity of pyridoxamine, Ac-Phe-Lys and Ac-Cys with various glycating carbonyl compounds. Amino Acids 2009;36:437–448.
- Mehansho H, Buss DD, Hamm MW, Henderson LM. Transport and metabolism of pyridoxine in rat liver. Biochim Biophys Acta 1980;631:112–123.
- Stitt A, Gardiner TA, Alderson NL, Canning P, Frizzell N, Duffy N, et al.. The AGE inhibitor pyridoxamine inhibits development of retinopathy in experimental diabetes. Diabetes 2002;51:2826–2832.
- Degenhardt TP, Alderson NL, Arrington DD, Beattie RJ, Basgen JM, Steffes MW, et al.. Pyridoxamine inhibits early renal disease and dyslipidemia in the streptozotocin-diabetic rat. Kidney Int 2002;61:939–950.
- Nakamura S, Li H, Adijiang A, Pischetsrieder M, Niwa T. Pyridoxal phosphate prevents progression of diabetic nephropathy. Nephrol Dial Transplant 2007;22: 2165–2174.
- Forbes JM, Thallas-Bonke V, Cooper ME, Merlin TC. Advanced glycation: how are we progressing to combat this web of sugar anomalies in diabetic nephropathy. Curr Pharm Des 2004;10:3361–3372.
- Williams ME, Bolton WK, Khalifah RG, Degenhardt TP, Schotzinger RJ, McGill JB. Effects of pyridoxamine in combined phase 2 studies of patients with type 1 and type 2 diabetes and overt nephropathy. Am J Nephrol 2007;27: 605–614.
- Sasaki NA, Garcia-Alvarez MC, Wang Q, Ermolenko L, Franck G, Nhiri N, et al.. N-Terminal 2,3-diaminopropionic acid (Dap) peptides as efficient methylglyoxal scavengers to inhibit advanced glycation endproduct (AGE) formation. Bioorg Med Chem 2009;17:2310–2320.
- Audic N, Potier G, Sasaki NA. New 2,3-diaminopropionic acid inhibitors of AGE and ALE formation. Org Biomol Chem 2012; Dec 17 [Epub ahead of print].
- Soulis T, Sastra S, Thallas V, Mortensen SB, Wilken M, Clausen JT, et al.. A novel inhibitor of advanced glycation end-product formation inhibits mesenteric vascular hypertrophy in experimental diabetes. Diabetologia 1999;42:472–479.
- Oturai PS, Christensen M, Rolin B, Pedersen KE, Mortensen SB, Boel E. Effects of advanced glycation end-product inhibition and cross-link breakage in diabetic rats. Metabolism 2000;49:996–1000.
- Silva PJ. Inductive and resonance effects on the acidities of phenol, enols, and carbonyl alpha-hydrogens. J Org Chem 2009;74:914–916.
- Lo CY, Hsiao WT, Chen XY. Efficiency of trapping methylglyoxal by phenols and phenolic acids. J Food Sci 2011; 76:H90–H96.
- Wang Y, Ho CT. Flavour chemistry of methylglyoxal and glyoxal. Chem Soc Rev 2012;41:4140–4149.
- Wu CH, Yen GC. Inhibitory effect of naturally occurring flavonoids on the formation of advanced glycation endproducts. J Agric Food Chem 2005;53:3167–3173.
- Lo CY, Li S, Tan D, Pan MH, Sang S, Ho CT. Trapping reactions of reactive carbonyl species with tea polyphenols in simulated physiological conditions. Mol Nutr Food Res 2006;50:1118–1128.
- Sang S, Shao X, Bai N, Lo CY, Yang CS, Ho CT. Tea polyphenol (-)-epigallocatechin-3-gallate: a new trapping agent of reactive dicarbonyl species. Chem Res Toxicol 2007;20:1862–1870.
- Lv L, Shao X, Chen H, Ho CT, Sang S. Genistein inhibits advanced glycation end product formation by trapping methylglyoxal. Chem Res Toxicol 2011;24:579–586.
- Shao X, Bai N, He K, Ho CT, Yang CS, Sang S. Apple polyphenols, phloretin and phloridzin: new trapping agents of reactive dicarbonyl species. Chem Res Toxicol 2008; 21:2042–2050.
- Matsuda H, Wang T, Managi H, Yoshikawa M. Structural requirements of fl avonoids for inhibition of protein glycation and radical scavenging activities. Bioorg Med Chem 2003; 11:5317–5323.
- Beretta G, Furlanetto S, Regazzoni L, Zarrella M, Facino RM. Quenching of alpha,beta-unsaturated aldehydes by green tea polyphenols: HPLC-ESI-MS/MS studies. J Pharm Biomed Anal 2008;48:606–611.
- Lv L, Shao X, Wang L, Huang D, Ho CT, Sang S. Stilbene glucoside from Polygonum multiflorum Thunb.: a novel natural inhibitor of advanced glycation end product formation by trapping of methylglyoxal. J Agric Food Chem 2010;58:2239–2245.
- Nakajima Y, Inokuchi Y, Shimazawa M, Otsubo K, Ishibashi T, Hara H. Astaxanthin, a dietary carotenoid, protects retinal cells against oxidative stress in-vitro and in mice in-vivo. J Pharm Pharmacol 2008;60:1365–1374.
- Peng X, Ma J, Chao J, Sun Z, Chang RC, Tse I, et al.. Beneficial effects of cinnamon proanthocyanidins on the formation of specific advanced glycation endproducts and methylglyoxal-induced impairment on glucose consumption. J Agric Food Chem 2010;58:6692–6696.
- Fodor G, Arnold R, Mohacsi T, Karle I, Flippen-Anderson J. A new role for l-ascorbic acid: Michael donor to alpha, beta-unsaturated carbonyl compounds. Tetrahedron 1983;39: 2137–2145.
- Miranda CL, Reed RL, Kuiper HC, Alber S, Stevens JF. Ascorbic acid promotes detoxification and elimination of 4-hydroxy-2(E)-nonenal in human monocytic THP-1 cells. Chem Res Toxicol 2009;22:863–874.
- Kesinger NG, Langsdorf BL, Yokochi AF, Miranda CL, Stevens JF. Formation of a vitamin C conjugate of acrolein and its paraoxonase-mediated conversion into 5,6,7,8- tetrahydroxy-4-oxooctanal. Chem Res Toxicol 2010;23: 836–844.
- Huby R, Harding JJ. Non-enzymic glycosylation (glycation) of lens proteins and protection by aspirin and reduced glutathione. Exp Eye Res 1988;47:53–59.
- Swamy MS, Abraham EC. Inhibition of lens crystallin glycation and high molecular weight aggregate formation by aspirin in vitro and in vivo. Invest. Ophthalmol Visual Sci 1989;30:1120–1126.
- Ajiboye R, Harding JJ. The non-enzymic glycosylation of bovine lens proteins by glucosamine and its inhibition by aspirin, ibuprofen and glutathione. Exp Eye Res 1989;49: 31–41.
- Crompton M, Rixon KC, Harding JJ. Aspirin prevents carbamylation of soluble lens proteins and prevents cyanate-induced phase separation opacities in vitro: a possible mechanism by which aspirin could prevent cataract. Exp Eye Res 1985;40:297–311.
- Rao GN, Lardis MP, Cotlier E. Acetylation of lens crystallins: a possible mechanism by which aspirin could prevent cataract formation. Biochem Biophys Res Commun 1985;128:1125–1132.
- Rao GN, Cotlier E. Aspirin prevents the nonenzymic glycosylation and carbamylation of the human eye lens crystallins in vitro. Biochem Biophys Res Commun 1988;151:991–996.
- Blakytny R, Harding JJ. Prevention of cataract in diabetic rats by aspirin, paracetamol (acetaminophen) and ibuprofen. Exp Eye Res 1992;54:509–518.
- Zhao W, Devamanoharan PS, Varma SD. Fructose induced deactivation of glucose-6-phosphate dehydrogenase activity and its prevention by pyruvate: implications in cataract prevention. Free Radic Res Commun 1998;29:315–320.
- Zhao W, Devamanoharan PS, Varma SD. Fructose-mediated damage to lens α-crystallin: prevention by pyruvate. Biochim Biophys Acta 2000a;1500:161–168.
- Devamanoharan PS, Ali AH, Varma SD. Non-enzymatic glycation of lens proteins and haemoglobin-inhibition by pyruvate: an in-vivo study. Diabetes Obes Metab 1999;1: 159–164.
- Hegde KR, Varma SD. Prevention of cataract by pyruvate in experimentally diabetic mice. Mol Cell Biochem 2005;269:115–120.
- Yabe-Nishimura C. Aldose reductase in glucose toxicity: a potential target for the prevention of diabetic complications. Pharmacol Rev 1998;50:21–33.
- Tsukushi S, Katsuzaki T, Aoyama I, Takayama F, Miyazaki T, Shimokata K, Niwa T. Increased erythrocyte 3-DG and AGEs in diabetic hemodialysis patients: role of the polyol pathway. Kidney Int 1999;55:1970–1976.
- Hamada Y, Nakamura J, Naruse K, Komori T, Kato K, Kasuya Y, et al.. Epalrestat, an aldose reductase ihibitor, reduces the levels of Nepsilon-(carboxymethyl)lysine protein adducts and their precursors in erythrocytes from diabetic patients. Diabetes Care 2000;23:1539–1544.
- Nakamura N, Yamazaki K, Satoh A, Urakaze M, Kobayashi M, Yamabe H, et al.. Effects of eparlestat on plasma levels of advanced glycation end products in patients with type 2 diabetes. In Vivo 2003;17:177–180.
- Kawai T, Takei I, Tokui M, Funae O, Miyamoto K, Tabata M, et al.. Effects of epalrestat, an aldose reductase inhibitor, on diabetic peripheral neuropathy in patients with type 2 diabetes, in relation to suppression of N(epsilon)-carboxymethyl lysine. J Diabetes Complications 2010;24: 424–432.
- Hallam KM, Li Q, Ananthakrishnan R, Kalea A, Zou YS, Vedantham S, et al.. Aldose reductase and AGE-RAGE pathways: central roles in the pathogenesis of vascular dysfunction in aging rats. Aging Cell 2010;9:776–784.
- Delpierre G, Rider MH, Collard F, Stroobant V, Vanstapel F, Santos H, Van Schaftingen E. Identification, cloning, and heterologous expression of a mammalian fructosamine-3-kinase. Diabetes 2000;49:1627–1634.
- Szwergold BS, Howell S, Beisswenger PJ. Human fructosamine-3-kinase. Purification, sequencing, substrate specificity, and evidence of activity in vivo. Diabetes 2001;50: 2139–2147.
- Delpierre G, Collard F, Fortpied J, Van Schaftingen E. Fructosamine 3-kinase is involved in an intracellular deglycation pathway in human erythrocytes. Biochem J 2002;365:801–808.
- Delpierre G, Van Schaftingen E. Fructosamine 3-kinase, an enzyme involved in protein deglycation. Biochem Soc Trans 2003;31:1354–1357.
- Delpierre G, Vertommen D, Communi D, Rider MH, Van Schaftingen E. Identification of fructosamine redues deglycated by fructosamine-3-kinase in human hemoglobin. J Biol Chem 2004;279:27613–27620.
- Veiga-da-Cunha M, Jacquemin P, Delpierre G, Godfraind C, Theate I, Vertommen D, et al.. Increased protein glycation in fructosamine 3-kinase-deficient mice. Biochem J 2006;399:257–264.
- Pascal SMA, Veiga-da-Cunha M, Gilon P, Van Schaftingen E, Jonas JC. Effects of fructosamine-3-kinase deficiency on function and survival of mouse pancreatic islets after prolonged culture in high glucose or robose concentrations. Am J Physiol Endocrinol Metab 2010;298:E586–E596.
- Van Schaftingen E, Collard F, Wiame E, Veiga-da-Cunha M. Enzymatic repair of Amadori products. Amino Acids 2012; 42:1143–1150.
- Delplanque J, Delpierre G, Opperdoes FR, Van Schaftingen E. Tissue distribution and evolution of fructosamine 3-kinase and fructosamine 3-kinase-related protein. J Biol Chem 2004;279:46606–46613.
- Van Schaftingen E, Delpierre G, Collard F, Fortpied J, Gemayel R, Wiame E, Veiga-da-Cunha M. Fructosamine 3-kinase and other enzymes involved in protein deglycation. Adv Enzyme Regul 2007;47:261–269.
- Deppe VM, Bongaerts J, O’Connell T, Maurer KH, Meinhardt F. Enzymatic deglycation of Amadori products in bacteria: mechanisms, occurrence and physiological functions. Appl Microbiol Biotechnol 2011;90:399–406.
- Thornalley PJ. Dicarbonyl intermediates in the Maillard reaction. Ann NY Acad Sci 2005;1043:111–117.
- Thornalley PJ. Protein and nucleotide damage by glyoxal and methylglyoxal in physiological systems – role in ageing and disease. Drug Metabol Drug Interact 2008;23:125–150.
- Hydman D, Bauman DR, Heredia VV, Penning TV. The aldo-keto reductase superfamily homepage. Chem Biol Interact 2003;143–144:621–631.
- Petrash JM. All in the family:aldose reductase and closely related aldo-keto reductases. Cell Mol Life Sci 2004;61: 737–749.
- Ansari NH, Bhatnagar A., Liu SQ, Srivastava SK. Purification and characterization of aldose reductase and aldehyde reductase from human kidney. Biochem Int 1991; 25:755–765.
- Das B, Srivastava SK. Purification and properties of aldose reductase and aldehyde reductase II from human erythrocyte. Arch Biochem Biophys 1985;238:670–679.
- Morjana NA, Flynn TG. Aldose reductase from human psoas muscle. Purification, substrate specificity, immunological characterization, and effect of drugs and inhibitors. J Biol Chem 1989;264:2906–2911.
- Srivastava SK, Ansari NH, Hair GA, Das B. Aldose and aldehyde reductases in human tissues. Biochim Biophys Acta 1984;800:220–227.
- van der Jagt DL, Hunsaker LA, Robinson B, Stangebye LA, Deck LM. Aldehyde and aldose reductases from human placenta. Heterogenous expression of multiple enzyme forms. J Biol Chem 1990;265:10912–10918.
- van der Jagt DL, Robinson B, Taylor KK, Hunsaker LA. Aldose reductase from human skeletal and heart muscle. Interconvertible forms related by thiol-disulfide exchange. J Biol Chem 1990;265:20982–20987.
- Wermuth B, Burgisser H, Bohren K, von Wartburg JP. Purification and characterization of humain brain aldose reductase. Eur J Biochem 1982;127:279–284.
- Kumar PA, Reddy GB. Focus on molecules: aldose reductase. Exp Eye Res 2007;85:739–740.
- Borhani DW, Harter TM, Petrash JM. The crystal structure of the aldose reductase NADPH binary complex. J Biol Chem 1992;267:24841–24847.
- Wilson DK, Bohren KM, Gabbay KH, Quiocho FA. An unlikely sugar substrate site in the 1.65 A structure of the human aldose reductase holoenzyme implicated in diabetic complications. Science 1992;257:81–84.
- Srivastava S, Watowich SJ, Petrash JM, Srivastava SK, Bhatnagar A. Structural and kinetic determinants of aldehyde reduction by aldose reductase. Biochemistry 1999;38: 42–54.
- Shen YI, Zhong L, Johnson S, Cao D. Human aldo-keto reductases 1B1 and 1B10; a comparative study of their enzyme activity toward electrophilic carbonyl compounds. Chem Biol Interact 2011;191:192–198.
- Vander Jagt DL, Robinson B, Taylor KK, Hunsaker LA. Reduction of trioses by NADPH-dependent aldo-keto reductases. Aldose reductase, methylglyoxal and diabetic complications. J Biol Chem 1992;267:4364–4369.
- Vander Jagt DL, Hassebrook RK, Hunsaker LA, Brown WM, Royer R. Metabolism of the 2-oxoaldehyde methylglyoxal by aldose reductase and by glyoxalase-I: roles for glutathione in both enzymes and implications for diabetic complications. Chem Biol Interact 2001;130–132:549–562.
- Yabe-Nishimura C, Nishinaka T, Iwata K, Seo HG. Up-regulation of aldose reductase by the substrate, methylglyoxal. Chem Biol Interact 2003;143–144:317–323.
- Baba SP, Barski OA, Ahmed Y, O’Toole TE, Conklin DJ, Bhatnagar A, Srivastava S. Reductive metabolism of AGE precursors: a metabolic route for preventing AGE accumulation in cardiovascular tissue. Diabetes 2009;58:2486–2497.
- Baba SP, Hellmann J, Srivastava S, Bhatnagar A. Aldose reductase (AKR1B3) regulates the accumulation of advanced glycosylation end products (AGEs) and the expression of AGE receptor (RAGE). Chem Biol Interact 2011;191: 357–363.
- Sakiyama H, Takahashi M, Yamamoto T, Teshima T, Lee SH, Miyamoto Y, et al.. The internalization and metabolism of 3-deoxyglucosone in human umbilical vein endothelial cells. J Biochem 2006;139:245–253.
- Thornalley PJ. Glyoxalase I – structure, function and a critical role in the enzymatic defence against glycation. Biochem Soc Trans 2003;31:1343–1348.
- Grillo MA, Colombatto S. Advanced glycation end-products (AGEs): involment in aging and in neurodegenerative diseases. Amino Acids 2008;35:29–36.
- Xue M, Rabbani N, Thornalley PJ. Glyoxalase in ageing. Semin Cell Dev Biol 2011;22:293–301.
- Marasinghe GPK, Sander IM, Bennett B, Periyannan G, Yang KW, Makaroff CA, Crowder MW. Structural studies on a mitochondrial glyoxalase II. J Biol Chem 2005;280: 40668–40675.
- Shinohara M, Thornalley PJ, Giardino I, Beisswenger P, Thorpe SR, Onorato J, Brownlee M. Overexpression of glyoxalase-I in bovine endothelial cell inhibits intracellular advanced glycation endproduct formation and prevents hyperglycemia-induced increases in macromolecular endocytosis. J Clin Invest 1998;101:1142–1147.
- Brouwers O, Niessen PM, Ferreira I, Miyata T, Scheffer PG, Teerlink T, et al.. Overexpression of glyoxalase-I reduces hyperglycemia-induced levels of advanced glycation end products and oxidative stress in diabetic rats. J Biol Chem 2011;286:1374–1380.
- Miller AG, Smith DG, Bhat M, Nagaraj RH. Glyoxalase I is critical for human retinal capillary pericyte survival under hyperglycemic conditions. J Biol Chem 2006;281: 11864–11887.
- Kumagai T, Nangaku M, Kojima I, Nagai R, Ingelfinger JR, Miyata T, et al.. Glyoxalase I overexpression ameliorates renal ischemia-reperfusion injury in rats. Am J Physiol Renal Physiol 2009;296:F912–F921.
- Inagi R, Kumagai T, Nangaku M. The role of glyoxalase system in renal hypoxia. Adv Exp Med Biol 2010;662: 49–55.
- Mailankot M, Padmanabha S, Pasupuleti N, Major D, Howell S, Nagaraj RH. Glyoxalase I activity and immunoreactivity in the aging human lens. Biogerontology 2009;10:711–720.
- Piec I, Listrat A, Alliot J, Chambon C, Taylor RG, Bechet D. Differential proteome analysis of aging in rat skeletal muscle. FASEB J 2005;19:1143–1145.
- Kuhla B, Boeck K, Luth HJ, Schmidt A, Weigle B, Schmitz M, et al.. Age-dependent changes of glyoxalase I expression in human brain. Neurobiol Aging 2006;27: 815–822.
- Kuhla B, Luth HJ, Haferburg D, Boeck K, Arendt T, Munch G. Methylglyoxal, glyoxal and their detoxification in Alzheimer's disease. Ann NY Acad Sci 2005;1043:211–216.
- Kuhla B, Boeck K, Schmidt A, Ogunlade V, Arendt T, Munch G, Luth HJ. Age-and stage-dependent glyoxalase I expression and its activity in normal and Alzheimer's disease. Neurobiol Aging 2007;28:29–41.
- Rabbani N, Thornalley PJ. Glycation research in amino acids: a place to call home. Amino Acids 2012;42:1087–1096.
- Singh R, Barden A, Mori T, Beilin L. Advanced glycation end-products: a review. Diabetologia 2001;44:129–146.
- Goldberg T, Cai W, Peppa M, Dardaine V, Baliga BS, Uribarri J, Vlassara H. Advanced glycoxidation end products in commonly consumed foods. J Am Diet Assoc 2004;104: 1287–1291.
- Foerster A, Henle T. Glycation in food and metabolic transit of dietary AGEs (advanced glycation end-products): studies on the urinary excretion of pyrraline. Biochem Soc Trans 2003;31:1383–1385.
- Ravikumar B, Sarkar S, Davies JE, Futter M, Garcia- Arencibia M, Green-Thompson ZW, et al.. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev 2010;90:1383–1435.
- Grimm S, Höhn A, Grune T. Oxidative protein damage and the proteasome. Amino Acids 2012a;42:23–38.
- Jung T, Höhn A, Catalgol B, Grune T. Age-related differences in oxidative protein damage in young and senescent fibroblasts. Arch Biochem Biophys 2009;483:127135.
- Castro JP, Ott C, Jung T, Grune T, Almeida H. Carbonylation of the cytoskeletal protein actin leads to aggregate formation. Free Radic Biol Med 2012;53:916–925.
- Kueper T, Grune T, Prahl S, Lenz H, Welge V, Biernoth T, et al.. Vimentin is a specific target in skin glycation. J Biol Chem 2007;282:23427–23436.
- Dunlop RA, Brunk UT, Rodgers KJ. Oxidized proteins: mechanisms of removal and consequences of accumulation. IUBMB Life 2009;61:522–527.
- Rajawat YS, Hilioti Z, Bossis I. Aging: central role for autophagy and the lysosomal degradative system. Ageing Res Rev 2009;8:199–213.
- De Duve C, Wattiaux R. Functions of lysosomes. Annu Rev Physiol 1966;28:435–492.
- Luzio JP, Pryor PR, Bright NA. Lysosomes: fusion and function. Nat Rev Mol Cell Biol 2007;8:622–632.
- Mellman I, Fuchs R, Helenius AW. Acidification of the endocytic and exocytic pathways. Annu Rev Biochem 1986; 55:663–700.
- Horiuchi S, Sakamoto Y, Sakai M. Scavenger receptors for oxidized and glycated proteins. Amino Acids 2003;25: 283–292.
- Bierhaus A, Humpert PM, Morcos M, Wendt T, Chavakis T, Arnold B, et al.. Understanding RAGE, the receptor for advanced glycation end products. J Mol Med 2005;83: 876–886.
- Ramasamy R, Yan SF, Schmidt AM. Arguing for the motion: yes, RAGE is a receptor for advanced glycation endproducts. Mol. Nutr. Food Res. 2007;51:1111–1115.
- Ohgami N, Nagai R, Ikemoto M, Arai H, Kuniyasu A, Horiuchi S, Nakayama H. Cd36, a member of the class b scavenger receptor family, as a receptor for advanced glycation end products. J Biol Chem 2001;276:3195–3202.
- Vlassara H, Li YM, Imani F, Wojciechowicz D, Yang Z, Liu FT, Cerami A. Identification of galectin-3 as a high- affinity binding protein for advanced glycation end products (AGE): a new member of the AGE-receptor complex. Mol Med 1995;1:634–646.
- Stolzing A, Grune T. Neuronal apoptotic bodies: phagocytosis and degradation by primary microglial cells. FASEB J 2004;18:743–745.
- Araki N, Higashi T, Mori T, Shibayama R, Kawabe Y, Kodama T, et al.. Macrophage scavenger receptor mediates the endocytic uptake and degradation of advanced glycation end products of the Maillard reaction. Eur J Biochem 1995;230:408–415.
- Grimm S, Ott C, Hörlacher M, Weber D, Höhn A, Grune T. Advanced glycation end products-induced formation of immunoproteasomes: involvement of the receptor for AGEs and Jak2/STAT1. Biochem J 2012b;448:127–139.
- Miyata S, Liu BF, Shoda H, Ohara T, Yamada H, Suzuki K, Kasuga M. Accumulation of pyrraline-modified albumin in phagocytes due to reduced degradation by lysosomal enzymes. J Biol Chem 1997;272:4037–4042.
- Saito A, Nagai R, Tanuma A, Hama H, Cho K, Takeda T, et al.. Role of megalin in endocytosis of advanced glycation end products: implications for a novel protein binding to both megalin and advanced glycation end products. J Am Soc Nephrol 2003;14:1123–1131.
- Stolzing A, Widmer R, Jung T, Voss P, Grune T. Degradation of glycated bovine serum albumin in microglial cells. Free Radic Biol Med 2006;40:1017–1027.
- Grimm S, Ernst L, Grötzinger N, Höhn A, Breusing N, Reinheckel T, Grune T. Cathepsin D is one of the major enzymes involved in intracellular degradation of AGE- modified proteins. Free Radic Res 2010;44:1013–1026.
- Grimm S, Horlacher M, Catalgol B, Hoehn A, Reinheckel T, Grune T. Cathepsins D and L reduce the toxicity of advanced glycation end products. Free Radic Biol Med 2012c;52: 1011–1023.
- Grune T, Jung T, Merker K, Davies KJ. Decreased proteolysis caused by protein aggregates, inclusion bodies, plaques, lipofuscin, ceroid, and ‘aggresomes’ during oxidative stress, aging, and disease. Int J Biochem Cell Biol 2004;36: 2519–2530.
- Grune T, Merker K, Sandig G, Davies KJ. Selective dregradation of oxidatively modified protein substrates by the proteasome. Biochem Biophys Res Commun 2003;305: 709–718.
- Kopito RR. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol 2000;10:524–530.
- Liscic RM, Grinberg LT, Zidar J, Gitcho MA, Cairns NJ. ALS and FTLD: two faces of TDP-43 proteinopathy. Eur J Neurol 2008;15:772–780.
- Bennett EJ, Shaler TA, Woodman B, Ryu KY, Zaitseva TS, Becker CH, et al.. Global changes to the ubiquitin system in Huntington's disease. Nature 2007;448:704–708.
- Tan JM, Wong ES, Kirkpatrick DS, Pletnikova O, Ko HS, Tay SP, et al.. Lysine 63-linked ubiquitination promotes the formation and autophagic clearance of protein inclusions associated with neurodegenerative diseases. Hum Mol Genet 2008;17:431–439.
- Yamamoto A, Simonsen A. The elimination of accumulated and aggregated proteins: a role for aggrephagy in neurodegeneration. Neurobiol Dis 2011;43:17–28.
- Chondrogianni N, Gonos ES. Proteasome function determines cellular homeostasis and the rate of aging. Adv Exp Med Biol 2010;694:38–46.
- Chondrogianni N, Gonos ES. Structure and function of the ubiquitin-proteasome system: modulation of components. Prog Mol Biol Transl Sci 2012;109:41–74.
- Goldberg AL. Functions of the proteasome: from protein degradation and immune surveillance to cancer therapy. Biochem Soc Trans 2007;35:12–17.
- Sijts EJ, Kloetzel PM. The role of the proteasome in the generation of MHC class I ligands and immune responses. Cell Mol Life Sci 2011;68:1491–1502.
- Angeles A, Fung G, Luo H. Immune and non-immune functions of the immunoproteasome. Front Biosci 2012;17: 1904–1916.
- Pickering AM, Koop AL, Teoh CY, Ermak G, Grune T, Davies KJ. The immunoproteasome, the 20S proteasome and the PA28αβ proteasome regulator are oxidative-stress-adaptive proteolytic complexes. Biochem J 2010;432:585–594.
- Seifert U, Bialy LP, Ebstein F, Bech-Otschir D, Voigt A, Schröter F, et al.. Immunoproteasomes preserve protein homeostasis upon interferon-induced oxidative stress. Cell 2010;142:613–624.
- Ciechanover A. Intracellular protein degradation: from a vague idea thru the lysosome and the ubiquitin-proteasome system and onto human diseases and drug targeting. Cell Death Differ 2005;12:1178–1190.
- Bulteau AL, Verbeke P, Petropoulos I, Chaffotte AF, Friguet B. Proteasome inhibition in glyoxal-treated fibroblasts and resistance of glycated glucose-6-phosphate dehydrogenase to 20 S proteasome degradation in vitro. J Biol Chem 2001;276:45662–45668.
- Cervantes-Laurean D, Roberts MJ, Jacobson EL, Jacobson MK. Nuclear proteasome activation and degradation of carboxymethylated histones in human keratinocytes following glyoxal treatment. Free Radic Biol Med 2005; 38:786–795.
- Uchiki T, Weikel KA, Jiao W, Shang F, Caceres A, Pawlak D, et al.. Glycation-altered proteolysis as a pathobiologic mechanism that links dietary glycemic index, aging, and age-related disease (in nondiabetics). Aging Cell 2012; 11:1–13.
- Moheimani F, Morgan PE, van Reyk DM, Davies MJ. Deleterious effects of reactive aldehydes and glycated proteins on macrophage proteasomal function: possible links between diabetes and atherosclerosis. Biochim Biophys Acta 2010;1802:561–571.
- Chondrogianni N, Gonos ES. Proteasome activation as a novel antiaging strategy. IUBMB Life 2008;60:651–655.
- Chondrogianni N, Petropoulos I, Grimm S, Georgila K, Catalgol B, Friguet B, et al.. Protein damage, repair and proteolysis. Mol Aspects Med 2012;in press.
- Gaczynska M, Rock KL, Spies T, Goldberg AL. Peptidase activities of proteasomes are differentially regulated by the major histocompatibility complex-encoded genes for LMP2 and LMP7. Proc Natl Acad Sci USA 1994;91:9213–9217.
- Gaczynska M, Goldberg AL, Tanaka K, Hendil KB, Rock KL. Proteasome subunits X and Y alter peptidase activities in opposite ways to the interferon-gamma-induced subunits LMP2 and LMP7. J Biol Chem 1996;271: 17275–17280.
- Chondrogianni N, Tzavelas C, Pemberton AJ, Nezis IP, Rivett AJ, Gonos ES. Overexpression of proteasome beta5 assembled subunit increases the amount of proteasome and confers ameliorated response to oxidative stress and higher survival rates. J Biol Chem 2005;280:11840–11850.
- Liu Y, Liu X, Zhang T, Luna C, Liton PB, Gonzalez P. Cytoprotective effects of proteasome beta5 subunit overexpression in lens epithelial cells. Mol Vis 2007;13:31–38.
- Kwak MK, Cho JM, Huang B, Shin S, Kensler TW. Role of increased expression of the proteasome in the protective effects of sulforaphane against hydrogen peroxide-mediated cytotoxicity in murine neuroblastoma cells. Free Radic Biol Med 2007;43:809–817.
- Hwang JS, Chang I, Kim S. Age-associated decrease in proteasome content and activities in human dermal fibroblasts: restoration of normal level of proteasome subunits reduces aging markers in fibroblasts from elderly persons. J Gerontol A Biol Sci Med Sci 2007;62:490–499.
- Chondrogianni N, Gonos ES. Overexpression of hUMP1/POMP proteasome accessory protein enhances proteasome-mediated antioxidant defence. Exp Gerontol 2007;42: 899–903.
- Chen Q, Thorpe J, Dohmen JR, Li F, Keller JN. Ump1 extends yeast lifespan and enhances viability during oxidative stress: central role for the proteasome?Free Radic Biol Med 2006;40:120–126.
- Hirano Y, Kaneko T, Okamoto K, Bai M, Yashiroda H, Furuyama K, et al.. Dissecting beta-ring assembly pathway of the mammalian 20S proteasome. EMBO J 2008;27: 2204–2213.
- Rosenzweig R, Glickman MH. Forging a proteasome alpha-ring with dedicated proteasome chaperones. Nat Struct Mol Biol 2008;15:218–220.
- Mannhaupt G, Schnall R, Karpov V, Vetter I, Feldmann HW. Rpn4p acts as a transcription factor by binding to PACE, a nonamer box found upstream of 26S proteasomal and other genes in yeast. FEBS Lett 1999;450:27–34.
- Ju D, Wang L, Mao X, Xie Y. Homeostatic regulation of the proteasome via an Rpn4-dependent feedback circuit. Biochem Biophys Res Commun 2004;321:51–57.
- Hahn JS, Neef DW, Thiele DJ. A stress regulatory network for co-ordinated activation of proteasome expression mediated by yeast heat shock transcription factor. Mol Microbiol 2006;60:240–251.
- Kruegel U, Robison B, Dange T, Kahlert G, Delaney JR, Kotireddy S, et al.. Elevated proteasome capacity extends replicative lifespan in Saccharomyces cerevisiae. PLoS Genet 2011;7:e1002253.
- Dahlmann B, Rutschmann M, Kuehn L, Reinauer H. Activation of the multicatalytic proteinase from rat skeletal muscle by fatty acids or sodium dodecyl sulphate. Biochem J 1985;228:171–177.
- Watanabe N, Yamada S. Activation of 20S proteasomes from spinach leaves by fatty acids. Plant Cell Physiol 1996;37: 147–151.
- Kohler A, Cascio P, Leggett DS, Woo KM, Goldberg AL, Finley D. The axial channel of the proteasome core particle is gated by the Rpt2 ATPase and controls both substrate entry and product release. Mol Cell 2001;7:1143–1152.
- Kisselev AF, Kaganovich D, Goldberg AL. Binding of hydrophobic peptides to several non-catalytic sites promotes peptide hydrolysis by all active sites of 20 S proteasomes. Evidence for peptide-induced channel opening in the alpha-rings. J Biol Chem 2002;277:22260–22270.
- Wilk S, Chen WE. Synthetic peptide-based activators of the proteasome. Mol Biol Rep 1997;24:119–124.
- Matsumura K, Aketa K. Proteasome (multicatalytic proteinase) of sea urchin sperm and its possible participation in the acrosome reaction. Mol Reprod Dev 1991;29:189–199.
- Ohkubo I, Gasa S, Namikawa C, Makita A, Sasaki M. Human erythrocyte multicatalytic proteinase: activation and binding to sulfated galacto- and lactosylceramides. Biochem Biophys Res Commun 1991;174:1133–1140.
- Ruiz de Mena I, Mahillo E, Arribas J, Castano JG. Kinetic mechanism of activation by cardiolipin (diphosphatidylglycerol) of the rat liver multicatalytic proteinase. Biochem J 1993;296:93–97.
- Silva GM, Netto LE, Simões V, Santos LF, Gozzo FC, Demasi MA, et al.. Redox control of 20S proteasome gating. Antioxid Redox Signal 2012;16:1183–1194.
- Katsiki M, Chondrogianni N, Chinou I, Rivett AJ, Gonos ES. The olive constituent oleuropein exhibits proteasome stimulatory properties in vitro and confers life span extension of human embryonic fibroblasts. Rejuvenation Res 2007; 10:157–172.
- Bulteau AL, Moreau M, Saunois A, Nizard C, Friguet B. Algae extract-mediated stimulation and protection of proteasome activity within human keratinocytes exposed to UVA and UVB irradiation. Antioxid Redox Signal 2006;8:136–143.
- Huang L, Ho P, Chen CH. Activation and inhibition of the proteasome by betulinic acid and its derivatives. FEBS Lett 2007;581:4955–4959.
- Dang Z, Lin A, Ho P, Soroka D, Lee KH, Huang L, Chen CH. Synthesis and proteasome inhibition of lithocholic acid derivatives. Bioorg Med Chem Lett 2011; 21:1926–1928.
- Kwak MK, Wakabayashi N, Greenlaw JL, Yamamoto M, Kensler TW. Antioxidants enhance mammalian proteasome expression through the Keap1-Nrf2 signaling pathway. Mol Cell Biol 2003;23:8786–8794.
- Lewis KN, Mele J, Hayes JD, Buffenstein R. Nrf2, a guardian of healthspan and gatekeeper of species longevity. Integr Comp Biol 2010;50:829–843.
- Nguyen T, Yang CS, Pickett CB. The pathways and molecular mechanisms regulating Nrf2 activation in response to chemical stress. Free Radic Biol Med 2004;37:433–441.
- Wasserman WW, Fahl WE. Functional antioxidant responsive elements. Proc Natl Acad Sci USA 1997;94: 5361–5366.
- D’Autréaux B, Toledano MB. ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat Rev Mol Cell Biol 2007;8:813–824.
- Furukawa M, Xiong Y. BTB protein Keap1 targets antioxidant transcription factor Nrf2 for ubiquitination by the Cullin 3-Roc1 ligase. Mol Cell Biol 2005;25:162–171.
- Boutten A, Goven D, Artaud-Macari E, Boczkowski J, Bonay M. NRF2 targeting: a promising therapeutic strategy in chronic obstructive pulmonary disease. Trends Mol Med 2011;17:363–371.
- Surh YJ, Kundu JK, Na HK. Nrf2 as a master redox switch in turning on the cellular signaling involved in the induction of cytoprotective genes by some chemopreventive phytochemicals. Planta Med 2008;74:1526–1539.
- Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, et al.. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun 1997;236:313–322.
- Venugopal R, Jaiswal AK. Nrf2 and Nrf1 in association with Jun proteins regulate antioxidant response element-mediated expression and coordinated induction of genes encoding detoxifying enzymes. Oncogene 1998;17:3145–3156.
- Eggler AL, Gay KA, Mesecar AD. Molecular mechanisms of natural products in chemoprevention: induction of cytoprotective enzymes by Nrf2. Mol Nutr Food Res 2008;52: S84–S94.
- Lau A, Villeneuve NF, Sun Z, Wong PK, Zhang DD. Dual roles of Nrf2 in cancer. Pharmacol Res 2008;58:262–270.
- Kapeta S, Chondrogianni N, Gonos ES. Nuclear erythroid factor 2-mediated proteasome activation delays senescence in human fibroblasts. J Biol Chem 2010;285:8171–8184.
- Chondrogianni N, Kapeta S, Chinou I, Vassilatou K, Papassideri I, Gonos ES. Anti-ageing and rejuvenating effects of quercetin. Exp Gerontol 2010;45:763–771.
- Tanigawa S, Fujii M, Hou DX. Action of Nrf2 and Keap1 in ARE-mediated NQO1 expression by quercetin. Free Radic Biol Med 2007;42:1690–1703.
- Kampkotter A, Timpel C, Zurawski RF, Ruhl S, Chovolou Y, Proksch P, Watjen W. Increase of stress resistance and lifespan of Caenorhabditis elegans by quercetin. Comp Biochem Physiol B Biochem Mol Biol 2008;149:314–323.
- Pietsch K, Saul N, Menzel R, Stürzenbaum SR, Steinberg CE. Quercetin mediated lifespan extension in Caenorhabditis elegans is modulated by age-1, daf-2, sek-1 and unc-43. Biogerontology 2009;10:565–578.
- Ishii T, Itoh K, Ruiz E, Leake DS, Unoki H, Yamamoto M, Mann GE. Role of Nrf2 in the regulation of CD36 and stress protein expression in murine macrophages: activation by oxidatively modified LDL and 4-hydroxynonenal. Circ Res 2004;94:609–616.
- Reiser K, McCormick RJ, Rucker RB. Enzymatic and nonenzymatic cross-linking of collagen and elastin. FASEB J 1992;6:2439–2449.
- Rees MD, Kennett EC, Whitelock JM, Davies MJ. Oxidative damage to extracellular matrix and its role in human pathologies. Free Radic Biol Med 2008;44:1973–2001.
- Kuzuya M, Asai T, Kanda S, Maeda K, Cheng XW, Iguchi A. Glycation cross-links inhibit matrix metalloproteinase-2 activation in vascular smooth muscle cells cultured on collagen lattice. Diabetologia 2001;44:433–436.
- Chellan P, Nagaraj RH. Protein crosslinking by the Maillard reaction: dicarbonyl-derived imidazolium crosslinks in aging and diabetes. Arch Biochem Biophys 1999;368:98–104.
- Sell DR, Biemel KM, Reihl O, Lederer MO, Strauch CM, Monnier VM. Glucosepane is a major protein cross-link of the senescent human extracellular matrix. Relationship with diabetes. J Biol Chem 2005;280:12310–12315.
- Biemel KM, Friedl DA, Lederer MO. Identification and quantification of major maillard cross-links in human serum albumin and lens protein. Evidence for glucosepane as the dominant compound. J Biol Chem 2002;277:24907–24915.
- Nasiri R, Field MJ, Zahedi M, Moosavi-Movahedi AA. Cross-linking mechanisms of arginine and lysine with α,β-dicarbonyl compounds in aqueous solution. J Phys Chem A 2011;115:13542–13555.
- Vasan S, Zhang X, Zhang X, Kapurniotu A, Bernhagen J, Teichberg S, et al.. An agent cleaving glucose-derived protein crosslinks in vitro and in vivo. Nature 1996;382:275–278.
- Thornalley PJ, Minhas HS. Rapid hydrolysis and slow alpha,beta-dicarbonyl cleavage of an agent proposed to cleave glucose-derived protein cross-links. Biochem Pharmacol 1999;57:303–307.
- Wolffenbuttel BH, Boulanger CM, Crijns FR, Huijberts MS, Poitevin P, Swennen GN, et al.. Breakers of advanced glycation end products restore large artery properties in experimental diabetes. Proc Natl Acad Sci USA 1998;95:4630–4634.
- Cheng G, Wang LL, Qu WS, Long L, Cui H, Liu HY, et al.. C16, a novel advanced glycation endproduct breaker, restores cardiovascular dysfunction in experimental diabetic rats. Acta Pharmacol Sin 2005;26:1460–1466.
- Cheng G, Wang LL, Long L, Liu HY, Cui H, Qu WS, Li S. Beneficial effects of C36, a novel breaker of advanced glycation endproducts cross-links, on the cardiovascular system of diabetic rats. Br J Pharmacol 2007;152:1196–1206.
- Joshi D, Gupta R, Dubey A, Shiwalkar A, Pathak P, Gupta RC, et al.. TRC4186, a novel AGE-breaker, improves diabetic cardiomyopathy and nephropathy in Ob-ZSF1 model of type 2 diabetes. J Cardiovasc Pharmacol 2009;54:72–81.
- Chandra KP, Shiwalkar A, Kotecha J, Thakkar P, Srivastava A, Chauthaiwale V, et al.. Phase I clinical studies of the advanced glycation end-product (AGE)-breaker TRC4186: safety, tolerability and pharmacokinetics in healthy subjects. Clin Drug Investig 2009;29:559–575.
- Nagai R, Murray DB, Metz TO, Baynes JW. Chelation: a fundamental mechanism of action of AGE inhibitors, AGE breakers, and other inhibitors of diabetes complications. Diabetes 2012;61:549–559.
- Yang S, Litchfield JE BJ. AGE-breakers cleave model compounds, but do not break Maillard crosslinks in skin and tail collagen from diabetic rats. Arch Biochem Biophys 2003;412:42–46.
- Mentink CJ, Hendriks M, Levels AA, Wolffenbuttel BH. Glucose-mediated cross-linking of collagen in rat tendon and skin. Clin Chim Acta 2002;321:69–76.
- Price DL, Rhett PM, Thorpe SR, Baynes JW. Chelating activity of advanced glycation end-product inhibitors. J Biol Chem 2001;276:48967–48972.
- Rahbar S, Figarola JL. Novel inhibitors of advanced glycation endproducts. Arch Biochem Biophys 2003;419:63–79.
- Kim T, Spiegel DA. The unique reactivity of N-phenacyl-derived thiazolium salts toward alpha-dicarbonyl compounds. Rejuvenation Res 2013;16:43–50.
- Li YM, Tan AX, Vlassara H. Antibacterial activity of lysozyme and lactoferrin is inhibited by binding of advanced glycation-modified proteins to a conserved motif. Nat Med 1995;1:1057–1061.
- Zheng F, Cai W, Mitsuhashi T, Vlassara H. Lysozyme enhances renal excretion of advanced glycation endproducts in vivo and suppresses adverse age-mediated cellular effects in vitro: a potential AGE sequestration therapy for diabetic nephropathy?Mol Med 2001;7:737–747.
- Mitsuhashi T, Li YM, Fishbane S, Vlassara H. Depletion of reactive advanced glycation endproducts from diabetic uremic sera using a lysozyme-linked matrix. J Clin Invest 1997;100:847–854.
- Cocchietto M, Zorzin L, Toffoli B, Candido R, Fabris B, Stebel M, Sava G. Orally administered microencapsulated lysozyme downregulates serum AGE and reduces the severity of early-stage diabetic nephropathy. Diabetes Metab 2008;34:587–594.
- Liu H, Zheng F, Cao Q, Ren B, Zhu L, Striker G, Vlassara H. Amelioration of oxidant stress by the defensin lysozyme. Am J Physiol Endocrinol Metab 2006;290: E824–E832.
- Liu H, Zheng F, Li Z, Uribarri J, Ren B, Hutter R, et al.. Reduced acute vascular injury and atherosclerosis in hyperlipidemic mice transgenic for lysozyme. Am J Pathol 2006;169:303–313.
- Pettersson C, Karlsson H, Ståhlman M, Larsson T, Fagerberg B, Lindahl M, et al.. LDL-associated apolipoprotein J and lysozyme are associated with atherogenic properties of LDL found in type 2 diabetes and the metabolic syndrome. J Intern Med 2011;269:306–321.
- Ueda S, Yamagishi S, Takeuchi M, Kohno K, Shibata R, Matsumoto Y, et al.. Oral adsorbent AST-120 decreases serum levels of AGEs in patients with chronic renal failure. Mol Med 2006;12:180–184.
- Yamagishi S, Nakamura K, Matsui T, Inoue H, Takeuchi M. Oral administration of AST-120 (Kremezin) is a promising therapeutic strategy for advanced glycation end product (AGE)-related disorders. Med Hypotheses 2007;69: 666–668.
- Nakamura T, Sato E, Fujiwara N, Kawagoe Y, Suzuki T, Ueda Y, Yamagishi S. Oral adsorbent AST-120 ameliorates tubular injury in chronic renal failure patients by reducing proteinuria and oxidative stress generation. Metabolism 2011;60:260–264.
- Hayashino Y, Fukuhara S, Akizawa T, Asano Y, Wakita T, Onishi Y, Kurokawa K. CAP-KD study group.Cost-effectiveness of administering oral adsorbent AST-120 to patients with diabetes and advance-stage chronic kidney disease. Diabetes Res Clin Pract 2010;90:154–159.
- Maeda K, Hamada C, Hayashi T, Shou I, Wakabayashi M, Fukui M, et al.. Long-term effects of the oral adsorbent, AST-120, in patients with chronic renal failure. J Int Med Res 2009;37:205–213.
- Uribarri J, Woodruff S, Goodman S, Cai W, Chen X, Pyzik R, et al.. Advanced glycation end products in foods and a practical guide to their reduction in the diet. J Am Diet Assoc 2010;110:911–916.
- Sgarbieri VC, Amaya J, Tanaka M, Chichester CO. Nutritional consequences of the Maillard reaction. Amino acid availability from fructose-leucine and fructose-tryptophan in the rat. J Nutr 1973;103:657–663.
- Koschinsky T, He CJ, Mitsuhashi T, Bucala R, Liu C, Buenting C, et al.. Orally absorbed reactive glycation products (glycotoxins): an environmental risk factor in diabetic nephropathy. Proc Natl Acad Sci USA 1997;94: 6474–6479.
- Huebschmann AG, Regensteiner JG, Vlassara H, Reusch JE. Diabetes and advanced glycoxidation end products. Diabetes Care 2006;29:1420–1432.
- Zheng F, He C, Li J, Vlassara H. Restriction of AGE content of food without lowering protein intake prevents diabetic nephropathy in mice. Diabetes 2000;49:A161.
- Vlassara H, Fuh H, Makita Z, Krungkai S, Cerami A, Bucala R. Exogenous advanced glycosylation end products induce complex vascular dysfunction in normal animals: a model for diabetic and aging complications. Proc Natl Acad Sci USA 1992;89:12043–12047.
- Goldin A, Beckman JA, Schmidt AM, Creager MA. Advanced glycation end products: sparking the development of diabetic vascular injury. Circulation 2006;114:597–605.
- Goh SY, Cooper ME. Clinical review: the role of advanced glycation end products in progression and complications of diabetes. J Clin Endocrinol Metab 2008;93:1143–1152.
- Yamagishi S. Role of advanced glycation end products (AGEs) and receptor for AGEs (RAGE) in vascular damage in diabetes. Exp Gerontol 2011;46:217–224.
- Ramasamy R, Vannucci SJ, Yan SS, Herold K, Yan SF, Schmidt AM. Advanced glycation end products and RAGE: a common thread in aging, diabetes, neurodegeneration, and inflammation. Glycobiology 2005;15:16R–28R.
- Ramasamy R, Yan SF, Schmidt AM. The diverse ligand repertoire of the receptor for advanced glycation endproducts and pathways to the complications of diabetes. Vascul Pharmacol 2012;57:160–167.
- Stogsdill JA, Stogsdill MP, Porter JL, Hancock JM, Robinson AB, Reynolds PR. Embryonic overexpression of receptors for advanced glycation end-products by alveolar epithelium induces an imbalance between proliferation and apoptosis. Am J Respir Cell Mol Biol 2012;47:60–66.
- Sakatani S, Yamada K, Homma C, Munesue S, Yamamoto Y, Yamamoto H, Hirase H. Deletion of RAGE causes hyperactivity and increased sensitivity to auditory stimuli in mice. PLoS One 2009;4:e8309.
- Sorci G, Riuzzi F, Giambanco I, Donato R. RAGE in tissue homeostasis, repair and regeneration. Biochim Biophys Acta 2013;1833:101–109.
- Han YT, Choi GI, Son D, Kim NJ, Yun H, Lee S, et al.. Ligand-based design, synthesis, and biological evaluation of 2-aminopyrimidines, a novel series of receptor for advanced glycation end products (RAGE) inhibitors. J Med Chem 2012;55:9120–9135.
- Sparvero LJ, Asafu-Adjei D, Kang R, Tang D, Amin N, Im J, et al.. RAGE (Receptor for Advanced Glycation Endproducts), RAGE ligands, and their role in cancer and inflammation. J Transl Med 2009;7:17.
- Barlovic DP, Soro-Paavonen A, Jandeleit-Dahm KA. RAGE biology, atherosclerosis and diabetes. Clin Sci (Lond) 2011;121:43–55.
- Yamamoto Y, Kato I, Doi T, Yonekura H, Ohashi S, Takeuchi M, et al.. Development and prevention of advanced diabetic nephropathy in RAGE-overexpressing mice. J Clin Invest 2001;108:261–268.
- Chen CY, Abell AM, Moon YS, Kim KH. An advanced glycation end product (AGE)-receptor for AGEs (RAGE) axis restores adipogenic potential of senescent preadipocytes through modulation of p53 protein function. J Biol Chem 2012;287:44498–44507.
- Fritz G. RAGE: a single receptor fits multiple ligands. Trends Biochem Sci 2011;36:625–632.
- Xue J, Rai V, Singer D, Chabierski S, Xie J, Reverdatto S, et al.. Advanced glycation end product recognition by the receptor for AGEs. Structure 2011;19:722–732.
- Schalkwijk CG, Miyata T. Early- and advanced non- enzymatic glycation in diabetic vascular complications: the search for therapeutics. Amino Acids 2012;42:1193–1204.
- Touré F, Fritz G, Li Q, Rai V, Daffu G, Zou YS, et al.. Formin mDia1 mediates vascular remodeling via integration of oxidative and signal transduction pathways. Circ Res 2012;110:1279–1293.
- Shang L, Ananthakrishnan R, Li Q, Quadri N, Abdillahi M, Zhu Z, et al.. RAGE modulates hypoxia/reoxygenation injury in adult murine cardiomyocytes via JNK and GSK-3beta signaling pathways. PLoS One 2010;5:e10092.
- Kumano-Kuramochi M, Ohnishi-Kameyama M, Xie Q, Niimi S, Kubota F, Komba S, Machida S. Minimum stable structure of the receptor for advanced glycation end product possesses multi ligand binding ability. Biochem Biophys Res Commun 2009;386:130–134.
- Basta G. Receptor for advanced glycation endproducts and atherosclerosis: From basic mechanisms to clinical implications. Atherosclerosis 2008;196:9–21.
- Jiao L, Chen L, Alsarraj A, Ramsey D, Duan Z, El-Serag HB. Plasma soluble receptor for advanced glycation end-products and risk of colorectal adenoma. Int J Mol Epidemiol Genet 2012;3:294–304.
- Falcone C, Emanuele E, D’Angelo A, Buzzi MP, Belvito C, Cuccia M, Geroldi D. Plasma levels of soluble receptor for advanced glycation end products and coronary artery disease in nondiabetic men. Arterioscler Thromb Vasc Biol 2005;25:1032–1037.
- Katakami N, Matsuhisa M, Kaneto H, Matsuoka TA, Sakamoto K, Nakatani Y, et al.. Decreased endogenous secretory advanced glycation end product receptor in type 1 diabetic patients: its possible association with diabetic vascular complications. Diabetes Care 2005;28:2716–2721.
- Koyama H, Shoji T, Fukumoto S, Shinohara K, Shoji T, Emoto M, et al.. Low circulating endogenous secretory receptor for AGEs predicts cardiovascular mortality in patients with end-stage renal disease. Arterioscler Thromb Vasc Biol 2007;27:147–153.
- Nakamura K, Yamagishi S, Adachi H, Kurita-Nakamura Y, Matsui T, Yoshida T, et al.. Elevation of soluble form of receptor for advanced glycation end products (sRAGE) in diabetic subjects with coronary artery disease. Diabetes Metab Res Rev 2007;23:368–371.
- Yang SJ, Kim S, Hwang SY, Kim TN, Choi HY, Yoo HJ, et al.. Association between sRAGE, esRAGE levels and vascular inflammation: analysis with (18)F-fluorodeoxyglucose positron emission tomography. Atherosclerosis 2012; 220:402–406.
- Qian L, Ding L, Cheng L, Zhu X, Zhao H, Jin J, et al.. Early biomarkers for post-stroke cognitive impairment. J Neurol 2012;259:2111–2118.
- Wittkowski H, Hirono K, Ichida F, Vogl T, Ye F, Yanlin X, et al.. Acute Kawasaki disease is associated with reverse regulation of soluble receptor for advance glycation end products and its proinflammatory ligand S100A12. Arthritis Rheum 2007;56:4174–4181.
- Greco R, Amantea D, Mangione AS, Petrelli F, Gentile R, Nappi G, et al.. Modulation of RAGE isoforms expression in the brain and plasma of rats exposed to transient focal cerebral ischemia. Neurochem Res 2012;1508–1516.
- Liang F, Jia J, Wang S, Qin W, Liu G. Decreased plasma levels of soluble low density lipoprotein receptor-related protein-1 (sLRP) and the soluble form of the receptor for advanced glycation end products (sRAGE) in the clinical diagnosis of Alzheimer's disease. J Clin Neurosci 2012;[Epub ahead of print].
- Marsche G, Weigle B, Sattler W, Malle E. Soluble RAGE blocks scavenger receptor CD36-mediated uptake of hypochlorite-modified low-density lipoprotein. FASEB J 2007;21:3075–3082.
- Kotani K, Caccavello R, Taniguchi N, Gugliucci A. Circulating soluble receptor for advanced glycation end products is inversely correlated to oxidized low-density lipoproteins in asymptomatic subjects. J Int Med Res 2012;40:1878–1883.
- Rai V, Touré F, Chitayat S, Pei R, Song F, Li Q, et al.. Lysophosphatidic acid targets vascular and oncogenic pathways via RAGE signaling. J Exp Med 2012;209:2339–2350.
- Lanati N, Emanuele E, Brondino N, Geroldi D. Soluble RAGE-modulating drugs: state-of-the-art and future perspectives for targeting vascular inflammation. Curr Vasc Pharmacol 2010;8:86–92.
- Basta G, Navarra T, De Simone P, Del Turco S, Gastaldelli A, Filipponi F. What is the role of the receptor for advanced glycation end products-ligand axis in liver injury?Liver Transpl 2011;17:633–640.
- Wendt TM, Tanji N, Guo J, Kislinger TR, Qu W, Lu Y, et al.. RAGE drives the development of glomerulosclerosis and implicates podocyte activation in the pathogenesis of diabetic nephropathy. Am J Pathol 2003;162:1123–1137.
- Chen Y, Yan SS, Colgan J, Zhang HP, Luban J, Schmidt AM, et al.. Blockade of late stages of autoimmune diabetes by inhibition of the receptor for advanced glycation end products. J Immunol 2004;173:1399–1405.
- Deane R, Singh I, Sagare AP, Bell RD, Ross NT, LaRue B, et al.. A multimodal RAGE-specific inhibitor reduces amyloid β-mediated brain disorder in a mouse model of Alzheimer disease. J Clin Invest 2012;122:1377–1392.
- Zlokovic BV. New therapeutic targets in the neurovascular pathway in Alzheimer's disease. Neurotherapeutics 2008; 5:409–414.
- Sabbagh MN, Agro A, Bell J, Aisen PS, Schweizer E, Galasko D. PF-04494700, an oral inhibitor of receptor for advanced glycation end products (RAGE), in Alzheimer's disease. Alzheimer Dis Assoc Disord 2011;25:206–212.
- Feng L, Xu YH, Wang SS, Au-Yeung W, Zheng ZG, Zhu Q, Xiang P. Competitive binding between 4,4’-diphenylmethane- bis(methyl) carbamate and RAGE ligand MG-H1 on human umbilical vein endothelial cell by cell membrane chromatography. J Chromatogr B Analyt Technol Biomed Life Sci 2012;881–882:55–62.
- Jung DH, Kim YS, Kim JS. Screening system of blocking agents of the receptor for advanced glycation endproducts in cells using fluorescence. Biol Pharm Bull 2012;35: 1826–1830.
- Win MT, Yamamoto Y, Munesue S, Saito H, Han D, Motoyoshi S, et al.. Regulation of RAGE for attenuating progression of diabetic vascular complications. Exp Diabetes Res 2012;2012:894605.
- Gospodarska E, Kupniewska-Kozak A, Goch G, Dadlez M. Binding studies of truncated variants of the Aβ peptide to the V-domain of the RAGE receptor reveal Aβ residues responsible for binding. Biochim Biophys Acta 2011;1814:592–609.
- Hearst SM, Walker LR, Shao Q, Lopez M, Raucher D, Vig PJ. The design and delivery of a thermally responsive peptide to inhibit S100B-mediated neurodegeneration. Neuroscience 2011;197:369–380.
- Arumugam T, Ramachandran V, Gomez SB, Schmidt AM, Logsdon CD. S100P-derived RAGE antagonistic peptide reduces tumor growth and metastasis. Clin Cancer Res 2012;18:4356–4364.
- Rao NV, Argyle B, Xu X, Reynolds PR, Walenga JM, Prechel M, et al.. Low anticoagulant heparin targets multiple sites of inflammation, suppresses heparin-induced thrombocytopenia, and inhibits interaction of RAGE with its ligands. Am J Physiol Cell Physiol 2010;299:C97–C110.
- Bansal S, Siddarth M, Chawla D, Banerjee BD, Madhu SV, Tripathi AK. Advanced glycation end products enhance reactive oxygen and nitrogen species generation in neutrophils in vitro. Mol Cell Biochem 2012;361:289–296.
- Zhao LM, Zhang W, Wang LP, Li GR, Deng XL. Advanced glycation end products promote proliferation of cardiac fibroblasts by upregulation of K(Ca)3.1 channels. Pflugers Arch 2012;464:613–621.
- Cheng CL, Tang Y, Zheng Z, Liu X, Ye ZC, Wang C, Lou TQ. Advanced glycation end-products activate the renin-angiotensin system through the RAGE/PI3-K signaling pathway in podocytes. Clin Invest Med 2012;35:E282.
- Mizumoto S, Takahashi J, Sugahara K. Receptor for advanced glycation end products (RAGE) functions as receptor for specific sulfated glycosaminoglycans, and anti-RAGE antibody or sulfated glycosaminoglycans delivered in vivo inhibit pulmonary metastasis of tumor cells. J Biol Chem 2012; 287:18985–18994.
- Clynes R, Herold K, Schmidt AM. RAGE: exacting a toll on the host in response to polymicrobial sepsis and Listeria monocytogenes. Crit Care 2007;11:183.
- Webster SJ, Mruthinti S, Hill WD, Buccafusco JJ, Terry AVJ. An aqueous orally active vaccine targeted against a RAGE/AB complex as a novel therapeutic for Alzheimer's disease. Neuromolecular Med 2012;14:119–130.
- Zhang Q, O’Hearn S, Kavalukas SL, Barbul A. Role of high mobility group box 1 (HMGB1) in wound healing. J Surg Res 2012;176:343–347.
- Vugmeyster Y, DeFranco D, Pittman DD, Xu X. Pharmacokinetics and lung distribution of a humanized anti-RAGE antibody in wild-type and RAGE-/- mice. MAbs 2010;2:571–575.
- Christaki E, Opal SM, Keith JCJ, Kessimian N, Palardy JE, Parejo NA, et al.. A monoclonal antibody against RAGE alters gene expression and is protective in experimental models of sepsis and pneumococcal pneumonia. Shock 2011;35:492–498.
- Müller-Krebs S, Kihm LP, Madhusudhan T, Isermann B, Reiser J, Zeier M, Schwenger V. Human RAGE antibody protects against AGE-mediated podocyte dysfunction. Nephrol Dial Transplant 2012;27:3129–3136.
- Li F, Cai Z, Chen F, Shi X, Zhang Q, Chen S, et al.. Pioglitazone attenuates progression of aortic valve calcification via down-regulating receptor for advanced glycation end products. Basic Res Cardiol 2012;107:306.
- Yamagishi S, Nakamura K, Matsui T. Regulation of advanced glycation end product (AGE)-receptor (RAGE) system by PPAR-gamma agonists and its implication in cardiovascular disease. Pharmacol Res 2009;60:174–178.
- Marx N, Walcher D, Ivanova N, Rautzenberg K, Jung A, Friedl R, et al.. Thiazolidinediones reduce endothelial expression of receptors for advanced glycation end products. Diabetes 2004;53:2662–2668.
- Liu X, Luo D, Zheng M, Hao Y, Hou L, Zhang S. Effect of pioglitazone on insulin resistance in fructose-drinking rats correlates with AGEs/RAGE inhibition and block of NADPH oxidase and NF kappa B activation. Eur J Pharmacol 2010;629:153–158.
- Wang K, Zhou Z, Zhang M, Fan L, Forudi F, Zhou X, et al.. Peroxisome proliferator-activated receptor gamma down-regulates receptor for advanced glycation end products and inhibits smooth muscle cell proliferation in a diabetic and nondiabetic rat carotid artery injury model. J Pharmacol Exp Ther 2006;317:37–43.
- Matsui T, Yamagishi S, Takeuchi M, Ueda S, Fukami K, Okuda S. Nifedipine inhibits advanced glycation end products (AGEs) and their receptor (RAGE) interaction-mediated proximal tubular cell injury via peroxisome proliferator- activated receptor-gamma activation. Biochem Biophys Res Commun 2010;398:326–330.
- Ishibashi Y, Matsui T, Takeuchi M, Yamagishi S. Metformin inhibits advanced glycation end products (AGEs)-induced renal tubular cell injury by suppressing reactive oxygen species generation via reducing receptor for AGEs (RAGE) expression. Horm Metab Res 2012;44:891–895.
- Matsui T, Yamagishi S, Takeuchi M, Ueda S, Fukami K, Okuda S. Irbesartan inhibits advanced glycation end product (AGE)-induced proximal tubular cell injury in vitro by suppressing receptor for AGEs (RAGE) expression. Pharmacol Res 2010;61:34–39.
- Ishibashi Y, Matsui T, Takeuchi M, Yamagishi S. Beneficial effects of metformin and irbesartan on advanced glycation end products (AGEs)-RAGE-induced proximal tubular cell injury. Pharmacol Res 2012;65:297–302.
- Yamagishi S, Matsui T, Nakamura K, Takeuchi M, Inoue H. Telmisartan inhibits advanced glycation end products (AGEs)-elicited endothelial cell injury by suppressing AGE receptor (RAGE) expression via peroxisome proliferator-activated receptor-gammaactivation. Protein Pept Lett 2008;15:850–853.
- Yamagishi S, Matsui T, Nakamura K, Inoue H, Takeuchi M, Ueda S, et al.. Olmesartan blocks advanced glycation end products (AGEs)-induced angiogenesis in vitro by suppressing receptor for AGEs (RAGE) expression. Microvasc Res 2008;75:130–134.
- Ishibashi Y, Matsui T, Takeuchi M, Yamagishi S. Vardenafil, an inhibitor of phosphodiesterase-5, blocks advanced glycation end product (AGE)-induced up-regulation of monocyte chemoattractant protein-1 mRNA levels in endothelial cells by suppressing AGE receptor (RAGE) expression via elevation of cGMP. Clin Exp Med 2011;11:131–135.
- Ishibashi Y, Matsui T, Takeuchi M, Yamagishi S. Glucagon-like peptide-1 (GLP-1) inhibits advanced glycation end product (AGE)-induced up-regulation of VCAM-1 mRNA levels in endothelial cells by suppressing AGE receptor (RAGE) expression. Biochem Biophys Res Commun 2010;391:1405–1408.
- Ishibashi Y, Matsui T, Ohta K, Tanoue R, Takeuchi M, Asanuma K, et al.. PEDF inhibits AGE-induced podocyte apoptosis via PPAR-gamma activation. Microvasc Res 2013;85:54–58.
- Ishibashi Y, Yamagishi S, Matsui T, Ohta K, Tanoue R, Takeuchi M, et al.. Pravastatin inhibits advanced glycation end products (AGEs)-induced proximal tubular cell apoptosis and injury by reducing receptor for AGEs (RAGE) level. Metabolism 2012;61:1067–1072.
- Wu J, Zhao MY, Zheng H, Zhang H, Jiang Y. Pentoxifylline alleviates high-fat diet-induced non-alcoholic steatohepatitis and early atherosclerosis in rats by inhibiting AGE and RAGE expression. Acta Pharmacol Sin 2010;31:1367–1375.
- Jing YH, Chen KH, Yang SH, Kuo PC, Chen JK. Resveratrol ameliorates vasculopathy in STZ-induced diabetic rats: role of AGE-RAGE signalling. Diabetes Metab Res Rev 2010;26:212–222.
- Yu W, Wu J, Cai F, Xiang J, Zha W, Fan D, et al.. Curcumin alleviates diabetic cardiomyopathy in experimental diabetic rats. PLoS One 2012;7:e52013.
- Li JZ, Wu JH, Yu SY, Shao QR, Dong XM. Inhibitory effects of paeoniflorin on lysophosphatidylcholine-induced inflammatory factor production in human umbilical vein endothelial cells. Int J Mol Med 2013;31:493–497.
- Chen F, Zhang HQ, Zhu J, Liu KY, Cheng H, Li GL, et al.. Puerarin enhances superoxide dismutase activity and inhibits RAGE and VEGF expression in retinas of STZ-induced early diabetic rats. Asian Pac J Trop Med 2012;5:891–896.
- Liu R, Wu CX, Zhou D, Yang F, Tian S, Zhang L, et al.. Pinocembrin protects against β-amyloid-induced toxicity in neurons through inhibiting receptor for advanced glycation end products (RAGE)-independent signaling pathways and regulating mitochondrion-mediated apoptosis. BMC Med 2012;10:105.
- Xiang M, Wang J, Zhang Y, Ling J, Xu X. Attenuation of aortic injury by ursolic acid through RAGE-Nox-NFκB pathway in streptozocin-induced diabetic rats. Arch Pharm Res 2012;35:877–886.
- Lee SH, Kim YS, Lee SJ, Lee BC. The protective effect of Salvia miltiorrhiza in an animal model of early experimentally induced diabetic nephropathy. J Ethnopharmacol 2011;137:1409–1414.
- Peng CH, Chyau CC, Chan KC, Chan TH, Wang CJ, Huang CN. Hibiscus sabdariffa polyphenolic extract inhibits hyperglycemia, hyperlipidemia, and glycation-oxidative stress while improving insulin resistance. J Agric Food Chem 2011;59:9901–9909.
- Xu L, Li B, Cheng M, Zhang W, Pan J, Zhang C, Gao H. Oral administration of grape seed proanthocyanidin extracts downregulate RAGE dependant nuclear factor- kappa BP65 expression in the hippocampus of streptozotocin induced diabetic rats. Exp Clin Endocrinol Diabetes 2008;116: 215–224.
- Sakuraoka Y, Sawada T, Okada T, Shiraki T, Miura Y, Hiraishi K, et al.. MK615 decreases RAGE expression and inhibits TAGE-induced proliferation in hepatocellular carcinoma cells. World J Gastroenterol 2010;16:5334–5341.
- Song MK, Salam NK, Roufogalis BD, Huang TH. Lycium barbarum (Goji Berry) extracts and its taurine component inhibit PPAR-γ-dependent gene transcription in human retinal pigment epithelial cells: Possible implications for diabetic retinopathy treatment. Biochem Pharmacol 2011; 82:1209–1218.
- Yan FL, Zheng Y, Zhao FD. Effects of Ginkgo biloba extract EGb761 on expression of RAGE and LRP-1 in cerebral microvascular endothelial cells under chronic hypoxia and hypoglycemia. Acta Neuropathol 2008;116:529–535.
- Baek GH, Jang YS, Jeong SI, Cha J, Joo M, Shin SW, et al.. Rehmannia glutinosa suppresses inflammatory responses elicited by advanced glycation end products. Inflammation 2012;35:1232–1241.
- de Bittencourt Pasquali MA, Gelain DP, Zeidán-Chuliá F, Pires AS, Gasparotto J, Terra SR, Moreira JC. Vitamin A (retinol) downregulates the receptor for advanced glycation endproducts (RAGE) by oxidant-dependent activation of p38 MAPK and NF-kB in human lung cancer A549 cells. Cell Signal 2013;25:939–954.
- Pillai SS, Sugathan JK, Indira M. Selenium downregulates RAGE and NFκB expression in diabetic rats. Biol Trace Elem Res 2012;149:71–77.
- Wei W, Chen M, Zhu Y, Wang J, Zhu P, Li Y, Li J. Down-regulation of vascular HMGB1 and RAGE expression by n-3 polyunsaturated fatty acids is accompanied by amelioration of chronic vasculopathy of small bowel allografts. J Nutr Biochem 2012;23:1333–1340.
- Chen SA, Chen HM, Yao YD, Hung CF, Tu CS, Liang YJ. Topical treatment with anti-oxidants and Au nanoparticles promote healing of diabetic wound through receptor for advance glycation end-products. Eur J Pharm Sci 2012;47:875–883.
- González I, Romero J, Rodríguez BL, Pérez-Castro R, Rojas A. The immunobiology of the receptor of advanced glycation end-products: Trends and challenges. Immunobiology 2012;Epub ahead of print.
- Basta G, Schmidt AM, De Caterina R. Advanced glycation end products and vascular inflammation: implications for accelerated atherosclerosis in diabetes. Cardiovasc Res 2004; 63:582–592.
- Baumann M. Role of advanced glycation end products in hypertension and cardiovascular risk: human studies. J Am Soc Hypertens 2012;6:427–435.
- Basta G, Lazzerini G, Massaro M, Simoncini T, Tanganelli P, Fu C, et al.. Advanced glycation end products activate endothelium through signal-transduction receptor RAGE: a mechanism for amplification of inflammatory responses. Circulation 2002;105:816–822.
- Farmer DG, Kennedy S. RAGE, vascular tone and vascular disease. Pharmacol Ther 2009;124:185–194.
- Sakaguchi M, Murata H, Yamamoto K, Ono T, Sakaguchi Y, Motoyama A, et al.. TIRAP, an adaptor protein for TLR2/4, transduces a signal from RAGE phosphorylated upon ligand binding. PLoS One 2011;6:e23132.
- Fukami K, Ueda S, Yamagishi S, Kato S, Inagaki Y, Takeuchi M, et al.. AGEs activate mesangial TGF-beta-Smad signaling via an angiotensin II type I receptor interaction. Kidney Int 2004;66:2137–2147.
- Xu Y, Toure F, Qu W, Lin L, Song F, Shen X, et al.. Advanced glycation end product (AGE)-receptor for AGE (RAGE) signaling and up-regulation of Egr-1 in hypoxic macrophages. J Biol Chem 2010;285:23233–23240.
- Hudson BI, Kalea AZ, Del Mar Arriero M, Harja E, Boulanger E, D’Agati V, Schmidt AM. Interaction of the RAGE cytoplasmic domain with diaphanous-1 is required for ligand-stimulated cellular migration through activation of Rac1 and Cdc42. J Biol Chem 2008;283:34457–34468.
- Soman S, Raju R, Sandhya VK, Advani J, Khan AA, Harsha HC, et al.. A multicellular signal transduction network of AGE/RAGE signaling. J Cell Commun Signal 2012; Epub ahead of print.
- Matsunaga-Irie S, Maruyama T, Yamamoto Y, Motohashi Y, Hirose H, Shimada A, Murata M, Saruta T. Relation between development of nephropathy and the p22phox C242T and receptor for advanced glycation end product G1704T gene polymorphisms in type 2 diabetic patients. Diabetes Care 2004;27:303–307.
- Wautier MP, Chappey O, Corda S, Stern DM, Schmidt AM, Wautier JL. Activation of NADPH oxidase by AGE links oxidant stress to altered gene expression via RAGE. Am J Physiol Endocrinol Metab 2001;280:E685–E694.
- Yin QQ, Dong CF, Dong SQ, Dong XL, Hong Y, Hou XY, et al.. AGEs induce cell death via oxidative and endoplasmic reticulum stresses in both human SH-SY5Y neuroblastoma cells and rat cortical neurons. Neurobiol Aging 2012;32: 1299–1309.
- Kim JM, Lee EK, Kim DH, Yu BP, Chung HY. Kaempferol modulates pro-inflammatory NF-kappaB activation by suppressing advanced glycation endproducts-induced NADPH oxidase. Age (Dordr) 2010;32:197–208.
- Hu P, Lai D, Lu P, Gao J, He H. ERK and Akt signaling pathways are involved in advanced glycation end product-induced autophagy in rat vascular smooth muscle cells. Int J Mol Med 2012;29:613–618.
- Zhu P, Ren M, Yang C, Hu YX, Ran JM, Yan L. Involvement of RAGE, MAPK and NF-κB pathways in AGEs-induced MMP-9 activation in HaCaT keratinocytes. Exp Dermatol 2012;21:123–129.
- Bansal S, Siddarth M, Chawla D, Banerjee BD, Madhu SV, Tripathi AK. Advanced glycation end products enhance reactive oxygen and nitrogen species generation in neutrophils in vitro. Mol Cell Biochem 2012;361:289–296.
- Gill PS, Wilcox CS. NADPH oxidases in the kidney. Antioxid Redox Signal 2006;8:1597–1607.
- Yan SD, Schmidt AM, Anderson GM, Zhang J, Brett J, Zou YS, et al.. Enhanced cellular oxidant stress by the interaction of advanced glycation end products with their receptors/binding proteins. J Biol Chem 1994;269:9889–9897.
- Zhang J, Slevin M, Duraisamy Y, Gaffney JA, Smith C, Ahmed N. Comparison of protective effects of aspirin, D-penicillamine and vitamin E against high glucose- mediated toxicity in cultured endothelial cells. Biochim Biophys Acta 2006;1762:551–557.
- Yamagishi S, Takeuchi M. Nifedipine inhibits gene expression of receptor for advanced glycation end products (RAGE) in endothelial cells by suppressing reactive oxygen species generation. Drugs Exp Clin Res 2004;30:169–175.
- Yamagishi S, Matsui T, Nakamura K, Inoue H, Takeuchi M, Ueda S, et al.. Olmesartan blocks inflammatory reactions in endothelial cells evoked by advanced glycation end products by suppressing generation of reactive oxygen species. Ophthalmic Res 2008;40:10–15.
- Marx N, Wöhrle J, Nusser T, Walcher D, Rinker A, Hombach V, et al.. Pioglitazone reduces neointima volume after coronary stent implantation: a randomized, placebo-controlled, double-blind trial in nondiabetic patients. Circulation 2005;112:2792–2798.
- Liu R, Zhang TT, Zhou D, Bai XY, Zhou WL, Huang C, et al.. Quercetin protects against the Aβ(25–35)-induced amnesic injury: Involvement of inactivation of RAGE-mediated pathway and conservation of the NVU. Neuropharmacology 2012;Dec 8. pii: S0028-3908(0012)00565-00565 [Epub ahead of print].
- Liu CA, Wang YJ, Na S. Effects soybean isoflavones on RAGE mediated signal transduction in the hippocampus of rats with Alzheimer's disease. Zhongguo Zhong Xi Yi Jie He Za Zhi 2012;32:797–800.
- Huang JS, Chuang LY, Guh JY, Yang YL, Hsu MS. Effect of taurine on advanced glycation end products-induced hypertrophy in renal tubular epithelial cells. Toxicol Appl Pharmacol 2008;233:220–226.
- Liu R, Zhang TT, Zhou D, Bai XY, Zhou WL, Huang C, et al.. Quercetin protects against the Aβ(25–35)-induced amnesic injury: Involvement of inactivation of RAGE- mediated pathway and conservation of the NVU. Neuropharmacology 2013;67:419–431.
- Lu J, Wu DM, Zheng YL, Hu B, Zhang ZF, Ye Q, et al.. Ursolic acid attenuates D-galactose-induced inflammatory response in mouse prefrontal cortex through inhibiting AGEs/RAGE/NF-κB pathway activation. Cereb Cortex 2010;20:2540–2548.
- Golubev A. How could the Gompertz-Makeham law evolve. J Theor Biol 2009;258:1–17.
- Zarkovic N, Ilic Z, Jurin M, Schaur RJ, Puhl H, Esterbauer H. Stimulation of HeLa cell growth by physiological concentrations of 4-hydroxynonenal. Cell Biochem Funct 1993;11: 279–286.
- Dwivedi S, Sharma A, Patrick B, Sharma R, Awasthi YC. Role of 4-hydroxynonenal and its metabolites in signaling. Redox Rep 2007;12:4–10.