Abstract
Advanced glycation end-products (AGEs) are a heterogeneous group of compounds formed by the Maillard chemical process of non- enzymatic glycation of free amino groups of proteins, lipids and nucleic acids. This chemical modification of biomolecules is triggered by endogeneous hyperglycaemic or oxidative stress-related processes. Additionally, AGEs can derive from exogenous, mostly diet-related, sources. Considering that AGE accumulation in tissues correlates with ageing and is a hallmark in several age-related diseases it is not surprising that the role of AGEs in ageing and pathology has become increasingly evident. The receptor for AGEs (RAGE) is a single transmembrane protein being expressed in a wide variety of human cells. RAGE binds a broad repertoire of extracellular ligands and mediates responses to stress conditions by activating multiple signal transduction pathways being mostly responsible for acute and/or chronic inflammation. RAGE activation has been implicated in ageing as well as in a number of age-related diseases, including atherosclerosis, neurodegeneration, arthritis, stoke, diabetes and cancer. Here we present a synopsis of findings that relate to AGEs-reported implication in cell signalling pathways and ageing, as well as in pathology. Potential implications and opportunities for translational research and the development of new therapies are also discussed.
Keywords::
| Abbreviations: | ||
| AD | = | Alzheimer's disease |
| AP-1 | = | Activator Protein 1 |
| AGEs | = | Advanced Glycation End-products |
| AGER1 | = | AGE advanced glycation receptor 1 |
| CML | = | Nϵ-(Carboxymethyl)lysine |
| CRP | = | C-reactive protein |
| ERK | = | extracellular signal-regulated kinase |
| ECM | = | extracellular matrix |
| FOXO1 | = | forkhead box O1 |
| GI | = | gastrointestinal |
| HMGB1 | = | high mobility group box-1 |
| IRF-1 | = | IFN regulatory factor-1 |
| JAK | = | Janus kinase |
| LOX-1 | = | Lipoprotein receptor-1 |
| LDL | = | low density lipoprotein |
| NF-κB | = | nuclear factor-kappa beta |
| RAGE | = | Receptor of AGEs |
| ROS | = | reactive oxygen species |
| STAT | = | Signal Transducer and Activator of Transcription |
| TNF-α | = | tumour necrosis factor-α |
| TGF-β | = | Transforming Growth Factor-beta |
| VEGF | = | vascular endothelial growth factor |
Introduction—chemistry and sources of AGEs
Advanced Glycation End-Products (AGEs) are a heterogeneous group of compounds formed by the Maillard chemical process, which refers to a non-enzymatic glycation of free amino groups of proteins, lipids, and nucleic acids, mostly, by reducing sugars and reactive aldehydes [Citation1,Citation2]. Although the “Maillard” reaction was initially studied in food chemistry [Citation2], it was later realised that it also occurs slowly in biological systems in vivo where it contributes to some of the tissue modifications that take place during ageing and in various pathologies [Citation1]. In biological systems, the process of AGE formation begins under hyperglycaemic and/or oxidative stress conditions and is marked by the conversion of reversibly formed Schiff-base adducts to covalently bound Amadori rearrangement products. In cases of sustained stress, these Amadori products undergo further rearrangement chemical reactions that result in the formation of irreversibly bound moieties known as AGEs that (among others) include Nϵ-(carboxymethyl)lysine (CML), pentosidine (a fluorescent crosslinker AGE), other fluorescent AGEs, and protein crosslinks [Citation3].
Apart from the endogenous biological processes, AGEs are also derived from exogenous sources that mostly relate to heat-processed AGE-rich diet [Citation4]. A major marker for AGEs in food analyses is CML that is generated by the oxidative cleavage of Amadori products and is an important biological marker of oxidative stress in vivo [Citation5]. The CML content in the same food can vary significantly as a result of different conditions and temperature of processing. Food that is eaten raw or processed at lower temperatures has low CML content, whereas foods with high lipid and protein content prepared at high temperatures are characterised by increased CML content [Citation6]. Other AGE-related chemical entities such as methylglyoxal (an intermediate product of the Maillard reaction) are also derived from food and are very reactive as they can readily modify lysine or arginine residues of proteins [Citation7]. Glucose-containing infusion and dialysis fluids may contain glucose degradation products, such as 3-deoxyglucosone which, besides being directly toxic, are direct promoters of AGEs formation [Citation8]; glucose degradation products are generated mostly upon heat sterilisation. Initially, the potential biological role of the exogenous AGEs was ignored as it was assumed that they undergo negligible gastrointestinal (GI) absorption. However, it soon became evident that diet-derived AGEs increase the tissue levels of AGEs and contribute to increased oxidative stress [Citation9]. They also induce inflammatory processes and correlate with age-related diseases [Citation4].
Herein, we will present a synopsis of findings that relate to molecular effects induced by AGEs and their plasma membrane binding receptors, in cell signalling pathways, ageing, and pathology. Also, potential implications and opportunities for translational research and the development of new therapies in relation to AGEs biology will be discussed.
Effects of AGEs on cell signalling pathways
Following cloning of the AGE receptor [Citation10], it became evident that AGEs activate a number of downstream signalling pathways which relate to increased oxidative stress and/or acute or chronic inflammation. These pathways often converge to nuclear factor-kappa beta (NF-κB) activation () and correlate with localised tissue damage [Citation11]. The plasma membrane receptor for AGEs (RAGE) is highly expressed in immune cells, neurons, skeletal cells, muscle, lung, and heart, and it is a single transmembrane multiligand receptor of the immunoglobulin superfamily that can initiate signalling by binding to distinct proinflammatory ligands [Citation12]. This multiligand receptor has one V (involved in most ligands binding) and two C type extracellular domains; a transmembrane domain and a cytoplasmic tail involved in cellular signalling [Citation13]. Binding ligands to RAGE, apart from AGEs, include CML, amyloid-β peptide, members of the S100 protein family, proteins of the high mobility group box-1 (HMGB1) and prions [Citation12]. Additional inflammatory mediators that are induced through AGEs-mediated activation of RAGE and downstream signalling are tumour necrosis factor-α (TNF-α), interleukin-6 and C-reactive protein (CRP) [Citation14]. It is not thus surprising that RAGE has been implicated in ageing, as well as in a number of pathological conditions, including atherosclerosis, arthritis, metabolic syndromes, stroke, neurodegeneration and cancer [Citation14].
Figure 1. Main signalling pathways known to be induced by AGEs binding to RAGE; other RAGE binding ligands include HMGB1 and S100 proteins. At gene expression level, inflammatory responses are mostly mediated by the transcription factors STAT3, NF-κB and Activator Protein 1 (AP-1), although AGEs may also activate Insulin Receptor, TGF-β, VEGF and FOXO1 signalling (not shown here). Additionally, binding of AGEs on RAGE activates JAK2 which induces the phosphorylation of STAT1. Then, homodimerised phosphorylated STAT1, together with IFN regulatory factor-1 (IRF-1), binds to genomic regions of target genes. In a counteracting effect, binding of AGEs to AGER1, or to soluble forms of RAGE suppresses oxidative stress, Ras signalling and inflammation.
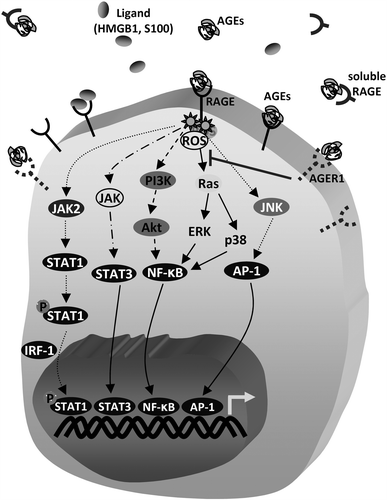
As the RAGE promoter contains sites for NF-κB binding, it seems that there is a positive feedback loop that results in increased RAGE expression thereby prolonging NF-kB activity [Citation15]. This might explain, at least in part, why RAGE is expressed in low levels in most normal adult tissues and increases under stress and pathological conditions [Citation16]. Transduction of signalling from RAGE seems also to relate to ligand-mediated formation of RAGE oligomers indicating that different adaptor proteins (e.g. TIRAP) can be recruited [Citation17].
In more recent studies it was shown that extracellular signal-regulated kinase (ERK)1/2 and Akt signalling pathways are involved in AGE-induced autophagy in rat vascular smooth muscle cells [Citation18] and that Ghrelin-inhibited AGE-induced apoptosis in human endothelial cells by involving ERK1/2 and phosphoinositide 3-kinase/Akt pathways [Citation19]. Moreover, AGEs increase migration and inflammatory responses of adventitial fibroblasts via RAGE, mitogen-activated protein kinases and NF-κB pathways [Citation20] and promoted apoptosis in fibroblasts through activation of reactive oxygen species (ROS), MAPK and the forkhead box O1 (FOXO1) transcription factor [Citation21]. Additionally, AGE (including CML) binding to RAGE has been found to stimulate vascular endothelial growth factor (VEGF) overexpression in retinal pericytes [Citation22] and mesangial Transforming Growth Factor-beta (TGF-β)-Smad signalling via an angiotensin II type I receptor interaction [Citation23]. Finally, studies in four generations of C57BL6 mice fed isocaloric diets with or without AGEs showed that feeding with AGEs promoted a pro-inflammatory phenotype associated with increased adiposity and premature insulin resistance [Citation24]. Interestingly, exogenously added bovine serum albumin-AGE and lysozyme-AGE preparations in human skin fibroblasts apart from similar effects in gene expression patterns also introduced preparation-specific gene expression signatures indicating the existence of different receptor-mediated transmission pathways that are activated by these two different preparations [Citation25].
Little is known about the proximal signalling events after AGEs binding to RAGE, but it seems that following ligand binding to RAGE, an induction of ROS, occurs that increases inflammation through various signalling pathways [Citation22,Citation26] (). It has been found that the mitochondrial electron transport chain and nicotinamide adenine dinucleotide phosphate oxidase pathway participate in RAGE-activation related oxidative stress [Citation27,Citation28].
Apart from RAGE, AGEs can also bind to additional receptors such as the AGE advanced glycation receptor 1 (AGER1). AGER1 counteracts RAGE activation signalling () resulting in decreased oxidative stress [Citation29]. It was found that stimulation of HEK293 cells with AGEs or CML increased phosphorylation of Akt by approximately three-fold in a redox-dependent manner. The use of p66shc mutants showed that the AGE-induced effects required Ser-36 phosphorylation of p66shc. These pro- oxidant responses were suppressed in AGER1 overexpressing cells and reappeared when AGER1 expression was reduced by small interfering RNA [Citation30]. Additional receptors that bind AGEs include macrophage class A and B scavenger receptors, lectin-like oxidised low-density lipoprotein receptor-1 (LOX-1), megalin, toll-like receptors and some other scavenger receptors [Citation31]. Macrophage class A scavenger receptors endocytose soluble AGE-modified proteins and modified LDL, and are involved in adhesion of macrophages with extracellular matrix (ECM) by recognising AGE-collagen, while LOX-1 was found to interact with oxidised LDL and AGE-modified proteins [Citation32]. On the other hand megalin is involved in re-absorption of native filtered proteins but also in endocytosis of AGE-modified proteins [Citation33], while Toll-like receptors participate in innate immune defences and their activation is achieved by different molecules shearing similar three-dimensional pattern [Citation34]. It is worth noting that apart from AGER1, AGEs-RAGE signalling can be also suppressed by the existence of several different molecular forms of RAGE that are formed via alternative gene splicing and post-translational proteolysis of full-length RAGE [Citation35,Citation36]. Soluble RAGE forms () are known as ‘decoy receptors’ since by interacting with RAGE deleterious ligands they diminish signalling that leads to inflammation and RAGE-mediated pathogenesis [Citation37,Citation38].
Contribution of AGEs to ageing and pathophysiology
Ageing refers to a multi-factorial process that is modulated by the interplay among environmental and genetic factors and it affects all tissues and organs of the body [Citation39,Citation40]. The major processes that contribute to ageing and to the emergence of age-related diseases are the accumulation of macromolecular damage, aberrant signalling and chronic inflammation [Citation41–44]. In relation to AGEs it has been shown that ageing correlates with high rates of AGEs formation and accumulation [Citation45] and that AGE accumulation is in fact a reliable biomarker of in vivo ageing [Citation46].
The amount of AGEs inside a cell and tissue reflects the extent of equilibrium between the rates of their formation and clearance. The rate of AGE formation depends on the amount of ROS and reducing sugars, while cathepsins D and L seem to represent two of the major enzymes involved in the degradation of AGEs at intracellular compartments [Citation47,Citation48]. Also, high amounts of intracellular AGEs inhibit both the proteasomal and lysosomal pathways [Citation49] and activate immunoproteasomes via the involvement of RAGE and Janus kinase 2 (JAK2)/Signal Transducer and Activator of Transcription 1 (STAT1) signalling [Citation50] (). Noteworthy, the concentration of immunoproteasome is higher in aged cells [Citation51] and the permanent presence of AGEs might be a potential reason for that.
There are two pathways recognised to participate in AGE contribution to ageing and age-related disorders. The first pathway includes AGE receptors whose activation induces complex intracellular reactions mainly related to stress and inflammatory processes (see above). At the other pathway, AGE-modified proteins (e.g. haemoglobin, albumin or collagen) have altered structure that leads to modified conformation and physico-chemical characteristics, aggregation and, most importantly, altered reactivity. For example, modified glycated haemoglobin (an intermediate in the production of AGEs; see also below) has an increased affinity for oxygen compared to non-modified haemoglobin; this characteristic was found to worsen the exercise capacity of diabetic patients [Citation52]. An AGE-albumin has altered affinity for many ligands, including hormones, vitamins, fatty acids, metal ions and pharmaceuticals, but due to the abundance of albumin and its fast turnover, the effect of the modified fraction of albumin is seemingly not pathologically significant [Citation53]. AGE-collagen molecules on the other hand have a very slow turnover and they form crosslinks that participate in vascular stiffening and circulatory complications [Citation54].
As half-lives of soluble proteins are in general shorter than half-lives of structural proteins (which are mainly affected by AGEs) the effects of their modifications may be limited. However, we assume that under conditions of sustained AGE accumulation the AGE-related modifications at even short-lived proteins and the downstream effects in related signalling pathways (e.g. via RAGE) may be significant [Citation14]. In addition, AGEs contribute in the generation of new antigenic determinants and thus, oxidative post-translational modifications of biomolecules are assumed to be part of the aetiology of age-related autoimmune disorders [Citation55,Citation56]. Native non-immunogenic self proteins may be modified in the backbone or have altered charge and/or conformation after modification by AGEs, resulting in a ‘new’ protein pattern, no longer recognised as self. These non-recognisable epitopes or structures may elicit autoimmune responses where the resultant antibodies interact with self-molecules resulting in the formation of immune complexes. Phagocytic cells then can bind to immune complexes, activating the mechanism of inflammation.
AGEs- and RAGE-mediated ROS production are involved in vascular ageing. Proteins of the extracellular matrix and vascular basement membrane have longer half-lives and they thus accumulate AGEs to a greater extent [Citation54]. Cross-linking of collagen and other proteins by AGEs in the arteries increased the stiffness of the vasculature as they affected the mechanical properties of the modified (mostly long-lived) proteins [Citation57]. Also, higher levels of AGEs have been found in erythrocytes during ageing contributing to alterations in their deformability [Citation58] as well as in the neurons of the aged human brain [Citation59]. Similarly, the amount of AGEs increase in the lens and the retina during ageing, and major structural proteins of the lens (like crystallins) are susceptible to glycation and AGE cross-linking [Citation60]. Furthermore, in skin fibroblasts from aged human donors, an enhanced accumulation of AGE-modified vimentin was observed [Citation61]. Reportedly, the modification of vimentin was site specific and although it was not directly demonstrated that the observed phenotypic effects were due to an AGE-related major impact on the function of the protein, fibroblasts with increased content of AGE-vimentin exhibited reduced contractile capacity, affecting tissue reorganisation and repair [Citation61]. Older adults have increased cross-linking of collagen and deposition of AGEs in skeletal muscle and these findings were correlated with increased muscle stiffness [Citation62]. AGEs may also affect sarcopenia through upregulation of inflammation and endothelial dysfunction in the microcirculation of skeletal muscle [Citation63]. Finally, serum AGEs are significantly higher in patients with osteoporosis compared with those of healthy controls [Citation64] and it has been shown that AGEs contribute to disturbed bone modelling and deterioration of bone tissue quality [Citation65], as well as that the levels of AGE in the bone correlate positively with age and associate negatively with bone density and mineralisation [Citation66,Citation67].
AGEs in pathophysiology
The most significant effects of AGE-induced protein alterations have been observed in patients with diabetes who reportedly have higher serum AGE concentrations compared with those of healthy controls [Citation68,Citation69]. Due to AGE accumulation in hyperglycaemia, AGEs are formed at an accelerated rate in poorly controlled diabetics compared to those of non-diabetics and many of the pathologies of diabetes correlate with increased AGEs, including renal and retinal disease, atherosclerosis, poor glycaemic control and microangiopathy [Citation68–72]. AGEs (particularly AGEs peptides) are mostly disposed in the kidney, this organ is also a major site for accumulation of AGEs and AGE-associated damage [Citation73,Citation74], and it has been found that AGEs concentration in the plasma associates with impaired renal function in a model of acute renal failure in rats [Citation74] or in non-diabetic adults [Citation75]. For years the glycated haemoglobin, HbA1c, has been used as a valuable clinical marker for the detection of hyperglycaemia. Notably however, the Amadori-product (1- deoxyfructosylated amino-terminus of the beta chain) in HbA1c is a glycation product that forms directly through Amadori-rearrangement from the initial Schiff base; thus, it does not qualify as an AGE and it is considered as an intermediate in the production of AGEs [Citation76,Citation77].
In relation to cardiovascular diseases, elevated serum AGEs have been described in patients with coronary artery disease [Citation78] and serum AGEs were associated with aortic stiffness [Citation79]. Elastin and collagen are long-living proteins (see also above) and their AGE-derivatives accumulate gradually during life [Citation54] contributing, along with the AGEs formed in the extracellular matrix [Citation80] to arterial stiffening. Besides their effects on elastin and collagen crosslinking, AGEs may also induce local inflammation and intracellular ROS which all together lead to vascular alterations and the expression of a variety of atherosclerosis-related genes, including VEGF and RAGE [Citation72]. AGE modification of low-density lipoprotein (LDL) results in reduced LDL plasma clearance contributing significantly to increased LDL in vivo, and potentially to atherosclerosis [Citation81]. Oxidised LDL is taken up by macrophages via scavenger receptors leading to formation of foam cells which are key mediators in the inflammatory reaction and atherogenesis [Citation82]. The AGE–RAGE interaction augments angiotensin II-induced smooth muscle cell proliferation and activation thus being involved in accelerated atherosclerosis in diabetes [Citation83]. Also, smooth muscle cell proliferation, migration and neointimal expansion upon arterial injury were strikingly suppressed in homozygous RAGE null mice compared with those observed in wild-type littermates [Citation84]. On the other hand, AGER1 over-expression in mice protected against injury-induced arterial intimal hyperplasia and inflammation by reducing tissue levels of AGEs and oxidative stress [Citation85].
Oxidative stress is an important parameter in the initiation and progression of neurodegenerative disorders in which a progressive decline in neural signal transmission is noted, together with neural loss and deposition of aggregated proteins in the brain. Diseases such as Alzheimer's, Parkinson's, Huntington's, Creutzfeld-Jakob or prion disease, and amyotrophic lateral sclerosis belong to this group of disorders. A common feature of these disorders is increased presence of AGEs in the brain, production of ROS, relatively low levels of antioxidant enzymes and elevated levels of iron that participates in redox reactions [Citation86,Citation87]. Expression of the amyloid precursor protein was found to be induced by AGEs [Citation87] and RAGE has been involved in amyloid β accumulation to the brain [Citation88]. Also, oxidative stress was found to increase expression of RAGE [Citation89] facilitating thus amyloid β accumulation in the brain. Furthermore, accumulation of AGEs in cells and tissues is accelerated in Alzheimer's disease (AD) where AGEs can be detected in pathological deposits such as amyloid plaques and neurofibrillary tangles [Citation90]. Glycoxidation may affect apolipoproteins (ApoB and ApoE), and these modifications have been proposed to impact on the function of the proteins causing impaired cholesterol and fat supply to neurons; axon damage and inappropriate transmission of neural signals, mitochondrial dysfunction, inflammation and further oxidative damage [Citation91–93]. Similar processes may underlie other neurodegenerative disorders, as accumulation of AGEs in proteins (together with Advanced Lipoxidation End products and various other protein oxidation modifications) will promote the accumulation of protease-resistant products, which by generating further oxidative stress and inflammatory reaction will lead to neuronal malfunctioning and death. Besides RAGE, a number of scavenger and other pattern recognition receptors may also participate in brain inflammatory processes and lesions [Citation94,Citation95].
Almost 25% of all cancers are estimated to be associated with inflammation [Citation96] as sustained inflammatory conditions stimulate tumorigenic signalling pathways and oncogenes activation [Citation97,Citation98]. Therefore, considering that sustained AGEs-mediated activation of RAGE signalling leads to increased oxidative stress and inflammation it is not surprising that RAGE ligands are formed and secreted by many types of cancer cells as well as that increased expression of RAGE has been detected in various tumours including colorectal carcinoma, gastric, pancreatic, prostate, breast, brain and ovarian tumours [Citation14]. RAGE-mediated activation of NF-κB has a pro-inflammatory effect as it promotes activation of genes for inducible nitric oxide synthase, TNF-α, interleukins-1, -6 and cyclooxygenase-2. Cyclooxygenase-2-derived prostaglandin E2 mediates cellular immune tolerance during tumorigenesis [Citation99] and the AGE–RAGE signalling enhances the angiogenic potential of hepatocellular carcinoma by inducing VEGF expression [Citation100]. NF-κB promotes activation of genes for X-linked inhibitor of apoptosis and some members of the B-cell lymphoma family (BcL-2 and BcL-X) involved in antiapoptotic signals [Citation96,Citation97]. Also, RAGE sustains autophagy and limits apoptosis promoting pancreatic tumour cell survival [Citation101], while low levels of the soluble ‘decoy’ RAGE forms have been found in patients with breast cancer [Citation102]. A more direct evidence for a pro- tumorigenic role of RAGE arises from mouse models. Specifically, RAGE−/− mice were almost resistant to chemical skin carcinogenesis showing reduced levels of inflammatory mediators, while the few tumours developed were highly differentiated [Citation103]. In the same study, it was shown that RAGE expression on immune cells, but not keratinocytes or endothelial cells, is essential for dermal infiltration and epidermal hyperplasia [Citation103]. In line with these data, in a mouse model of colitis-induced cancer it was found that RAGE plays essential roles in tumor–stromal interactions leading to inflammation-associated colon carcinogenesis [Citation104]. Interestingly, although RAGE is highly expressed in lung tissue, especially at the site of alveolar epithelium, its expression is significantly reduced in lung carcinomas [Citation105] indicating that RAGE may exhibit a tumour-suppressive function in a tissue-dependent manner.
Notably, however beyond the effect(s) of activated RAGE in accelerating age-related diseases it seems that RAGE signalling influences gross behavioural activity patterns in basal conditions, as it has been found that RAGE knockout in mice relates with an increase of home cage activity and in higher sensitivity to auditory signals [Citation106]; thus, systemic therapeutic treatment to occlude RAGE activation (see below) should be exercised cautiously as it may have adverse effects on several physiological functions.
Reduction of AGEs as a strategy for the improvement of healthspan
Strategies to reduce harmful effects of AGEs and increase healthspan (i.e. the disease-free period of life) involve prevention of AGEs formation and/or removal of those already formed. To prevent accumulation of AGEs in the organism it would be essential to adopt a life style that lowers the exposure to harmful agents such as obesity, smoking, physical inactivity and environmental pollution. Environmental pollutants seem to increase the expression of several markers of oxidative stress, including RAGE [Citation107], while, similarly, cigarette smoking was found to increase the levels of AGEs and AGE-ApoB (although the exact type of modification is not specified) in both the plasma [Citation108] and the urine of smokers [Citation109].
Food and the way it is prepared, along with the degree of AGEs intestinal absorption, seem to have the greatest health impact of all exogenous AGE sources as heat- treatment during cooking or roasting and/or irradiation during food sterilisation may increase significantly the content of AGEs and Advanced Lipoxidation End products in food [Citation110]. In relation to food contribution to AGEs accumulation it was found in human studies that approximately 10% of diet-derived AGEs were absorbed from GI and from these, a significant amount remained in the body within 3 days from ingestion [Citation111]. Also, human breast milk contains approximately 70-fold lower amounts of CML than commercial infant milk and breast-fed infants had significantly lower plasma CML compared to infants fed with commercial infant milk [Citation112]. Short-term restriction of AGEs in the diet in either healthy or diabetic subjects was associated with significant decrease of serum AGE levels [Citation4] and reduced expression of inflammation related markers (e.g. TNF-α or CRP) [Citation9,Citation113], while LDL particles were more glycated and oxidised in subjects fed with AGE-rich diet [Citation114]. Consumption of heated or unheated high protein diet (that was otherwise similar in regard to energy, protein and carbohydrate intake) correlated with high serum CML levels in those subjects fed with a high-AGE diet [Citation9].
In support to findings in human studies, prolonged administration of AGEs rich diet to healthy rodents increased plasma levels of AGEs and their related derivatives, as compared with the group on AGE-poor diet [Citation72] and induced glomerular hypertrophy [Citation115], focal glomerulosclerosis, mesangial expansion and albuminuria [Citation116]. Additionally, long-term AGE-rich diets in non-obese diabetic mice correlated with diabetic nephropathy [Citation117], whereas an AGE-poor diet delivered to mice for 2 months decreased serum levels of AGEs and reduced inflammatory responses [Citation118]. Finally, in a study where C57/BL6 mice were fed with an isocaloric high fat diet with either low or high AGEs content only those animals fed with the high fat high-AGEs diet developed diabetes [Citation119].
Thus, inhibition of absorption of dietary AGEs or oral administration of AGE crosslink breakers may be novel approaches for therapeutic intervention in AGEs-related disorders [Citation120]. Specifically, the oral absorbent AST-120 was found to bind to CML and it was shown that administration of AST-120 decreased serum levels of AGEs in patients with chronic renal failure [Citation121]. Similarly, Galectin-3, which is a high-affinity binding protein for AGEs, could serve as a paradigm of approaches that could contribute in the potential elimination and neutralisation of these pathogenic substances. Specifically, it was found that galectin-3 may serve in the endocytosis by macrophages of a heterogeneous pool of AGE moieties, thus contributing to the elimination of these pathogenic substances [Citation122]. Moreover, galectin-3 ablation is associated with increased susceptibility to diabetes- and AGE- induced glomerulopathy (thus indicating a protective role of galectin-3 as an AGE receptor) [Citation123] while, ageing galectin-3 knockout mice develop more pronounced changes in renal function and structure than wild-type mice, in parallel with a more marked degree of AGE accumulation, oxidative stress, and associated low-grade inflammation [Citation124]. In randomised clinical trials using aminoguanidine, a prototype therapeutic agent for the prevention of AGE formation, it was found that it prevented the progression of diabetic retinopathy in diabetic patients [Citation125]. Although due to reported side effects, further clinical trials of aminoguanidine were terminated due to safety concerns, this study was the first proof of the concept that AGEs formation contributes to serious complication of diabetes which can be reduced by administration of a specific therapeutic agent [Citation125]. Moreover, alagebrium chloride (ALT-711); an AGE-lowering agent with beneficial effects in renal structural and functional parameters in diabetes was found to decrease diabetes-accelerated atherosclerosis and age-related myocardial stiffening [Citation126]. Pyridoxamine inhibits Maillard reactions [Citation127,Citation128] and exerted a renoprotective effect in experimental chronic allograft nephropathy in rats [Citation129], while astragalosides isolated from the root of astragalus radix inhibited the formation of AGEs [Citation130]. Benfotiamine suppressed protein damage in renal glomeruli, retina, nerve, plasma and urine in a rat model of diabetes [Citation131] and it also alleviated macro- and micro-vascular endothelial dysfunction and oxidative stress following a meal rich in AGEs in individuals with type 2 diabetes [Citation132]; on a more recent study however, benfotiamine did not resulted in significant reductions (compared to placebo) in plasma or urinary AGEs or plasma markers of endothelial dysfunction and low-grade inflammation in patients with type 2 diabetes and nephropathy [Citation133]. Nevertheless, it has been suggested that natural products can be a promising source for the identification of AGEs inhibitors that could prevent (via daily nutritional intake) lifestyle-related diseases such as diabetic complications and atherosclerosis [Citation134].
Furthermore, inhibition of RAGE signalling may represent an alternative effective way to suppress the effects of AGEs as it has been reported that deletion of RAGE reduces glomerulosclerosis and preserves renal function in the diabetic OVE26 mouse [Citation135]; in support, FPS-ZM1 a potent multimodal RAGE blocker effectively reduced amyloid β-mediated brain disorder in a mouse model of AD [Citation136]. Alternatively, exogenously administered soluble RAGE may capture and eliminate circulating AGEs, thus protecting against the AGEs-elicited tissue damage by acting as a decoy receptor [Citation137].
Conclusion
On the basis of the aforementioned findings it is evident that restriction of AGEs production (and/or accumulation) along with the effective breaking of existing AGEs crosslinks represent preventive and/or therapeutic targets for ageing-associated organ damage and various age-related diseases. Particularly, food preparation by high-temperature processing and deep-frying, that is a relatively recent development in Western diet, most likely relates to an overall increase in the content of AGEs in the diet. Consumption of whole vegetables and fruits is more efficient in overcoming oxidative burden than the use of individual supplements [Citation138], while the so-called Mediterranean diet which is rich in ω-3 fatty acids, carotenoids, vitamins, folic acid and phenolic compounds is considered to contain reduced amounts of dietary AGEs [Citation138].
Nevertheless, a better understanding of the molecular basis and rate of AGEs absorption from the GI is needed. Specifically, it should be critically addressed how consumption of an AGE-rich diet would introduce accumulation of similar modifications in endogenous tissue and/or plasma proteins. It is highly unlikely that following degradation of AGE-modified proteins in the GI the modified amino acids are absorbed and are then used for protein synthesis. Alternatively, modified proteins (or peptides) are directly taken up by the cells raising the possibility of a disrupted intestinal barrier by AGE-modified proteins and initiation of the inflammatory response.
Also, additional knowledge and a direct relationship between AGEs oral administration and longevity should be addressed in, preferentially, large experimental cohorts as the extent of the AGEs harmful effect is still not fully evaluated and significant population diversity in response to AGEs-rich diet has been observed. These studies should be linked with interventions where reduction of AGEs accumulation (by dietary or pharmacological means) will be directly correlated with effects on health- and life-span.
Despite the existence of a number of AGEs and heavy-labelled AGE standards along with very sensitive fluorometric and mass spectrometric assays that are available for AGEs’ measurement, these cannot be used for routine analyses in clinical samples. Thus, an upgrade of the current methodological approaches in quantifying AGEs is needed, especially regarding reference material, referent method and the established analytical validation procedure. These approaches will, among others, help to adopt a universal terminology in the field as by the rather general term AGEs different preparations are usually assigned including AGE-modified proteins, free AGE-modified amino acids or even glyoxal/methylglyoxal (which are not AGEs). The clarification of the used nomenclature will help the non-expert reader to follow the various chemical categories and modifications that are generally grouped under the term AGEs.
Finally, in regard to AGEs biology additional points that need further understanding relate to the characterisation of single nucleotide polymorphisms in large cohorts that may associate with the amount of circulating AGEs and/or RAGE function, as well as to the determination of whether younger adults with high circulating AGE levels are at an increased risk for metabolic syndrome, or other health problems in later life.
In conclusion, the detailed analysis of the AGE-related effects in the overall organism physiology, ageing and pathology remain a very challenging research field with significant translational applications in different disciplines such as food chemistry and biomedical sciences.
Declaration of interest
The authors report no declarations of interest. The authors alone are responsible for the content and writing of the paper.
This work was supported by the EU COST Action CM1001. ON also acknowledges funding from the Ministry of Education and Science of Serbia (Grant number 173042) and IPT funding from INsPiRE/ REGPOT-CT-2011-284460, the Bodosakis Foundation and the Empeirikio Foundation.
References
- Ulrich P, Cerami A. Protein glycation, diabetes, and aging. Recent Prog Horm Res 2001;56:1–21.
- Cho SJ, Roman G, Yeboah F, Konishi Y. The road to advanced glycation end products: a mechanistic perspective. Curr Med Chem 2007;14:1653–1671.
- Takeuchi M, Yamagishi S. Involvement of toxic AGEs (TAGE) in the pathogenesis of diabetic vascular complications and Alzheimer's disease. J. Alzheimers Dis 2009;16:845–858.
- Uribarri J, Cai W, Sandu O, Peppa M, Goldberg T, Vlassara H. Diet-derived advanced glycation end products are major contributors to the body's AGE pool and induce inflammation in healthy subjects. Ann N Y Acad Sci 2005; 1043:461–466.
- Semba RD, Ang A, Talegawkar S, Crasto C, Dalal M, Jardack P, et al. Dietary intake associated with serum versus urinary carboxymethyl-lysine, a major advanced glycation end product, in adults: the Energetics Study. Eur J Clin Nutr 2012;66:3–9.
- Assar SH, Moloney C, Lima M, Magee R, Ames JM. Determination of Nϵ-(carboxymethyl)lysine in food systems by ultra performance liquid chromatography-mass spectrometry. Amino Acids 2009;36:317–326.
- Hipkiss AR. Can the beneficial effects of methionine restriction in rats be explained in part by decreased methylglyoxal generation resulting from suppressed carbohydrate metabolism?Biogerontology 2012;13:633–636.
- Bryland A, Broman M, Erixon M, Klarin B, Linden T, Friberg H, et al. Infusion fluids contain harmful glucose degradation products. Intensive Care Med 2010;36:1213–1220.
- Šebeková K, Somoza V. Dietary advanced glycation endproducts (AGEs) and their health effects – PRO. Mol Nutr Food Res 2007;51:1079–1084.
- Schmidt AM, Vianna M, Gerlach M, Brett J, Ryan J, Kao J, et al. Isolation and characterization of two binding proteins for advanced glycosylation end products from bovine lung which are present on the endothelial cell surface. J Biol Chem. 1992;267:14987–14997.
- Bierhaus A, Humpert PM, Morcos M, Wendt T, Chavakis T, Arnold B, et al. Understanding RAGE, the receptor for advanced glycation end products. J Mol Med 2005;83: 876–886.
- Basta G. Receptor for advanced glycation endproducts and atherosclerosis: from basic mechanisms to clinical implications. Atherosclerosis 2008;196:9–21.
- Sparvero LJ, Asafu-Adjei D, Kang R, Tang D, Amin N, Im J, et al. RAGE (Receptor for Advanced Glycation Endproducts), RAGE Ligands and their role in Cancer and Inflammation. J Translat Med 2009;7:17.
- Riehl A, Németh J, Angel P, Hess J. The receptor RAGE: Bridging inflammation and cancer. Cell Commun Signal 2009;7:12.
- Li J, Schmidt AM. Characterization and functional analysis of the promoter of RAGE, the receptor for advanced glycation end products. J Biol Chem 1997;272:16498–16506.
- Brett J, Schmidt AM, Yan SD, Zou YS, Weidman E, Pinsky D, et al. Survey of the distribution of a newly characterized receptor for advanced glycation end products in tissues. Am J Pathol 1993;143:1699–1712.
- Sakaguchi M, Murata H, Yamamoto K, Ono T, Sakaguchi Y, Motoyama A, et al. TIRAP, an adaptor protein for TLR2/4, transduces a signal from RAGE phosphorylated upon ligand binding. PLoS One 2011;6:e23132.
- Hu P, Lai D, Lu P, Gao J, He H. ERK and Akt signaling pathways are involved in advanced glycation end product-induced autophagy in rat vascular smooth muscle cells. Int J Mol Med 2012;29:613–618.
- Xiang Y, Li Q, Li M, Wang W, Cui C, Zhang J. Ghrelin inhibits AGEs-induced apoptosis in human endothelial cells involving ERK1/2 and PI3K/Akt pathways. Cell Biochem Funct 2011;29:149–155.
- Liu Y, Liang C, Liu X, Liao B, Pan X, Ren Y, et al. AGEs increased migration and inflammatory responses of adventitial fibroblasts via RAGE, MAPK and NF-kappaB pathways. Atherosclerosis 2010;208:34–42.
- Alikhani M, Maclellan CM, Raptis M, Vora S, Trackman PC, Graves DT. Advanced glycation end products induce apoptosis in fibroblasts through activation of ROS, MAP kinases, and the FOXO1 transcription factor. Am J Physiol Cell Physiol 2007;292:C850–C856.
- Yamagishi S, Inagaki Y, Okamoto T, Amano S, Koga K, Takeuchi M, et al. Advanced glycation end products induced apoptosis and overexpression of vascular endothelial growth factor in bovine retinal pericytes. Biochem Biophys Res Commun 2002;290:973–978.
- Fukami K, Ueda S, Yamagishi S, Kato S, Inagaki Y, Takeuchi M, et al. AGEs activate mesangial TGF-beta-Smad signaling via an angiotensin II type I receptor interaction. Kidney Int. 2004;66:2137–2147.
- Cai W, Ramdas M, Zhu L, Chen X, Striker GE, Vlassara H. Oral advanced glycation end products (AGEs) promote insulin resistance and diabetes by depleting the antioxidant defenses AGE receptor-1 and sirtuin 1. Proc Natl Acad Sci USA 2012;109:15888–15893.
- Molinari J, Ruszova E, Velebny V, Robert L. Effect of advanced glycation endproducts on gene expression profiles of human dermal fibroblasts. Biogerontology 2008;9: 177–182.
- Bansal S, Siddarth M, Chawla D, Banerjee BD, Madhu SV, Tripathi AK. Advanced glycation end products enhance reactive oxygen and nitrogen species generation in neutrophils in vitro. Mol Cell Biochem 2012;361:289–296.
- Wautier MP, Chappey O, Corda S, Stern DM, Schmidt AM, Wautier JL.Activation of NADPH oxidase by AGE links oxidant stress to altered gene expression via RAGE. Am J Physiol Endocrinol Metab 2001;280:E685–E694.
- Coughlan MT, Thorburn DR, Penfold SA, Laskowski A, Harcourt BE, Sourris KC, et al. RAGE-induced cytosolic ROS promote mitochondrial superoxide generation in diabetes. J Am Soc Nephrol 2009;20:742–752.
- Cai W, He JC, Zhu L, Lu C, Vlassara H. Advanced glycation end product (AGE) receptor 1 suppresses cell oxidant stress and activation signaling via EGF receptor. Proc Natl Acad Sci USA 2006;103:13801–13806.
- Cai W, He JC, Zhu L, Chen X, Striker GE, Vlassara H. AGE-receptor-1 counteracts cellular oxidant stress induced by AGEs via negative regulation of p66shc-dependent FKHRL1 phosphorylation. Am J Physiol Cell Physiol 2008;294:C145–C152.
- Nagai R, Mera K, Nakajou K, Fujiwara Y, Iwao Y, Imai H, et al. The ligand activity of AGE-proteins to scavenger receptors is dependent on their rate of modification by AGEs. Biochim Biophys Acta 2007;1772:1192–1198.
- Pluddemann A, Neyen C, Gordon S. Macrophage scavenger receptors and host-derived ligands. Methods 2007;43: 207–217.
- Saito A, Takeda T, Hama H, Oyama Y, Hosaka K, Tanuma A, et al. Role of megalin, a proximal tubular endocytic receptor, in the pathogenesis of diabetic and metabolic syndrome-related nephopathies: Protein metabolic overload hypothesis. Nephrology 2005;10:S26–S31.
- Rojas A, Delgado-Lopez F, Gonzalez I, Perez-Castro R, Romero J, Rojas I. The receptor for advanced glycation end-products: A complex signaling scenario for a promiscuous receptor. Cell Signal 2013;25:609–614.
- Ohe K, Watanabe T, Harada S, Munesue S, Yamamoto Y, Yonekura H, Yamamoto H. Regulation of alternative splicing of the receptor for advanced glycation endproducts (RAGE) through G-rich cis-elements and heterogenous nuclear ribonucleoprotein H. J Biochem 2010;147:651–659.
- Raucci A, Cugusi S, Antonelli A, Barabino SM, Monti L, Bierhaus A, et al. A soluble form of the receptor for advanced glycation endproducts (RAGE) is produced by proteolytic cleavage of the membrane-bound form by the sheddase a disintegrin and metalloprotease 10 (ADAM10). FASEB J 2008;22:3716–3727.
- Vazzana N, Santilli F, Cuccurullo C, Davi G. Soluble forms of RAGE in internal medicine. Intern Emerg Med 2009;4:389–401.
- Lertwittayapon T, Tencomnao T, Santiyanont R. Inhibitory effect of alternatively spliced RAGEv1 on the expression of NF-kB and TNF-α in hepatocellular carcinoma cells. Genet Mol Res 2012;11:1712–1720.
- Markaki M, Tavernarakis N. The role of autophagy in genetic pathways influencing ageing. Biogerontology 2011;12: 377–386.
- Rattan SI. Biogerontology: from here to where? The Lord Cohen Medal Lecture-2011. Biogerontology 2012;13:83–91.
- Rattan SI. Increased molecular damage and heterogeneity as the basis of aging. Biol Chem 2008;389:267–272.
- Trougakos IP, Gonos ES. Chapter 9: Oxidative stress in malignant progression: The role of Clusterin, a sensitive cellular biosensor of free radicals. Adv Cancer Res 2009; 104:171–210.
- Rattan SI. Synthesis, modification and turnover of proteins during aging. Adv Exp Med Biol 2010;694:1–13.
- Hubbard VM, Valdor R, Macian F, Cuervo AM. Selective autophagy in the maintenance of cellular homeostasis in aging organisms. Biogerontology 2012;13:21–35.
- Li SY, Du M, Dolence EK, Fang CX, Mayer GE, Ceylan-Isik AF, et al. Aging induces cardiac diastolic dysfunction, oxidative stress, accumulation of advanced glycation endproducts and protein modification. Aging Cell 2005;4:57–64.
- Jacobson J, Lambert AJ, Portero-Otín M, Pamplona R, Magwere T, Miwa S, et al. Biomarkers of aging in Drosophila. Aging Cell 2010;9:466–477.
- Grimm S, Ernst L, Grötzinger N, Höhn A, Breusing N, Reinheckel T, Grune T. Cathepsin D is one of the major enzymes involved in intracellular degradation of AGE- modified proteins. Free Radic Res 2010;44:1013–1026.
- Grimm S, Horlacher M, Catalgol B, Hoehn A, Reinheckel T, Grune T. Cathepsins D and L reduce the toxicity of advanced glycation end products. Free Radic Biol Med 2012;52: 1011–1023.
- Stolzing A, Widmer R, Jung T, Voss P, Grune T. Degradation of glycated bovine serum albumin in microglial cells. Free Radic Biol Med 2006;40:1017–1027.
- Grimm S, Ott C, Hörlacher M, Weber D, Höhn A, Grune T. Advanced-glycation-end-product-induced formation of immunoproteasomes: involvement of RAGE and Jak2/STAT1. Biochem J 2012;448:127–139.
- Stratford FL, Chondrogianni N, Trougakos IP, Gonos ES, Rivett AJ. Proteasome response to interferon-gamma is altered in senescent human fibroblasts. FEBS Lett 2006;580:3989–3994.
- Demir I, Ermiş C, Altunbaş H, Balci MK. Serum HbA1c levels and exercise capacity in diabetic patients. Jpn Heart J 2001;42:607–616.
- Vetter SW, Indurthi VSK. Moderate glycation of serum albumin affects folding, stability, and ligand binding. Clin Chim Acta 2011;412:2105–2116.
- Sell DR, Monnier VM. Molecular basis of arterial stiffening: Role of glycation – A mini-review. Gerontology 2012;58: 227–237.
- Virella G, Thorpe SR, Alderson NL, Stephan EM, Atchley D, Wagner F, Lopes-Virella MF; DCCT/EDIC Research Group. Autoimmune response to advanced glycosylation end- products of human LDL. J Lipid Res 2003;44:487–493.
- Vytásek R, Sedová L, Vilím V. Increased concentration of two different advanced glycation end-products detected by enzyme immunoassays with new monoclonal antibodies in sera of patients with rheumatoid arthritis. BMC Musculoskelet Disord 2010;11:83.
- Greenwald SE. Ageing of the conduit arteries. J Pathol 2007;211:157–172.
- Ando K, Beppu M, Kikugawa K, Nagai R, Horiuchi S. Membrane proteins of human erythrocytes are modified by advanced glycation end products during aging in the circulation. Biochem Biophys Res Commun 1999;258: 123–127.
- Kimura T, Takamatsu J, Ikeda K, Kondo A, Miyakawa T, Horiuchi S. Accumulation of advanced glycation end products of the Maillard reaction with age in human hippocampal neurons. Neurosci Lett 1996;208:53–56.
- Kumar PA, Kumar MS, Reddy GB. Effect of glycation on a-crystallin structure and chaperone-like function. Biochem J 2007;408:251–258.
- Kueper T, Grune T, Prahl S, Lenz H, Welge V, Biernoth T, et al. Vimentin is the specific target in skin glycation. Structural prerequisites, functional consequences, and role in skin aging. J Biol Chem 2007;282:23427–23436.
- Haus JM, Carrithers JA, Trappe SW, Trappe TA. Collagen, cross-linking and advanced glycation end products in aging human skeletal muscle. J Appl Physiol 2007; 103:2068–2076.
- Semba RD, Bandinelli S, Sun K, Guralnik JM, Ferrucci L. Relationship of an advanced glycation end product, plasma carboxymethyl-lysine, with slow walking speed in older adults: the InCHIANTI study. Eur J Appl Physiol 2010;108:191–195.
- Hein G, Wiegand R, Lehmann G, Stein G, Franke S. Advanced glycation end-products pentosidine and Nϵ-carboxymethyllysine are elevated in serum of patients with osteoporosis. Rheumatology 2003;42:1242–1246.
- Hein GE. Glycation endproducts in osteoporosis—is there a pathophysiologic importance?Clin Chim Acta 2006;371: 32–36.
- Odetti P, Rossi S, Monacelli F, Poggi A, Cirnigliaro M, Federici M, Federici A. Advanced glycation end-products and bone loss during aging. Ann N Y Acad Sci 2005;1043: 710–717.
- Tang SY, Zeenath U, Vashishth D. Effects of non-enzymatic glycation on cancellous bone fragility. Bone 2007;40: 1144–1151.
- Aso Y, Inukai T, Tayama K, Takemura Y. Serum concentrations of advanced glycation endproducts are associated with the development of atherosclerosis as well as diabetic microangiopathy in patients with type 2 diabetes. Acta Diabetol 2000;37:87–92.
- Bansal S, Chawla D, Siddarth M, Banerjee BD, Madhu SV, Tripathi AK. A study on serum advanced glycation end products and its association with oxidative stress and paraoxonase activity in type 2 diabetic patients with vascular complications. Clin Biochem 2013;46:109–114.
- Beisswenger PJ, Makita Z, Curphey TJ, Moore LL, Jean S, Brinck-Johnsen T, et al. Formation of immunochemical advanced glycosylation end products precedes and correlates with early manifestations of renal and retinal disease in diabetes. Diabetes 1995;44:824–829.
- Kiuchi K, Nejima J, Takano T, Ohta M, Hashimoto H. Increased serum concentrations of advanced glycation end products: a marker of coronary artery disease activity in type 2 diabetic patients. Heart 2001;85:87–91.
- Yamagishi S. Role of advanced glycation end products (AGEs) and receptor for AGEs (RAGE) in vascular damage in diabetes. Exp Gerontol 2011;46:217–224.
- Schinzel R, Münch G, Heidland A, Sebekova K. Advanced glycation end products in end-stage renal disease and their removal. Nephron 2001;87:295–303.
- Sebeková K, Blazícek P, Syrová D, Krivosíková Z, Spustová V, Heidland A, Schinzel R. Circulating advanced glycation end product levels in rats rapidly increase with acute renal failure. Kidney Int Suppl 2001;78:S58–S62.
- Stam F, Schalkwijk CG, van Guldener C, ter Wee PM, Stehouwer CDA. Advanced glycation end-product peptides are associated with impaired renal function, but not with biochemical markers of endothelial dysfunction and inflammation in non-diabetic individuals. Nephrol Dial Transplant 2006;21:677–682.
- Misciagna G, De Michele G, Trevisan M. Non enzymatic glycated proteins in the blood and cardiovascular disease. Curr Pharm Des 2007;13:3688–3695.
- Koga M, Kasayama S. Clinical impact of glycated albumin as another glycemic control marker. Endocr J 2010;57: 751–762.
- Kanauchi M, Tsujimoto N, Hashimoto T. Advanced glycation end products in nondiabetic patients with coronary artery disease. Diabetes Care 2001;24:1620–1623.
- McNulty M, Mahmud A, Feely J. Advanced glycation end-products and arterial stiffness in hypertension. Am J Hypertens 2007;20:242–247.
- Bucala R, Tracey KJ, Cerami A. Advanced glycosylation products quench nitric oxide and mediate defective endothelium-dependent vasodilatation in experimental diabetes. J Clin Invest 1991;87:432–438.
- Bucala R, Mitchell R, Arnold K, Innerarity T, Vlassara H, Cerami A. Identification of the major site of apolipoprotein B modification by advanced glycosylation end products blocking uptake by the low density lipoprotein receptor. J Biol Chem 1995;270:10828–10832.
- Hofnagel O, Luechtenborg B, Weissen-Plenz G, Robenek H. Statins and foam cell formation: impact on LDL oxidation and uptake of oxidized lipoproteins via scavenger receptors. Biochim Biophys Acta 2007;1771:1117–1124.
- Shaw SS, Schmidt AM, Banes AK, Wang X, Stern DM, Marrero MB. S100B-RAGE-mediated augmentation of angiotensin II-induced activation of JAK2 in vascular smooth muscle cells is dependent on PLD2. Diabetes 2003;52: 2381–2388.
- Sakaguchi T, Yan SF, Yan SD, Belov D, Rong LL, Sousa M, et al. Central role of RAGE-dependent neointimal expansion in arterial restenosis. J Clin Invest 2003;111:959–972.
- Torreggiani M, Liu H, Wu J, Zheng F, Cai W, Striker G, Vlassara H. Advanced glycation end product receptor-1 transgenic mice are resistant to inflammation, oxidative stress, and post-injury intimal hyperplasia. Am J Pathol 2009;175:1722–1732.
- Schulz JB, Lindenau J, Seyfried J, Dichgans J. Glutathione, oxidative stress and neurodegeneration. Eur J Biochem 2000;267:4904–4911.
- Li J, Liu D, Sun L, Lu Y, Zhang Z. Advanced glycation end products and neurodegenerative diseases: Mechanisms and perspective. J Neurol Sci 2012;317:1–5.
- Carrano A, Hoozemans JJ, Van der Vies SM, Rozemuller AJ, Van Horssen J, De Vries HJ. Amyloid beta induces oxidative stress-mediated blood-brain barrier changes in capilary amyloid angiopathy. Antioxid Redox Signal 2011;15:1167–1178.
- Cai Z, Zhao B, Ratka A. Oxidative stress and β-amyloid protein in Alzheimer's Disease. Neuromol Med 2011;13: 223–250.
- Srikanth V, Maczurek A, Phan T, Steele M, Westcott B, Juskiw D, Münch G. Advanced glycation endproducts and their receptor RAGE in Alzheimer's disease. Neurobiol Aging 2011;32:763–777.
- Laffont I, Shuvaev VV, Briand O, Lestavel S, Barbier A, Taniguchi N, et al. Early-glycation of apolipoprotein E: Effects on its binding to LDL receptor, scavenger receptor A and heparin sulfate. Biochim Biophys Acta 2002;1583: 99–107.
- Candore G, Bulati M, Caruso C, Castiglia L, Colonna- Romano G, Di Bona D, et al. Inflammation, cytokines, immune response, apolipoprotein E, cholesterol, and oxidative stress in Alzheimer disease: Therapeutic implications. Rejuv Res 2010;13:301–313.
- Seneff S, Wainwright G, Mascitelli L. Nutrition and Alzheimer's disease: The detrimental role of a high carbohydrate diet. Eur J Intern Med 2011;22:134–140.
- Salminen A, Ojala J, Kauppinen A, Kaarniranta K, Suuronen T. Inflammation in Alzheimer's disease: Amyloid-β oligomers trigger innate immunity defence via pattern recognition receptors. Progress Neurobiol 2009;87:181–194.
- Cho S. CD36 as a therapeutic target for endothelial dysfunction in stroke. Curr Pharmaceut Des 2012;18:3721–3730.
- Rojas A, Figueroa H, Morales E. Fueling inflammation at tumor microenvironment: The role of multiligand/rage axis. Carcinogenesis 2010;31:334–341.
- Sesti F, Tsitsilonis OE, Kotsinas A, Trougakos IP. Oxidative stress-mediated biomolecular damage and inflammation in tumorigenesis. In Vivo 2012;26:395–402.
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011;144:646–674.
- Greenhough A, Smartt HJM, Moore AE, Roberts HR, Williams AC, Paraskeva C, Kaidi A. The COX-2/PEG2 pathway: A key role in the hallmarks of cancer and adaptation to the tumor microenvironment. Carcinogenesis 2009;30: 377–386.
- Takino J, Yamagishi S, Takeuchi M. Glycer-AGEs-RAGE signaling enhances the angiogenic potential of hepatocellular carcinoma by upregulating VEGF expression. World J Gastroenterol 2012;18:1781–1788.
- Kang R, Tang D, Schapiro NE, Livesey KM, Farkas A, Loughran P, et al. The receptor for advanced glycation end products (RAGE) sustains autophagy and limits apoptosis, promoting pancreatic tumor cell survival. Cell Death Differ 2010;17:666–676.
- Tesarova P, Kalousova M, Jachymova M, Mestek O, Petruzelka L, Zima T. Receptor for advanced glycation end products (RAGE) – soluble form (sRAGE) and gene polymorphism in patients with breast cancer. Cancer Invest 2007;25:720–725.
- Gebhardt C, Riehl A, Durchdewald M, Nemeth J, Furstenberger G, Muller-Decker K, et al. RAGE signaling sustains inflammation and promotes tumor development. J Exp Med 2008;205:275–285.
- Turovskaya O, Foell D, Sinha P, Vogl T, Newlin R, Nayak J, et al. RAGE, carboxylated glycans and S100A8/A9 play essential roles in colitis-associated carcinogenesis. Carcinogenesis 2008;29:2035–2043.
- Stav D, Bar I, Sandbank J. Usefulness of CDK5RAP3, CCNB2, and RAGE genes for the diagnosis of lung adenocarcinoma. Int J Biol Markers 2007;22:108–113.
- Sakatani S, Yamada K, Homma C, Munesue S, Yamamoto Y, Yamamoto H, Hirase H. Deletion of RAGE causes hyperactivity and increased sensitivity to auditory stimuli in mice. PLoS One 2009;4:e8309.
- Kodavanti UP, Thomas R, Ledbetter AD, Schaldweiler MC, Shannahan JH, Wallenborn JG, et al. Vascular and cardiac impairment in rats inhaling ozone and diesel exhaust particles. Environm Health Perspect 2011;119:312–318.
- Giordano D. Tobacco smoke is a source of advanced glycation end products (AGEs): possible role in the accelerated vascular disease of smokers. J Invest Med 1996;44:200a.
- Campos C, Guzmán R, López-Fernández E, Casado A. Urinary biomarkers of oxidative/nitrosative stress in healthy smokers. Inh Toxicol 2011;23:148–156.
- Tessier FJ, Birlouez-Aragon I. Health effects of dietary Maillard reaction products: The results of ICARE and other studies. Amino Acids 2012;42:1119–1131.
- Koschinsky T, He CJ, Mitsuhashi T, Bucala R, Liu C, Buenting C, et al. Orally absorbed reactive glycation products (glycotoxins): an environmental risk factor in diabetic nephropathy. Proc. Natl Acad. Sci. USA 1997;94:6474–6479.
- Šebeková K, Saavedra G, Zumpe C, Somoza V, Klenovicsová K, Birlouez-Aragon I. Plasma concentration and urinary excretion of Ne-(carboxymethyl)lysine in breast milk- and formula-fed infants. Ann N Y Acad Sci. 2008;1126:177–180.
- Vlassara H, Cai W, Crandall J, Goldberg T, Oberstein R, Dardaine V, et al. Inflammatory mediators are induced by dietary glycotoxins, a major risk factor for diabetic angiopathy. Proc Natl Acad Sci USA 2002;99:15596–15601.
- Cai W, He JC, Zhu L, Peppa M, Lu C, Uribarri J, Vlassara H. High levels of dietary advanced glycation end products transform low-density lipoprotein into a potent redox- sensitive mitogen-activated protein kinase stimulant in diabetic patients. Circulation 2004;110:285–291.
- Yang CW, Vlassara H, Peten EP, He CJ, Striker GE, Striker LJ. Advanced glycation end products upregulate gene expression found in diabetic glomerular disease. Proc Natl Acad Sci USA 1994;91:9436–9440.
- Vlassara H, Striker LJ, Teichberg S, Fuh H, Li YM, Steffes M. 1994. Advanced glycation end products induce glomerular sclerosis and albuminuria in normal rats. Proc Natl Acad Sci USA 1994;91:11704–11708.
- Zheng F, He C, Cai W, Hattori M, Steffes M, Vlassara H. Prevention of diabetic nephropathy in mice by a diet low in glycoxidation products. Diab Metab Res Rev 2002;18: 224–237.
- Lin RY, Choudhury RP, Cai W, Lu M, Fallon JT, Fisher EA, Vlassara H. Dietary glycotoxins promote diabetic atherosclerosis in apolipoprotein E-deficient mice. Atherosclerosis 2003;168:213–220.
- Sandu O, Song K, Cai W, Zheng F, Uribarri J, Vlassara H. Insulin resistance and type 2 diabetes in high-fat-fed mice are linked to high glycotoxin intake. Diabetes 2005; 54:2314–2319.
- Susic D. Cross-link breakers as a new therapeutic approach to cardiovascular disease. Biochem Soc Trans 2007;35: 853–856.
- Ueda S, Yamagishi S, Takeuchi M, Kohno K, Shibata R, Matsumoto Y, et al. Oral adsorbent AST-120 decreases serum levels of AGEs in patients with chronic renal failure. Mol Med 2006;12:180–184.
- Vlassara H, Li YM, Imani F, Wojciechowicz D, Yang Z, Liu FT, Cerami A. Identification of galectin-3 as a high- affinity binding protein for advanced glycation end products (AGE): a new member of the AGE-receptor complex. Mol Med 1995;1:634–646.
- Iacobini C, Menini S, Oddi G, Ricci C, Amadio L, Pricci F, et al. Galectin-3/AGE-receptor 3 knockout mice show accelerated AGE-induced glomerular injury: evidence for a protective role of galectin-3 as an AGE receptor. FASEB J 2004;18:1773–1775.
- Iacobini C, Oddi G, Menini S, Amadio L, Ricci C, Di Pippo C, et al. Development of age-dependent glomerular lesions in galectin-3/AGE-receptor-3 knockout mice. Am J Physiol Renal Physiol 2005;289:F611–F621.
- Bolton WK, Cattran DC, Williams ME, Adler SG, Appel GB, Cartwright K, et al. Randomized trial of an inhibitor of formation of advanced glycation end products in diabetic nephropathy. Am J Nephrol 2004;24:32–40.
- Coughlan MT, Forbes JM, Cooper ME. Role of the AGE crosslink breaker, alagebrium, as a renoprotective agent in diabetes. Kidney Int Suppl 2007;106:S54–S60.
- Padival S, Nagaraj RH. Pyridoxamine inhibits maillard reactions in diabetic rat lenses. Ophthalmic Res 2006;38: 294–302.
- Adrover M, Vilanova B, Frau J, Muñoz F, Donoso J. The pyridoxamine action on Amadori compounds: A reexamination of its scavenging capacity and chelating effect. Bioorg Med Chem 2008;16:5557–5569.
- Waanders F, van den Berg E, Nagai R, van Veen I, Navis G, van Goor H. Renoprotective effects of the AGE-inhibitor pyridoxamine in experimental chronic allograft nephropathy in rats. Nephrol Dial Transplant. 2008;23:518–524.
- Motomura K, Fujiwara Y, Kiyota N, Tsurushima K, Takeya M, Nohara T, et al. Astragalosides isolated from the root of astragalus radix inhibit the formation of advanced glycation end products. J Agric Food Chem 2009;57:7666–7672.
- Karachalias N, Babaei-Jadidi R, Rabbani N, Thornalley PJ. Increased protein damage in renal glomeruli, retina, nerve, plasma and urine and its prevention by thiamine and benfotiamine therapy in a rat model of diabetes. Diabetologia 2010;53:1506–1516.
- Stirban A, Negrean M, Stratmann B, Gawlowski T, Horstmann T, Götting C, et al. Benfotiamine prevents macro- and microvascular endothelial dysfunction and oxidative stress following a meal rich in advanced glycation end products in individuals with type 2 diabetes. Diabetes Care 2006;29:2064–2071.
- Alkhalaf A, Kleefstra N, Groenier KH, Bilo HJ, Gans RO, Heeringa P, et al. Effect of benfotiamine on advanced glycation endproducts and markers of endothelial dysfunction and inflammation in diabetic nephropathy. PLoS One 2012;7:e40427.
- Nagai R, Shirakawa JI, Ohno RI, Moroishi N, Nagai M. Inhibition of AGEs formation by natural products. Amino Acids 2013; [Epub ahead of print]
- Reiniger N, Lau K, McCalla D, Eby B, Cheng B, Lu Y, et al. Deletion of the receptor for advanced glycation end products reduces glomerulosclerosis and preserves renal function in the diabetic OVE26 mouse. Diabetes 2010; 59:2043–2054.
- Deane R, Singh I, Sagare AP, Bell RD, Ross NT, LaRue B, et al. A multimodal RAGE-specific inhibitor reduces amyloid β-mediated brain disorder in a mouse model of Alzheimer disease. J Clin Invest 2012;122:1377–1392.
- Yamagishi S, Matsui T, Nakamura K. Kinetics, role and therapeutic implications of endogenous soluble form of receptor for advanced glycation end products (sRAGE) in diabetes. Curr Drug Targets 2007;8:1138–1143.
- Steele M, Stuchbury G, Münch G. The molecular basis of the prevention of Alzeimer's disease through healthy nutrition. Exp Gerontol 2007;42:28–36.