Abstract
The review deals with impairment of Ca2+-ATPases by high glucose or its derivatives in vitro, as well as in human diabetes and experimental animal models. Acute increases in glucose level strongly correlate with oxidative stress. Dysfunction of Ca2+-ATPases in diabetic and in some cases even in nondiabetic conditions may result in nitration of and in irreversible modification of cysteine-674. Nonenyzmatic protein glycation might lead to alteration of Ca2+-ATPase structure and function contributing to Ca2+ imbalance and thus may be involved in development of chronic complications of diabetes. The susceptibility to glycation is probably due to the relatively high percentage of lysine and arginine residues at the ATP binding and phosphorylation domains. Reversible glycation may develop into irreversible modifications (advanced glycation end products, AGEs). Sites of SERCA AGEs are depicted in this review. Finally, several mechanisms of prevention of Ca2+-pump glycation, and their advantages and disadvantages are discussed.
Introduction
Calcium ATPases, sarco/endoplasmic reticulum ATPase (SERCA), and plasma membrane calcium ATPase (PMCA) play key roles in calcium homeostasis and cell signaling. These calcium pumps have been suggested to represent major targets that are readily damaged by ROS/RNS [Citation1,Citation2]. In the majority of cases the oxidation of one or two amino acids has a minimal effect on protein function, altering neither the stability nor the function of the protein [Citation3,Citation4]. In contrast, some proteins are selectively oxidized at critical sites that regulate their function in a reversible manner. In this respect, SERCA and PMCA belong to proteins, with selectively oxidized unique sites associated with alterations their function observed in many human diseases and experimental models on animals [Citation5–7].
This review is focused on the impairment of Ca2+-pumps by high glucose (HG) and diabetes. Hyperglycemia is the most important factor in the onset and progress of diabetic complications. The mechanisms by which hyperglycemia may lead to complications of long-term diabetes include polyol activation, formation of glycated proteins resulting in advanced glycation end products (AGEs), and an increase in oxidative stress. Inactivation of Ca2+ pumps contributes to variety of pathologies. Modulation of calcium pumps may offer a new therapeutic tool in the treatment of several human diseases, including diabetes.
Physiological importance of sarco/endoplasmic reticulum Ca2+-ATPase
SERCA is an intracellular membrane-bound enzyme that utilizes the free energy of ATP to transport Ca2+ against a concentration gradient. The physiological role of SERCA is to sequester cytosolic Ca2+ into membrane-bound intracellular compartments. SERCA-mediated Ca2+ uptake into stores plays a key role in the maintenance of intracellular free Ca2+ levels within the physiological range. Ca2+ uptake into SR via SERCA is responsible for the removal of 90% of Ca2 +from the cytoplasm [Citation8]. In addition, following contractions caused by elevated levels of intracellular Ca2+, accelerated sequestration of Ca2+ by SERCA mediates smooth, cardiac, and skeletal muscle relaxation. In noncontractile cells such as proliferating smooth muscle cells or endothelium, SERCA modulates many cellular processes via alteration of intracellular Ca2+, including cell growth, apoptosis, and migration [Citation9,Citation10].
Sarcoplasmic reticulum Ca2+-ATPase isoforms
SERCA is a 97–115 kDa membrane protein expressed in the ER/SR of all cells as the product of one of three genes [Citation11]. SERCA1 is present in fast twitch skeletal muscle, SERCA2a in heart and slow twitch skeletal muscle, SERCA2b in all cells including smooth muscle cells (SMCs), endothelial cells (ECs), and platelets, and SERCA3 in nonmuscle cells including plateles and EC. SERCA2a is also expressed in proliferating SMCs [Citation12,Citation13]. The major SMC isoform, SERCA2b, is an alternatively spliced form that differs from SERCA2a by having an elongated C-terminus within the SR lumen. SERCA3 isoforms differ with respect to SERCA1 only slightly in many functional aspects, including those determining the apparent affinity for cytoplasmic Ca2+. However, with respect to the dephosphorylation of E2P, which could be critical for the lumenal Ca2+ concentration, a modulatory role of the C terminus was found: the longer the C-terminal extension, the faster dephosphorylation proceeded [Citation14]. Individual pumps differ in their regulatory and kinetic properties, allowing for optimized function in the tissue where they are expressed [Citation15].
SERCA dysfunction
Ca2+-ATPase dysfunction has been detected in experimental and human pathology [Citation16,Citation17] including heart failure [Citation18], diabetes [Citation19], atherosclerosis [Citation20], and restenosis [Citation21], as well as in the aging skeletal muscle [Citation22] reviewed by Jia Ying et al. [Citation23]. A common feature of these pathological situations is the increased and prolonged production of reactive oxygen species (ROS) [Citation24], to which SERCA is susceptible owing to critical active site of cysteines.
Previous studies showed that oxidative stress reduced SERCA activity by nitration of tyrosine [Citation6,Citation25] and oxidative modification of Cys674 leading to a reduced level of GSS-SERCA, the activated form of SERCA [Citation20,Citation26]. As mentioned above, two susceptible SERCA tyrosines (294, 295) were also nitrated. Mass spectrometry and antibody studies indicate that these tyrosines are nitrated under the same conditions in which Cys674 is oxidized [Citation23,Citation27]. Posttranslational modification of amino acid residues of SERCA [Citation28] by ROS may affect the structure of the Ca2+ transporter and consequently affect its function resulting in elevation of cytosolic Ca2 + concentration [Citation29].
Hyperglycemia and diabetes
Hyperglycemia is the most important factor in the onset and progression of diabetic complications. Mechanisms of hyperglycemia underlying complications of long-term diabetes include polyol activation, formation of glycated proteins resulting in AGEs, and an increase in oxidative stress. HG levels are known to cause impairment of Ca2+-ATPase activity which can be prevented by compounds that are also known to be protective against diabetic pathology.
Polyol pathway and cardiac dysfunction in relation to SERCA
Polyol pathway in diabetic patients and animals.
The polyol pathway has been implicated in the pathogenesis of various diabetic complications [Citation30,Citation31]. In this metabolic pathway, glucose is reduced to sorbitol by aldose reductase (AR; EC 1.1.1.21) with the oxidation of its co-factor NADPH to NADP+, and sorbitol is then converted to fructose by sorbitol dehydrogenase (SDH: EC 1.1.1.14) with concomitant reduction of NAD+ to NADH [Citation32]. In hyperglycemia, increased flux of glucose through the polyol pathway leads to the depletion of NADPH and NAD+. The decrease in the level of NADPH is thought to lead to a decreased level of reduced glutathione (GSH) because NADPH is also the co-factor for glutathione reductase (GR), which regenerates GSH from glutathione disulfide (GSSG) [Citation33].
Studies in acute hyperglycemic hearts also showed that activation of the polyol pathway increases the NADH/NAD ratio, which was found to stimulate the NADH oxidase (EC 1.6.99.3) to generate ROS [Citation34,Citation35] and the increased ROS concentration can inhibit SERCA by oxidizing the cysteine thiols, interfering with the ATP-binding site and inhibiting hydrolysis of ATP [Citation36].
Further evidence for interaction of the polyol pathway and SERCA comes from work by Tang et al. [Citation16], who found that high-glucose perfusion led to increased nitration of SERCA and an increased level of SERCA Cys674-SO3H (sulfonic acid), suggesting that reduction of SERCA activity was due to high glucose-induced oxidative stress. Addition of either aldose AR or SDH inhibitor to the high-glucose perfusate reduced the levels of nitrated SERCA and SERCA Cys674-SO3H and normalized SERCA activity, indicating that the polyol pathway is the major contributor to high glucose-induced oxidative stress and the cause of high glucose-induced inactivation of SERCA, leading to contractile dysfunction.
Polyol pathway in nondiabetic subjects.
The polyol pathway has been reported to be activated in hearts following ischemia/reperfusion injury (I/R) even in nondiabetic animals [Citation37], which may relate to the observation in humans that up to half of nondiabetic patients with acute myocardial infarction have high blood glucose levels when admitted to hospital, presumably as a consequence of stress [Citation38]. A recent study reported that in the I/R-treated hearts of nondiabetic rats the polyol pathway-mediated depletion of NAD+ led to increased oxidative damage [Citation39], resulting in cardiac dysfunction [Citation40]. These authors showed that in this model the polyol pathway contributed to the impairment of SERCA and the ryanodine receptor (RyR), two players in Ca2 + signaling that regulate cardiac contraction. Polyol pathway activities increased the level of peroxynitrite, which enhanced the tyrosine nitration of SERCA and irreversibly modified it to form SERCACys674/SO3H. Rat hearts perfused in high-glucose medium (33.3 mM) for 2 h compared with those perfused with normal glucose medium (11.1 mM) showed increased apoptosis and increased superoxide and nitrotyrosine levels, indicating that oxidative stress occurs within a short period of hyperglycemia [Citation41]. In general, acute increases in glucose level strongly correlated with oxidative stress in diabetic patients and animals [Citation41,Citation42].
Thus, bringing together this evidence, it appears that oxidative stress induced by the polyol pathway can reduce SERCA activity either by irreversible modification of cysteine-674 to form the sulfonic acid or nitration of tyrosine, and this could also play a role in nondiabetic rats and patients with cardiovascular I/R.
Vascular complications related to SERCA impairment in the presence of high glucose
Under normal conditions, a major action of NO in all cells is to decrease intracellular calcium, which can inhibit cellular functions, including growth, proliferation, adhesion, and contractility. An immediate consequence of the reduction of intracellular Ca2+ in smooth muscle cells (SMCs) by NO is relaxation. NO relaxes vascular SMCs via both cyclic GMP-dependent and GMP-independent mechanisms. The second mechanism includes direct modification (activation) of SERCA protein by S-glutathiolation of the most reactive thiol, Cys674 [Citation43].
Nitric oxide has an important effect on SERCA in SMCs, as illustrated by the review of Tong et al. [Citation44]. NO can stimulate the uptake of cytosolic Ca2+ via SERCA by adducting glutathione to the reactive Cys674. In SMC's exposed to high glucose, NO produced less glutathione adducts on SERCA Cys674 thiol. Thiol labeling studies showed that the Cys674 thiol was oxidized by high glucose. In addition, the sequence-specific antibody that stained sulfonic acid at Cys674 SERCA also detected that the thiol was oxidized in SMC. These studies indicate that failure of NO to inhibit migration in SMCs exposed to HG is due to irreversible oxidation of the SERCA reactive Cys674 [Citation45].
The mechanisms of abnormal vascular SMC migration in type 2 diabetes were studied also in the obese Zucker rat, a model of obesity and insulin resistance [Citation46], SERCA nitrotyrosine-294, 295 and cysteine-674 (Cys674)-SO3H were found to be increased in Zucker rat SMCs, indicating oxidative or nitrative stress. The authors propose that the abnormal response to NO in Zucker rat SMCs is caused by redox regulation of SERCA, in the same way as described above for cultured cell studies. Nitration of tyrosine 294 and 295 is known to occur under the same conditions as oxidation of Cys674 [Citation23,Citation27], but may be less important in mediating effects of NO, as a Cys674S mutant SERCA has normal calcium uptake activity and only lacks stimulation of activity by NO. This indicates that oxidation of the single thiol Cys674 is sufficient for SMCs to be insensitive to NO and that its restitution can restore the response. Overall, these findings suggest that the responsiveness of SERCA to NO is of key importance in modulating SMC behaviour [Citation46].
NO function is impaired in a variety of cardiovascular diseases, including diabetes, hypercholesterolemia, and atherosclerosis, which are all associated with SERCA dysfunction caused by the increased oxidative balance in these diseases [Citation44]. In these pathological states, SERCA dysfunction could be attributed to excess production of peroxynitrite, which may result in both tyrosine nitration and thiol oxidation. In the diabetic and insulin-treated Wistar rat aorta, an increase in tyrosine nitration of aortic SERCA2b correlated with impaired aortic relaxation to acetylcholine, which is characteristic of the diabetic state [Citation47]. Interestingly, prolonged treatment of diabetic rats with insulin also impaired aortic relaxations attributed to dysfunction of the smooth muscle, and were caused, at least in part, by SERCA dysfunction [Citation47]. Similarly, in established diabetes, the presence of excess insulin may cause both ONOO- formation and an increase in nitrotyrosine in SERCA protein via increased production of superoxide O2 −and NO. Enhancement of vascular disease can occur independently through high-insulin and high-glucose levels [Citation48,Citation49].
The diabetic hypercholesterolemic (HC) pig aorta has been reported to contain more sulfonated SERCA2b (Cys674 in sulfonic acid) than healthy control aorta, an increase that was prevented by insulin [Citation23]. Interestingly, the irreversible oxidation of SERCA in the diabetic pig aorta was not primarily associated with a 110 kDa SERCA, but with a lower molecular mass SERCA protein of approximately 70 kDa and by higher molecular mass aggregates [Citation23]. These studies suggest that SERCA oxidation may result in degradation, which could play a role in the progression of diabetic vascular disease [Citation23]. A number of studies support the concept that increased aggregation of platelets contributes to the pathogenesis and progression of vascular complications of diabetes. SERCA2 in platelets from patients with type 2 diabetes mellitus showed increased platelet-free Ca2+ levels and elevated tyrosine nitration accompanied by inactivation of SERCA. In platelets from healthy volunteers, tyrosine nitration of SERCA2 could be evoked by peroxynitrite in vitro, suggesting the importance of the oxidation of SERCA in diabetic patients [Citation50].
A major finding of Randriamboavonjy et al. [Citation50], is that type 2 diabetes mellitus is associated with the degradation of platelets through a mechanism involving tyrosine nitration of SERCA2, inactivation of SERCA2, and increase in intracellular Ca2+ level. The tyrosine nitration of SERCA2 in vitro in platelets from healthy volunteers could be evoked by peroxynitrite. In vitro, the effects of peroxynitrite on SERCA activity appear to be biphasic because short-term application of low concentrations of peroxynitrite increased platelet Ca2+-ATPase activity. These observations are in line with a previous report in native vascular smooth muscle cells in which peroxynitrite (10–50 μM) increased SERCA activity via S-glutathiolation of critical cysteine residues [Citation20]. Higher concentrations of peroxynitrite, on the other hand, enhanced tyrosine nitration of SERCA2 in platelets from nondiabetic individuals and decreased platelet Ca2+-ATPase activity.
Ca2+-ATPase glycation
ATPase activity of erythrocyte membranes is known to be decreased in diabetic patients with elevated blood glucose [Citation51,Citation52] and in red blood cells treated ex vivo with glucose [Citation53], and there is evidence to suggest that such effects relate to glycation of the Ca2+-ATPase. Gonzales-Flecha [Citation54] demonstrated incorporation of radioactively labeled glucose into PMCA in erythrocyte membranes and formation of covalent interaction between glucose and PMCA. Glycation of only one out of the 80 lysine residues accessible outside the transmembrane domains of PMCA was suggested to decrease the Vmax of ATP hydrolysis without affecting PMCA affinities for Ca2+, ATP, or calmodulin [Citation55]. Glycation of the corresponding lysine residue in the ATP binding domain of SERCA in hearts of diabetic rats has been observed by Bidasee et al. [Citation56] ().
Figure 1. Simplified scheme of PMCA structure with sites modified by high glucose. Transmembrane regions are numbered 1–10. Approximate positions of the phosphoryl-aspartate (D*) and lysine (K) crucial for ATP binding are shown. In the C-terminal region, tyrosine (Y) residue is indicated. Its phosphorylation results in inactivation of the pump. Positions of particular residues are numbered according to the human PMCA4b sequence.
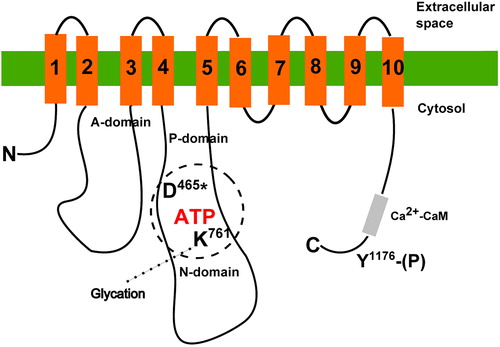
On the contrary to glycation of PMCA, normal extrusion of calcium by the intact red blood cells incubated with 30 mM glucose ex vivo was reported [Citation57], and later, Bookchin et al. concluded that PMCA is not glycated in diabetes, based on a lack of change of calcium extrusion in intact erythrocytes of diabetic and nondiabetic persons [Citation58]. In fact, no change in calcium extrusion could be caused by switch of calcium extrusion from PMCA to sodium/calcium exchanger [Citation59,Citation60]. ATPase activity was not measured, and no direct assessment of glycation, for example by mass spectrometry or glucose labeling studies was made in intact RBC in addition to Ca extrusion experiments. Thus, there is little evidence whether PMCA glycation occurs in intact RBC.
Advanced glycation end products, in sarco/endoplasmic reticulum Ca2+-ATPase and plasma membrane Ca2+-ATPase
Several studies have reported that biomolecules like proteins, lipids, and nucleic acids are able to react in vivo with glucose to generate advanced glycation end- products (AGEs), which seems to be an important pathogenic mechanism of almost all diabetes complications and of aging [Citation61,Citation62]. The first steps of this reaction are reversible and begin with the addition of a primary amino group of the biomolecule to the carbonyl group of glucose, and the rearrangement of the Schiff base to form an Amadori product. The Amadori products undergo a series of irreversible intermolecular reactions to produce AGEs [Citation63]. Both the early products and the end-products of glycation affect the physicochemical and biological properties of the target molecules [Citation64], and they also interact with other biomolecules, resulting in alteration of their structure and function. Potential targets for glycation include the amine-containing lipids and proteins present in biological membranes such as the Ca2+-pumps SERCA and PMCA.
AGEs include products formed by protein–protein crosslinking between Lys (lysine) and Lys or Arg (arginine) residues, as well as noncrosslinking modifications of Lys, Arg, and His (histidine) residues of protein [Citation62]. The most frequent noncrosslinking AGEs formed on Ca2+-pumps are pyrraline, Nϵ-(carboxymethyl)-lysine, 1-carboxy alkyllysine at Lys residue, AFGP (1-alkyl-2-formyl-3,4-glycosyl pyrrole) at Lys or His residues, and imidazolone A and B at Arg residues [Citation56]. Crosslinking of two Ca2+-pump proteins can occur between Lys residues as a crossline or as pentodilysine or between Lys and Arg as a pentosidine AGE [Citation56,Citation62] ().
Figure 2. Structures of noncrosslinking and crosslinking AGEs in Ca2+-pumps. Formation of advanced glycation end-products through glucose-protein Schiff base and Amadori rearrangement (a), and structures of noncrosslinking (b) and crosslinking (c) AGE molecules most frequently formed on Ca2+-pumps.
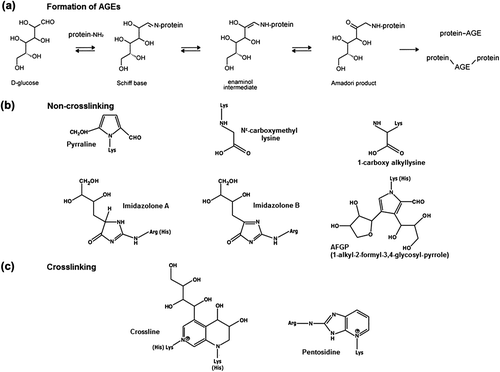
The group of Bidasee et al. [Citation56] was the first to show that AGEs were formed on intracellular SR Ca2+-ATPase (SERCA2a) during chronic diabetes, suggesting a mechanism by which cardiac relaxation may be impaired during diabetes. They were also first to pinpoint specific amino acid residues within the SERCA2a sequence where AGEs modifications occur, using MALDI-TOF MS analyses of heart SERCA2a from streptozotocin-induced diabetic rats to reveal several single noncrosslinked as well as crosslinked AGEs of Lys and Arg.
Single AGEs like imidazolone A, imidazolone B, or AFGP (1-alkyl-2-formyl-3,4-glycosyl pyrrole molecule) were found in three cytoplasmic domains, referred to as A (actuator), N (nucleotide binding), and P (phosphorylation) domains (on Arg164, Lys 476 and 481, His 526, and Arg 636). Lysine residues within A, N, and P domains were cross-linked to arginine residues within the A and P domains via pentosidine AGEs (between Lys 135 and Arg 164, between Lys 141 and Arg 677, between Lys 135 and Arg 655, between Lys 460 and Arg 636, and between Lys 683 and Arg 619). None of these AGE adducts were found within the transmembrane helixes domain (). Interestingly, in this study a two-week insulin treatment attenuated AGEs formation within the SERCA2a [Citation56]. This fact confirmed earlier findings that the structure and function of Ca2+-ATPase are restored to normal under good glycemic control by insulin [Citation65].
Figure 3. Planar model of the primary and estimated secondary structure of rat SERCA2b isoforms highlighting AGEs formation and posttranslational modifications. The model is adapted from Dode et al. [Citation108] and based on the crystal structure of SERCA1. Each circle corresponds to an amino acid residue. Amino acids arginines R164, 636, lysines K476, 481, and histidine H526 are targets for noncrosslinking AGEs like imidazolone A, imidazolone B and AFGP (red circles with black letters). Between amino acids K135 and R164, K141 and R677, K135 and R655, K460 and R636, and between K683 and R619 crosslinking AGEs-like pentosidine (dotted red circles with red letters) are formed. Tyrosines Y294 and Y295 are documented sites for nitration (presented by pink circles and pink letters). Cysteine C674 is glutathionylated in the presence of NO (blue letters in blue circles). Asparagine N1035 is a potential glycosylation target (orange circle with orange letter). Aspartic acid D351 (black circle and letter with the phosphate group indicated) represents the phosphorylation site. The nucleotide-binding domain spans from residue T357 to L599 (shown as green circles with white letter) and the most crucial amino acid residues for ATP binding are lysines K514, K492 and phenylalanine F487 (green circles with black letters). Amino acid residues contributing to Ca2+ ligand binding are shown as black circles with black letters in the high affinity Ca2+ binding site of the transmembrane domain on M4, M5, M6, and M8 helixes.
![Figure 3. Planar model of the primary and estimated secondary structure of rat SERCA2b isoforms highlighting AGEs formation and posttranslational modifications. The model is adapted from Dode et al. [Citation108] and based on the crystal structure of SERCA1. Each circle corresponds to an amino acid residue. Amino acids arginines R164, 636, lysines K476, 481, and histidine H526 are targets for noncrosslinking AGEs like imidazolone A, imidazolone B and AFGP (red circles with black letters). Between amino acids K135 and R164, K141 and R677, K135 and R655, K460 and R636, and between K683 and R619 crosslinking AGEs-like pentosidine (dotted red circles with red letters) are formed. Tyrosines Y294 and Y295 are documented sites for nitration (presented by pink circles and pink letters). Cysteine C674 is glutathionylated in the presence of NO (blue letters in blue circles). Asparagine N1035 is a potential glycosylation target (orange circle with orange letter). Aspartic acid D351 (black circle and letter with the phosphate group indicated) represents the phosphorylation site. The nucleotide-binding domain spans from residue T357 to L599 (shown as green circles with white letter) and the most crucial amino acid residues for ATP binding are lysines K514, K492 and phenylalanine F487 (green circles with black letters). Amino acid residues contributing to Ca2+ ligand binding are shown as black circles with black letters in the high affinity Ca2+ binding site of the transmembrane domain on M4, M5, M6, and M8 helixes.](/cms/asset/51aad77e-0644-4cdf-b78d-067facb5b73a/ifra_a_807923_f0003_b.jpg)
As yet, there appear to be no reports in the literature of AGE-modification of PMCA, but as SERCA and PMCA are functionally and structurally similar, it is likely that such modifications may occur in vivo during relevant pathological conditions. Further studies of PMCA are required to investigate this.
Prevention of ca2+-ATPase injury induced by glycation
Restoration of Ca2+-ATPase expression or activity
IGF-1.
There is some evidence that IGF-1 (insulin-like growth factor 1) may possess therapeutic potential in the treatment of diabetes complications. Exogenous IGF-1 treatment restored the diabetes-induced decline of SERCA protein levels [Citation66], and a beneficial role of overexpression of IGF-1 in aged mice has also been reported Li et al. [Citation67]. In the latter study, reduced SERCA expression and activity with age were normalized by the IGF-1 transgene, but interestingly the increased level of glycation end products and oxidative stress were not affected by IGF-1.
Taurine.
Taurine (2-aminoethanesulfonic acid) at physiological concentrations (50–100 μM) has been found to prevent significantly the reduction of Ca2+-ATPase activity in high glucose-treated red blood cells (RBCs) [Citation68]. Taurine stimulated the pumping rate of the Ca2+-activated ATPase pump, possibly by increasing the turnover rate of the pump. Several other investigators also observed positive effects of taurine in high glucose-treated cells. For example, taurine attenuated apoptosis of hyperglycemia-induced human umbilical vein endothelial cells via ROS inhibition and Ca2+ stabilization [Citation69]. Taurine also exhibited high reactivity with glucose and other aldehydes in vitro and may exert an inhibitory effect against protein modification in vivo [Citation70]. The effects described above for taurine could have relvance to mechanisms of diabetic pathology, as taurine is known to be decreased in plasma and platelets in diabetic patients, and supplementation can restore platelet function in this condition [Citation71].
Exendin-4.
Recently, Kim et al. [Citation72] demonstrated that pretreatment of beta-cells with Exendin-4, a glucagon-like peptide-1 analogue, had an anti-apoptotic effect through modulation of ER stress and ER Ca2+ replenishment by restoration of SERCA activity.
N-acetylcysteine.
N-acetylcysteine (NAC) administration has been found to ameliorate the effect of hyperglycemia on oxidative stress and activity of membrane transporting protein in the streptozotocin-induced rat model of diabetes type 1 [Citation73]. Treatment with NAC significantly improved lipid composition and lowered the hyperglycemia-induced lipid peroxidation, thereby restoring membrane fluidity and activity of plasma membrane Ca2+-ATPase and Na, K-ATPase.
Phospholamban
An increase in the amount of phospholamban, a protein that decreases the affinity of SERCA for calcium, was also reported to contribute to the decrease in SR function [Citation74]. A decrease in phospholamban expression may be another therapeutic approach to allow increased activity of SERCA in the diabetic cell, as overexpression of phospholamban or expression of a onphosphorylatable form of phospholamban leads to decreased SERCA activity [Citation75], while a knockout of phospholamban results in increased SERCA activity.
Modulation of SERCA expression
PPAR-γ (peroxisome proliferator-activated receptor-gamma) is a receptor for the class of antidiabetic drugs that include rosiglitazone and pioglitazone, which have been reported to have some effects on the expression on Ca2+ pumps [Citation76].
For example, activation of PPAR- γ by pioglitazone prevented the loss of SERCA2b expression in pancreatic INS-1 cells exposed to HG and the cytokine IL-1β, and was simultaneously able to prevent hyperglycemia and cytokine-induced elevations in cytosolic Ca2+ levels, insulin-secretory defects, and cell death [Citation77]. In light of these results, the authors suggested that pioglitazone modulates SERCA2b expression through direct transcriptional regulation of the gene and indirectly through prevention of CDK5-induced phosphorylation of PPAR-γ.
In patients with type 2 diabetes mellitus, rosiglitazone treatment for 12 weeks increased platelet SERCA2 expression and Ca2+-ATPase activity, decreased SERCA2 tyrosine nitration, normalized intracellular Ca2+ level, and restored platelet sensitivity to nitric oxide synthase inhibition [Citation50]. Rosiglitazone has also previously been reported to increase SERCA2 gene expression in cardiac myocytes [Citation78] and in CD34 [Citation79] bone marrow cells. The increase in SERCA2 expression in platelets from diabetic individuals was associated with decreased plasma nitrotyrosine levels and tyrosine nitration of SERCA2. The action of rosiglitazone is most probably a combination of its effects on plasma glucose levels, ROS production, and platelet Ca2+ signaling.
Treatment by enzymes or enzyme inhibitors
Catalase and superoxide dismutase.
Studies of the therapeutic potential of cardiac-specific overexpression of catalase in the treatment of age-related diseases in mice have found significantly reduced formation of AGEs, improved intracellular Ca2+ dysfunction through restoration of Na+/Ca+2 + exchanger level, but did not repair expression of SERCA2a in aged FVB mice hearts [Citation80]. Interestingly, overexpression of another antioxidant enzyme, superoxide dismutase, has been reported to prevent defective responses to NO in SMCs exposed to high glucose, where the oxidation of cysteine-674 was found to be a key factor the lack of response to NO. As superoxide dismutase detoxifies superoxide and can also inhibit the expression of NADH oxidase it is feasible that overexpression of superoxide dismutase would protect against oxidation of SERCA2a and therefore deleterious effects of hyperglycemia. However, further research is required to confirm a direct link in these processes.
Inhibitors of aldose reductase and sorbitol dehydrogenase.
As described above the polyol pathway, involving AR and SDH, have been reported to adversely affect Ca2 + pump function, and could be important in the mechanisms of I/R injury, hyperglycemia, and diabetes. There is evidence that treatment with AR and SDH inhibitors can significantly ameliorate these conditions, and therefore AR and SDH present novel targets for pharmacological protection against I/R-induced injuries of the heart [Citation40]. For example, inhibition of AR, the first and rate-limiting enzyme of the polyol pathway, has been reported to attenuate contractile dysfunction in diabetic animals [Citation16]. Cardiomyocytes incubated in high-glucose medium showed abnormal Ca2+ signaling due to decreased activity of SERCA, but inhibition of AR and SDH ameliorated contractile dysfunction, attenuated oxidative stress, and normalized Ca2+ signaling and SERCA activity.
Treatment by low molecular weight antioxidants
Butylated hydroxytoluene.
Preventing SERCA from being oxidized and/or increasing expression of SERCA may limit disease development and thus SERCA may be considered a potential target for treatment. An example may be the effect of the antioxidant butylated hydroxytoluene (BHT) in the HC rabbit aorta. BHT dramatically improved the smooth muscle response to NO as well as endothelium-dependent relaxation to acetylcholine, and in parallel it restored aortic SERCA activity which was decreased in hypercholesterolemia and decreased tyrosine-nitrated SERCA without changing SERCA protein expression. It is known that improvement in NO bioactivity can limit the progression of atherosclerosis [Citation44].
Green tea extract.
Babu et al. [Citation81] explored the cardioprotective potential of green tea extract (GTE) in diabetes complications. The sum of epigallocatechin (EGC) and epigallocatechin-3-gallate (EGCG) represented more than 70% of the catechin mixture in the GTE extract. Oral treatment of diabetic rats with GTE lowered the level of blood glucose, lipid peroxidation, and protein glycation in the heart, improved the decrease of Ca2+-ATPase activity and normalized the Ca2+ concentration in the heart.
Lipoic acid.
Lipoic acid is an organosulfur compound containing two sulfur atoms connected by disulfide bond. It is reduced intracellularly to dihydrolipoic acid, which is more bioactive and responsible for most of the antioxidant effects, probably due to the two free sulfhydryls. At least two cytosolic enzymes, glutathione reductase (GR) and thioredoxin reductase(Trx1), are able to reduce lipoic acid [Citation82,Citation83].
Lipoic acid supplementation has been found to be beneficial in preventing neurovascular abnormalities in diabetic neuropathy. HG treatment can cause glycosylation of proteins, resulting in significantly lowered activities of Na+/K+-ATPase and Ca2+-ATPase of red blood cells (RBCs). This can in turn affect the intracellular concentrations of Na+, K+, and Ca2+. Lipoic acid can lower protein glycosylation, lipid peroxidation and increase (Na+ and K+) Ca2+-ATPase activities in RBCs exposed to high glucose, suggesting a potential mechanism by which lipoic acid may delay or inhibit the development of neuropathy in diabetes [Citation84].
Breviscapine.
Breviscapine, a flavonoid extracted from Erigeron breviscapus, ameliorated cardiac dysfunction, and regulated myocardial Ca2+-cycling proteins in streptozotocin-induced diabetic rats. Myocardium contraction and relaxation are directly regulated by Ca2+-cycling proteins such as the RyR, the SR Ca2+ ATPase pump (SERCA) and the Na+/Ca2+ exchanger. Heart failure is associated with alterations in Ca2+ handling via many of these proteins, including decreased expression of SERCA2 [Citation85–87]. Diabetic rats showed impaired cardiac structure and function, and decreased expression of SERCA2. Breviscapine had a protective effect on diabetic cardiomyopathy and dose dependently regulated the expression of SERCA2 [Citation87].
Vitamin E and stobadine.
Controversial effects have been reported for stobadine [Citation88]. Stobadine alone significantly increased microsomal Ca2+-ATPase activity in the heart of normal rats. However, neither treatment with stobadine nor vitamin E alone, nor their combination did change cardiac Ca2+-ATPase activity in the diabetic heart. In normal rats, neither antioxidant had a significant effect on hepatic Ca2+-ATPase activity. Hepatic ATPase activity of diabetic rats was not changed by single treatment with stobadine, while vitamin E alone completely prevented diabetes-induced inhibition in microsomal ATPase activity in the liver [Citation88].
In other studies, the Ca2+-ATPase activity was reduced not only in the heart of diabetic animals but also in the brain [Citation89] and retina [Citation90]. Ten-week vitamin E treatment (500 IU/kg/day, orally) prevented the decrease of Ca2+-ATPase activity in the brain of streptozotocin (STZ)- induced diabetic rats and led to a significant inhibition in blood glucose, protein glycosylation, and lipid peroxidation [Citation89]. Administration of supplemental dietary alpha-tocopherol acetate for 2 months prevented the elevation of retinal TBARS and the decrease of Ca2+-ATPase activity in retinas of diabetic animals without exerting any beneficial effect on plasma TBARS [Citation90].
Tempol, NOX-101.
In SMCs exposed to HG the antioxidant Tempol prevented the effects of HG. Experiments with SERCA C674 mutants indicated that the redox regulation of cysteine-674 was the key to the restoration of NO function. Overexpression of SERCA may achieve part of its therapeutic effect by mechanisms involving redox regulation of Cys674, which can be preserved by antioxidants such as Tempol and BHT [Citation45,Citation91]. Long-term administration of NOX-101, an .NO scavenger, prevented the impairment of endothelial function seen in aortas from streptozotocin (S)-induced diabetic rats [Citation91].
Enalapril.
In the streptozotocin-induced diabetic rat model, enalapril (the angiotensin-converting enzyme inhibitor) helped to prevent the diabetes–related impairment of SERCA function in the aorta by controlling the excess ROS formation and normalizing the impaired aortic relaxation response to NO in diabetic rats [Citation92]. The angiotensin II level found in the diabetic aorta probably impaired SERCA function and this impairment led to a reduction in the relaxation normally induced by SERCA. Chronic treatment of diabetic rats with enalapril significantly improved relaxation in the diabetic aorta [Citation44].
Gene therapy.
Cardiomyopathy caused by reduced contractility is associated with changes in calcium handling within myocytes in the diabetic state. Most of the calcium involved in contraction of myocytes is derived from the sarcoplasmic reticulum, and SERCA activity has been reported to be reduced in diabetes. SR function/calcium handling can be improved by the expression of SERCA and various other proteins using gene tranfer (e.g. adenovirus backbone as a vector for gene transfer). The advantage of using transgenic overexpression is in the application of different promoters in front of the target gene (e.g. SERCA), which enhance expression of the target gene, independently of the natural promoter which may be subject to downregulation during the diabetic state. Improvement of the SERCA level/activity by using gene therapy may reduce/remove the contractile phenotype associated with the diabetic state.
SERCA2b protein and mRNA levels are dramatically reduced in the liver of obese mice. Overexpression of SERCA2b from Adenovirus-SERCA2b construct in the liver of obese and diabetic mice alleviates ER stress, increases glucose tolerance, and significantly reduces the blood glucose level [Citation93]. In a type 2 diabetes rat model, left ventricular diastolic dysfunction characterized by a slow rate of ventricular relaxation is accounted for by decreased SERCA2a protein expression. Transcoronary gene transfer with an adenoviral vector to overexpress SERCA2a increased coronary blood flow and decreased cardiomyocyte hypertrophy [Citation94]. Currently, a number of clinical trials of SERCA2a gene therapy are being performed for treatment of patients with heart failure [Citation95,Citation96].
Kallikrein.
Kallikrein-kinin system (KKS) components are locally expressed in the heart [Citation97], and streptozotocin-iduced diabetes results in a decrease of active cardiac tissue kallikrein levels [Citation98], in turn resulting in increased thickness of the left ventricle wall and cardiac hypertrophy [Citation99]. Expression of human tissue kallikrein from Adenovirus-SERCA2a construct in rats increases SERCA2a and phosphorylated phospholamban levels and reduces elevated blood glucose levels induced by streptozotocin treatment of rats.
Ca2+-ATPases modulate insulin secretion
Leucine, which induces insulin secretion in the absence of glucose, suppressed pancreatic islet Ca2+-ATPase activity [Citation100]. Kulkarni et al. [Citation101] reported that deletion of IRS-1 (insulin receptor substrate) in knockout mice islets dramatically reduced expression of SERCA2b and − 3 genes. Furthermore, some studies demonstrated that IRS-1 and SERCA were localized in ER vesicles from beta-cells and could interact directly with one another [Citation102]. These authors found that pharmacological inhibition of SERCA in beta-cells resulted in enhanced secretion of insulin [Citation102] and chronic activation of insulin receptor signaling by IRS-1 overexpression in beta-cells inhibited gene expression of SERCA [Citation103]. In insulin-dependent diabetes of acute and chronic streptozotocin rat models a 30% decrease of the SERCA mRNA level was also described [Citation104]. These findings support the hypothesis that Ca2+-ATPases are involved in the specificity of islet response and in insulin secretion.
Critical comments on preventing effects of hyperglycemia
To summarize, in the prevention against glucose-induced Ca2+-ATPase, several mechanisms can operate: 1. Restoration of Ca2+-ATPase level by SERCA expression (rosiglitazone and pioglitazone), 2. Increasing SERCA activity. SERCA activity may be increased by lowering lipid peroxidation, or by other antioxidant effects, furthermore also by knock out of phospholamban expression, which decreases the affinity of SERCA for calcium. 3. Ability of some agents to react with glucose, as found with taurine and rosiglitazone. In this respect it is necessary to take into consideration also the side effects of the compounds decreasing the glucose level. For example, increased vascular complications and mortality of rosiglitazone-treated patients were reported [Citation105]. Even glucose level lowered by insulin was shown to increase myocardial injury (cardiac troponin I) and inflammation (high sensitivity CRP) markers [Citation106]. The authors observed that when the glucose-lowering rate is too high, the inflammatory system may be overactivated and can induce myocardial injury and adverse effects on the cardiovascular system. Thus, vascular/cardiac complications should be considered before using of compounds decreasing the level of blood glucose as it is difficult to precisely control fluctuations of glucose concentration in blood. 4. Modulation of enzyme levels related to oxidative stress. Good examples are the expression of catalase and SOD, and inhibition or inhibited expression of AR, SDH, and NADH oxidase. However, a certain level of ROS is crucial to keep a normal function of enzymes. Imbalance in oxidative status of particular cells/tissue may result in metabolic and calcium distribution defects, which results in onset of pathological processess. Additionally, there is evidence that decreasing the level of ROS may not be beneficial in cancer cells, where increased ROS is crucial for suppression of proliferation. 5. Improvement of the SERCA level/activity by using gene therapy. The major advantage of the gene therapy is its relative inexpensiveness and the fact that high levels of target gene can be reached. However, possible integration of the transgene into the site in the genome which is crucial for tissue specificity or function belong to disadvantages of gene therapy. Moreover, levels of transgene expression may be affected by site of transgene integration. In addition, gene delivery systems carrying inserted genes can induce immune responses, which may result in nonefficient gene expression or side effects [Citation107]. So far, agents with different preventive mechanisms of action against Ca2+-ATPase glycation have not been not evaluated in the same experimental model. Therefore it is not possible to compare experimentally their preventive effects against glycation or the effects of HG treatment. It can be supposed that compounds with manifold effects may be most effective. An example of an agent with manifold effect is taurine, which prevented reduction of Ca2+-ATPase by stimulating the turnover rate of the pump, exhibited high reactivity with glucose and attenuated appoptosis via ROS inhibition and Ca2+ stabilization. Another example of a manifold effect may be GTE. Oral treatment of diabetic rats with GTE lowered the level of blood glucose, lipid peroxidation and protein glycation in the heart, improved the decrease of Ca2+-ATPase activity and normalized the Ca2+ concentration in the heart.
Declaration of interest
The authors report no declarations of interest. The authors alone are responsible for the content and writing of the paper.
This work was supported by COST action CM 1001, by VEGA 2/0038/11, and by The Agency of the Ministry of Education, Science, Research and Sport of the Slovak Republic for the Structural Funds of EU, OP R&D of ERDF by realization of the Project Evaluation of natural substances and their selection for prevention and treatment of lifestyle diseases (ITMS 26240220040).
References
- Squier TC. Oxidative stress and protein aggregation during biological aging. Exp Gerontol 2001;36:1539–1550.
- Squier TC, Bigelow DJ. Protein oxidation and age-dependent alterations in calcium homeostasis. Front Biosci 2000;5: 504–526.
- Levine RL, Mosoni L, Berlett BS, Stadtman ER. Methionine residues as endogenous antioxidants in proteins. Proc Natl Acad Sci U S A 1996;93:15036–15040.
- Moreau R, Heath SHD, Doneanu CE, Lindsay JG, Hagen TM. Age-related increase in 4-hydroxynonenal adduction to rat heart alpha-ketoglutarate dehydrogenase does not cause loss of its catalytic activity. Antioxid Redox Sign 2003;5:517–527.
- Gao J, Yin DH, Yao Y, Sun H, Qin Z, Schöneich C, et al. Loss of conformational stability in calmodulin upon methionine oxidation. Biophys J 1998;74:1115–1134.
- Viner RI, Ferrington DA, Williams TD, Bigelow DJ, Schoneich C. Protein modification during biological aging: selective tyrosine nitration of the SERCA2a isoform of the sarcoplasmic reticulum Ca2+-ATPase in skeletal muscle. Biochem J 1999;340:657–669.
- Chen B, Jones TE, Bigelow DJ. The nucleotide-binding site of the sarcoplasmic reticulum Ca-ATPase is conformationally altered in aged skeletal muscle. Biochemistry 1999; 38:14887–14896.
- Bers DM. Calcium fluxes involved in control of cardiac myocyte contraction. Circ Res 2000;87:275–281.
- Adachi T. Modulation of vascular sarco/endoplasmic reticulum calcium ATPase in cardiovascular pathophysiology. Adv Pharmacol 2010;59:165–195.
- Brini M, Carafoli E. Calcium pumps in health and disease. Physiol Rev 2009;89:1341–1378.
- MacLennan DH, Rice WJ, Green NM. The mechanism of Ca2+ transport by sarco(endo)plasmic reticulum Ca2+-ATPases. J Biol Chem 1997;272:28815–28818.
- Magnier-Gaubil C, Herbert JM, Quarck R, Papp B, Corvazier E, Wuytack F, et al. Smooth muscle cell cycle and proliferation. Relationship between calcium influx and sarco-endoplasmic reticulum Ca2+ATPase regulation. J Biol Chem 1996;271:27788–27794.
- Magnier C, Papp B, Corvazier E, Bredoux R, Wuytack F, Eggermont J, et al. Regulation of sarco-endoplasmic reticulum Ca(2+)-ATPases during platelet-derived growth factor-induced smooth muscle cell proliferation. J Biol Chem 1992;267:15808–15815.
- Dode L, Vilsen B, Van Baelen K, Wuytack F, Clausen JD, Andersen JP. Dissection of the functional differences between sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA) 1 and 3 isoforms by steady-state and transient kinetic analyses. J Biol Chem 2002;277:45579–45591.
- Hovnanian A. SERCA pumps and human diseases. Subcell Biochem 2007;45:337–363.
- Tang WH, Cheng WT, Kravtsov GM, Tong XY, Hou XY, Chung SK, Chung SS. Cardiac contractile dysfunction during acute hyperglycemia due to impairment of SERCA by polyol pathway-mediated oxidative stress. Am J Physiol Cell Physiol 2010;299:643–653.
- Inesi G, Hua S, Xu C, Ma H, Seth M, Prasad AM, Sumbilla C. Studies of Ca2+ ATPase (SERCA) inhibition. J Bioenerg Biomembr 2005;37:365–368.
- Díaz ME, Graham HK, O’Neill SC, Trafford AW, Eisner DA. The control of sarcoplasmic reticulum Ca content in cardiac muscle. Cell Calcium 2005;38:391–396.
- Belke DD, Dillmann WH. Altered cardiac calcium handling in diabetes. Curr Hypertens Rep 2004;6:424–429.
- Adachi T, Weisbrod RM, Pimentel DR, Ying J, Sharov VS, Schöneich C, Cohen RA. S-Glutathiolation by peroxynitrite activates SERCA during arterial relaxation by nitric oxide. Nat Med 2004;10:1200–1207.
- Lipskaia L, del Monte F, Capiod T, Yacoubi S, Hadri L, Hours M, et al. Sarco/endoplasmic reticulum Ca2+-ATPase gene transfer reduces vascular smooth muscle cell proliferation and neointima formation in the rat. Circ Res 2005;97: 488–495.
- Sharov VS, Dremina ES, Galeva NA, Williams TD, Schoneich C. Quantitative mapping of oxidation-sensitive cysteine residues in SERCA in vivo and in vitro by HPLC-electrospray-tandem MS: selective protein oxidation during biological aging. Biochem J 2006;394:605–615.
- Ying J, Sharov V, Xu S, Jiang B, Gerrity R, Schoneich C, Cohen RA. Cysteine-674 oxidation and degradation of sarcoplasmic reticulum Ca(2+) ATPase in diabetic pig aorta. Free Radic Biol Med 2008;45:756–762.
- Dhalla NS, Temsah RM, Netticadan T. Role of oxidative stress in cardiovascular diseases. J Hypertens 2000;18: 655–673.
- Knyushko TV, Sharov VS, Williams TD, Schoneich C, Bigelow DJ. 3-Nitrotyrosine modification of SERCA2a in the aging heart: a distinct signature of the cellular redox environment. Biochemistry 2005;44:13071–13081.
- Adachi T, Matsui R, Xu S, Kirber M, Lazar HL, Sharov VS, et al. Antioxidant improves smooth muscle sarco/endoplasmic reticulum Ca(2+)-ATPase function and lowers tyrosine nitration in hypercholesterolemia and improves nitric oxide-induced relaxation. Circ Res 2002;90:1114–1121.
- Xu S, Ying J, Jiang B, Guo W, Adachi T, Sharov V, et al. Detection of sequence-specific tyrosine nitration of manganese SOD and SERCA in cardiovascular disease and aging. Am J Physiol Heart Circ Physiol 2006;290: H2220–2227.
- Schoneich C, Sharov VS. Mass spectrometry of protein modifications by reactive oxygen and nitrogen species. Free Radic Biol Med 2006;41:1507–1520.
- Lounsbury KM, Hu Q, Ziegelstein RC. Calcium signaling and oxidant stress in the vasculature. Free Radic Biol Med 2000;28:1362–1369.
- Chung SSM, Chung SK. Aldose reductase in diabetic microvascular complications. Curr Drug Targets 2005;6: 475–486.
- Srivastava SK, Ramana KV, Bhatnagar A. Role of aldose reductase and oxidative damage in diabetes and the consequent potential for therapeutic options. Endocr Rev 2005;26:380–392.
- Chung SSM, Chung SK. Genetic analysis of aldose reductase in diabetic complications. Curr Med Chem 2003;10: 1375–1387.
- Lee AY, Chung SS. Contributions of polyol pathway to oxidative stress in diabetic cataract. FASEB J 1999;13: 23–30.
- Bassenge E, Sommer O, Schwemmer M, Bünger R. Antioxidant pyruvate inhibits cardiac formation of reactive oxygen species through changes in redox state. Am J Physiol Heart Circ Physiol 2000;279:2431–2438.
- Mohazzab-H KM, Kaminski PM, Wolin MS. Lactate and PO2 modulate superoxide anion production in bovine cardiac myocytes: potential role of NADH oxidase. Circulation 1997;96:614–620.
- Xu KY, Zweier JL, Becker LC. Hydroxyl radical inhibits sarcoplasmic reticulum Ca(2+)-ATPase function by direct attack on the ATP binding site. Circ Res 1997;80:76–81.
- Ramasamy R, Oates PJ, Schaefer S. Aldose reductase inhibition protects diabetic and nondiabetic rat hearts from ischemic injury. Diabetes 1997;46:292–300.
- Oswald GA, Smith CC, Betteridge DJ, Yudkin JS. Determinants and importance of stress hyperglycaemia in non-diabetic patients with myocardial infarction. Br Med J (Clin Res Ed) 1986;293:917–922.
- Tang WH, Wu S, Wong TM, Chung SK, Chung SSM. Polyol pathway mediates iron-induced oxidative injury in ischemic-reperfused rat heart. Free Radic Biol Med 2008; 45:602–610.
- Tang WH, Kravtsov GM, Sauert M, Tong XY, Hou XY, Wong TM, et al. Polyol pathway impairs the function of SERCA and RyR in ischemic-reperfused rat hearts by increasing oxidative modifications of these proteins. J Mol Cell Cardiol 2010;49:58–69.
- Ceriello A, Quagliaro L, D’Amico M, Di Filippo C, Marfella R, Nappo F, et al. Acute hyperglycemia induces nitrotyrosine formation and apoptosis in perfused heart from rat. Diabetes 2002;51:1076–1082.
- Monnier L, Mas E, Ginet C, Michel F, Villon L, Cristol JP, Colette C. Activation of oxidative stress by acute glucose fluctuations compared with sustained chronic hyperglycemia in patients with type 2 diabetes. JAMA 2006;295: 1681–1687.
- Ying J, Tong X, Pimentel DR, Weisbrod RM, Trucillo MP, Adachi T, Cohen RA. Cysteine-674 of the sarco/endoplasmic reticulum calcium ATPase is required for the inhibition of cell migration by nitric oxide. Arterioscler Thromb Vasc Biol 2007;27:783–790.
- Tong X, Evangelista A, Cohen RA. Targeting the redox regulation of SERCA in vascular physiology and disease. Curr Opin Pharmacol 2010;10:133–138.
- Tong X, Ying J, Pimentel DR, Trucillo M, Adachi T, Cohen RA. High glucose oxidizes SERCA cysteine-674 and prevents inhibition by nitric oxide of smooth muscle cell migration. J Mol Cell Cardiol 2008;44:361–369.
- Tong X, Hou X, Jourd'heuil D, Weisbrod RM, Cohen RA. Upregulation of Nox4 by TGF{beta}1 oxidizes SERCA and inhibits NO in arterial smooth muscle of the prediabetic Zucker rat. Circ Res 2010;107:975–983.
- Kobayashi T, Taguchi K, Takenouchi Y, Matsumoto T, Kamata K. Insulin-induced impairment via peroxynitrite production of endothelium-dependent relaxation and sarco/endoplasmic reticulum Ca(2+)-ATPase function in aortas from diabetic rats. Free Radic Biol Med 2007;43: 431–443.
- Pandolfi A, Iacoviello L, Capani F, Vitacolonna E, Donati MB, Consoli A. Glucose and insulin independently reduce the fibrinolytic potential of human vascular smooth muscle cells in culture. Diabetologia 1996;39:1425–1431.
- Kobayashi T, Oishi K, Hayashi Y, Matsumoto T, Kamata K. Changes in aortic endothelial gene expressions and relaxation responses following chronic short-term insulin treatment in diabetic rats. Atherosclerosis 2006;185: 47–57.
- Randriamboavonjy V, Pistrosch F, Bolck B, Schwinger RHG, Dixit M, Badenhoop K, et al. Platelet sarcoplasmic endoplasmic reticulum Ca2+-ATPase and mu-calpain activity are altered in type 2 diabetes mellitus and restored by rosiglitazone. Circulation 2008;117:52–60.
- Schaefer W, Beeker J, Gries FA. Influence of hyperglycemia on Ca2+-Mg2+ -ATPase of red blood cells from diabetic patients. Klin Wochenschr 1988;66:443–446.
- Schaefer W, Priessen J, Mannhold R, Gries AF. Ca2+-Mg2+-ATPase activity of human red blood cells in healthy and diabetic volunteers. Klin Wochenschr 1987;65:17–21.
- González Flecha FL, Bermúdez MC, Cédola N V, Gagliardino JJ, Rossi JP. Decreased Ca2(+)-ATPase activity after glycosylation of erythrocyte membranes in vivo and in vitro. Diabetes 1990;39:707–711.
- Gonzalez Flecha FL, Castello PR, Caride AJ, Gagliardino JJ, Rossi JP. The erythrocyte calcium pump is inhibited by non-enzymic glycation: studies in situ and with the purified enzyme. Biochem J 1993;293:369–375.
- González Flecha FL, Castello PR, Gagliardino JJ, Rossi JP. Molecular characterization of the glycated plasma membrane calcium pump. J Membr Biol 1999;171:25–34.
- Bidasee KR, Zhang Y, Shao CH, Wang M, Patel KP, Dincer UD, Besch HR Jr. Diabetes increases formation of advanced glycation end products on Sarco(endo)plasmic reticulum Ca2+-ATPase. Diabetes 2004;53:463–473.
- Raftos JE, Edgley A, Bookchin RM, Etzion Z, Lew VL, Tiffert T. Normal Ca2+ extrusion by the Ca2+ pump of intact red blood cells exposed to high glucose concentrations. Am J Physiol Cell Physiol 2001;280:1449–1454.
- Bookchin RM, Etzion Z, Lew VL, Tiffert T. Preserved function of the plasma membrane calcium pump of red blood cells from diabetic subjects with high levels of glycated haemoglobin. Cell Calcium 2009;45:260–263.
- Rossi JPFC, Villamil AM, Echarte MM, Alzugaray ME, Borelli MI, García ME, et al. Plasma membrane calcium pump activity in rat pancreatic islets: an accurate method to measure its calcium-dependent modulation. Cell Biochem Biophys 2006;46:193–200.
- Herchuelz A, Kamagate A, Ximenes H, Van Eylen F. Role of Na/Ca exchange and the plasma membrane Ca2+-ATPase in beta cell function and death. Ann N Y Acad Sci 2007; 1099:456–467.
- Levi V, Villamil Giraldo AM, Castello PR, Rossi JPFC, González Flecha FL. Effects of phosphatidylethanolamine glycation on lipid-protein interactions and membrane protein thermal stability. Biochem J 2008;416:145–152.
- Ulrich P, Cerami A. Protein glycation, diabetes, and aging. Recent Prog Horm Res 2001;56:1–21.
- Cho S-J, Roman G, Yeboah F, Konishi Y. The road to advanced glycation end products: a mechanistic perspective. Curr Med Chem 2007;14:1653–1671.
- Oliver CM, Melton LD, Stanley RA. Creating proteins with novel functionality via the Maillard reaction: a review. Crit Rev Food Sci Nutr 2006;46:337–350.
- Reddi AS, Dasmahapatra A, Jyothirmayi GN, Jayasundaramma B. Erythrocyte Ca, Na/K-ATPase in long-term streptozotocin diabetic rats. Effect of good glycemic control and a Ca antagonist. Am J Hypertens 1992;5:863–868.
- Norby FL, Wold LE, Duan J, Hintz KK, Ren J. IGF-I attenuates diabetes-induced cardiac contractile dysfunction in ventricular myocytes. Am J Physiol Endocrinol Metab 2002;283:658–666.
- Li Q, Wu S, Li S-Y, Lopez FL, Du M, Kajstura J, et al. Cardiac-specific overexpression of insulin-like growth factor 1 attenuates aging-associated cardiac diastolic contractile dysfunction and protein damage. Am J Physiol Heart Circ Physiol 2007;292:1398–1403.
- Nandhini TA, Anuradha CV. Inhibition of lipid peroxidation, protein glycation and elevation of membrane ion pump activity by taurine in RBC exposed to high glucose. Clin Chim Acta 2003;336:129–135.
- Wu QD, Wang JH, Fennessy F, Redmond HP, Bouchier-Hayes D. Taurine prevents high-glucose-induced human vascular endothelial cell apoptosis. Am J Physiol 1999;277:1229–1238.
- Ogasawara M, Nakamura T, Koyama I, Nemoto M, Yoshida T. Reactivity of taurine with aldehydes and its physiological role. Adv Exp Med Biol 1994;359:71–78.
- Franconi F, Bennardini F, Mattana A, Miceli M, Ciuti M, Mian M, et al. Plasma and platelet taurine are reduced in subjects with insulin-dependent diabetes mellitus: effects of taurine supplementation. Am J Clin Nutr 1995;61: 1115–1119.
- Kim JY, Lim DM, Park HS, Moon CI, Choi KJ, Lee SK, et al. Exendin-4 protects against sulfonylurea-induced β-cell apoptosis. J Pharmacol Sci 2012;118:65–74.
- Kamboj SS, Chopra K, Sandhir R. Hyperglycemia-induced alterations in synaptosomal membrane fluidity and activity of membrane bound enzymes: beneficial effect of N-acetylcysteine supplementation. Neuroscience 2009;162: 349–358.
- Kim HW, Ch YS, Lee HR, Park SY, Kim YH. Diabetic alterations in cardiac sarcoplasmic reticulum Ca2+-ATPase and phospholamban protein expression. Life Sci 2001;70: 367–379.
- Brittsan AG, Carr AN, Schmidt AG, Kranias EG. Maximal inhibition of SERCA2 Ca(2+) affinity by phospholamban in transgenic hearts overexpressing a non-phosphorylatable form of phospholamban. J Biol Chem 2000;275:12129–12135.
- Lehmann JM, Moore LB, Smith-Oliver TA, Wilkison WO, Willson TM, Kliewer SA. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor gamma (PPAR gamma). J Biol Chem 1995;270: 12953–12956.
- Kono T, Ahn G, Moss DR, Gann L, Zarain-Herzberg A, Nishiki Y, et al. PPAR-γ activation restores pancreatic islet SERCA2 levels and prevents β-cell dysfunction under conditions of hyperglycemic and cytokine stress. Mol Endocrinol 2012;26:257–271.
- Shah RD, Gonzales F, Golez E, Augustin D, Caudillo S, Abbott A, et al. The antidiabetic agent rosiglitazone upregulates SERCA2 and enhances TNF-alpha- and LPS-induced NF-kappaB-dependent transcription and TNF-alpha-induced IL-6 secretion in ventricular myocytes. Cell Physiol Biochem 2005;15:41–50.
- Jardin I, Redondo PC, Salido GM, Pariente JA, Rosado JA. Endogenously generated reactive oxygen species reduce PMCA activity in platelets from patients with non- insulin-dependent diabetes mellitus. Platelets 2006;17: 283–288.
- Ren J, Li Q, Wu S, Li SY, Babcock SA. Cardiac overexpression of antioxidant catalase attenuates aging-induced cardiomyocyte relaxation dysfunction. Mech Ageing Dev 2007;128:276–285.
- Babu PVA, Sabitha KE, Shyamaladevi CS. Therapeutic effect of green tea extract on oxidative stress in aorta and heart of streptozotocin diabetic rats. Chem Biol Interact 2006;162:114–120.
- Arnér ES, Nordberg J, Holmgren A. Efficient reduction of lipoamide and lipoic acid by mammalian thioredoxin reductase. Biochem Biophys Res Commun 1996;225:268–274.
- Constantinescu A, Pick U, Handelman GJ, Haramaki N, Han D, Podda M, et al. Reduction and transport of lipoic acid by human erythrocytes. Biochem Pharmacol 1995;50: 253–261.
- Jain SK, Lim G. Lipoic acid decreases lipid peroxidation and protein glycosylation and increases (Na(+) + K(+))- and Ca(++)-ATPase activities in high glucose-treated human erythrocytes. Free Radic Biol Med 2000;29:1122–1128.
- Vasanji Z, Cantor EJF, Juric D, Moyen M, Netticadan T. Alterations in cardiac contractile performance and sarcoplasmic reticulum function in sucrose-fed rats is associated with insulin resistance. Am J Physiol Cell Physiol 2006;291: 772–780.
- Belke DD, Swanson EA, Dillmann WH. Decreased sarcoplasmic reticulum activity and contractility in diabetic db/db mouse heart. Diabetes 2004;53:3201–3208.
- Wang M, Zhang WB, Zhu JH, Fu GS, Zhou BQ. Breviscapine ameliorates cardiac dysfunction and regulates the myocardial Ca(2+)-cycling proteins in streptozotocin- induced diabetic rats. Acta diabetol 2010;47:209–218.
- Pekiner B, Ulusu NN, Das-Evcimen N, Sahilli M, Aktan F, Stefek M, et al. In vivo treatment with stobadine prevents lipid peroxidation, protein glycation and calcium overload but does not ameliorate Ca2+ -ATPase activity in heart and liver of streptozotocin-diabetic rats: comparison with vitamin E. Biochim Biophys Acta 2002;1588:71–78.
- Das Evcimen N, Ulusu NN, Karasu C, Doğru B. Adenosine triphosphatase activity of streptozotocin-induced diabetic rat brain microsomes. Effect of vitamin E. Gen Physiol Biophys 2004;23:347–355.
- Kowluru RA, Kern TS, Engerman RL, Armstrong D. Abnormalities of retinal metabolism in diabetes or experimental galactosemia. III. Effects of antioxidants. Diabetes 1996;45:1233–1237.
- Pieper GM, Dembny K, Siebeneich W. Long-term treatment in vivo with NOX-101, a scavenger of nitric oxide, prevents diabetes-induced endothelial dysfunction. Diabetologia 1998;41:1220–1226.
- Taguchi K, Kobayashi T, Hayashi Y, Matsumoto T, Kamata K. Enalapril improves impairment of SERCA- derived relaxation and enhancement of tyrosine nitration in diabetic rat aorta. Eur J Pharmacol 2007;556:121–128.
- Park SW, Zhou Y, Lee J, Lee J, Ozcan U. Sarco(endo)plasmic reticulum Ca2+-ATPase 2b is a major regulator of endoplasmic reticulum stress and glucose homeostasis in obesity. Proc Natl Acad Sci U S A 2010;107:19320–19325.
- Sakata S, Lebeche D, Sakata Y, Sakata N, Chemaly ER, Liang L, et al. Transcoronary gene transfer of SERCA2a increases coronary blood flow and decreases cardiomyocyte size in a type 2 diabetic rat model. Am J Physiol Heart Circ Physiol 2007;292:1204–1207.
- Levitsky DO, Clergue M, Lambert F, Souponitskaya MV, Le Jemtel TH, Lecarpentier Y, et al. Sarcoplasmic reticulum calcium transport and Ca(2+)-ATPase gene expression in thoracic and abdominal aortas of normotensive and spontaneously hypertensive rats. J Biol Chem 1993;268: 8325–8331.
- Jaski BE, Jessup ML, Mancini DM, Cappola TP, Pauly DF, Greenberg B, et al; Calcium Upregulation by Percutaneous Administration of Gene Therapy in Cardiac Disease (CUPID) Trial Investigators. Calcium upregulation by percutaneous administration of gene therapy in cardiac disease (CUPID Trial), a first-in-human phase clinical trial. J Card Fail 2009;15:171–181.
- Nolly HL, Saed G, Scicli G, Carretero OA, Scicli AG. The kallikrein-kinin system in cardiac tissue. Agents Actions Suppl 1992;38:62–72.
- Tschope C, Walther T, Yu M, Reinecke A, Koch M, Seligmann C, et al. Myocardial expression of rat bradykinin receptors and two tissue kallikrein genes in experimental diabetes. Immunopharmacology 1999;44:35–42.
- Sharma JN, Uma K, Yusof AP. Left ventricular hypertrophy and its relation to the cardiac kinin-forming system in hypertensive and diabetic rats. Int J Cardiol 1998;63:229–235.
- Levin SR, Kasson BG, Driessen JF. Adenosine triphosphatases of rat pancreatic islets: comparison with those of rat kidney. J Clin Invest 1978;62:692–701.
- Kulkarni RN, Roper MG, Dahlgren G, Shih DQ, Kauri LM, Peters JL, et al. Islet secretory defect in insulin receptor substrate 1 null mice is linked with reduced calcium signaling and expression of sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA)-2b and -3. Diabetes 2004;53:1517–1525.
- Borge PD, Wolf BA. Insulin receptor substrate 1 regulation of sarco-endoplasmic reticulum calcium ATPase 3 in insulin-secreting beta-cells. J Biol Chem 2003;278:11359–11368.
- Xu GG, Gao ZY, Borge PD, Jegier PA, Young RA, Wolf BA. Insulin regulation of beta-cell function involves a feedback loop on SERCA gene expression, Ca(2+) homeostasis, and insulin expression and secretion. Biochemistry 2000;39: 14912–14919.
- Depre C, Young ME, Ying J, Ahuja HS, Han Q, Garza N, et al. Streptozotocin-induced changes in cardiac gene expression in the absence of severe contractile dysfunction. J Mol Cell Cardiol 2000;32:985–996.
- Chen X, Yang L, Zhai S. Risk of cardiovascular disease and all-cause mortality among diabetic patients prescribed rosiglitazone or pioglitazone: a meta-analysis of retrospective cohort studies. Chin Med J 2012;125:4301–4306.
- Wu W, Li Q, Xia J, Wang M, Sun Z, Miao J, Zheng Z. Effects of the glucose-lowering rate on cTnI and hs-CRP serum levels in type 2 diabetics. Hum Immunol 2013;74: 379–382.
- Zhou H, Liu D, Liang C. Challenges and strategies: the immune responses in gene therapy. Med Res Rev 2004;24:748–761.
- Dode L, Andersen JP, Leslie N, Dhitavat J, Vilsen B, Hovnanian A. Dissection of the functional differences between sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA) 1 and 2 isoforms and characterization of Darier disease (SERCA2) mutants by steady-state and transient kinetic analyses. J Biol Chem 2003;278:47877–47889.